
Sesquiterpene Lactones: Promising Natural Compounds to Fight Inflammation


Abstract
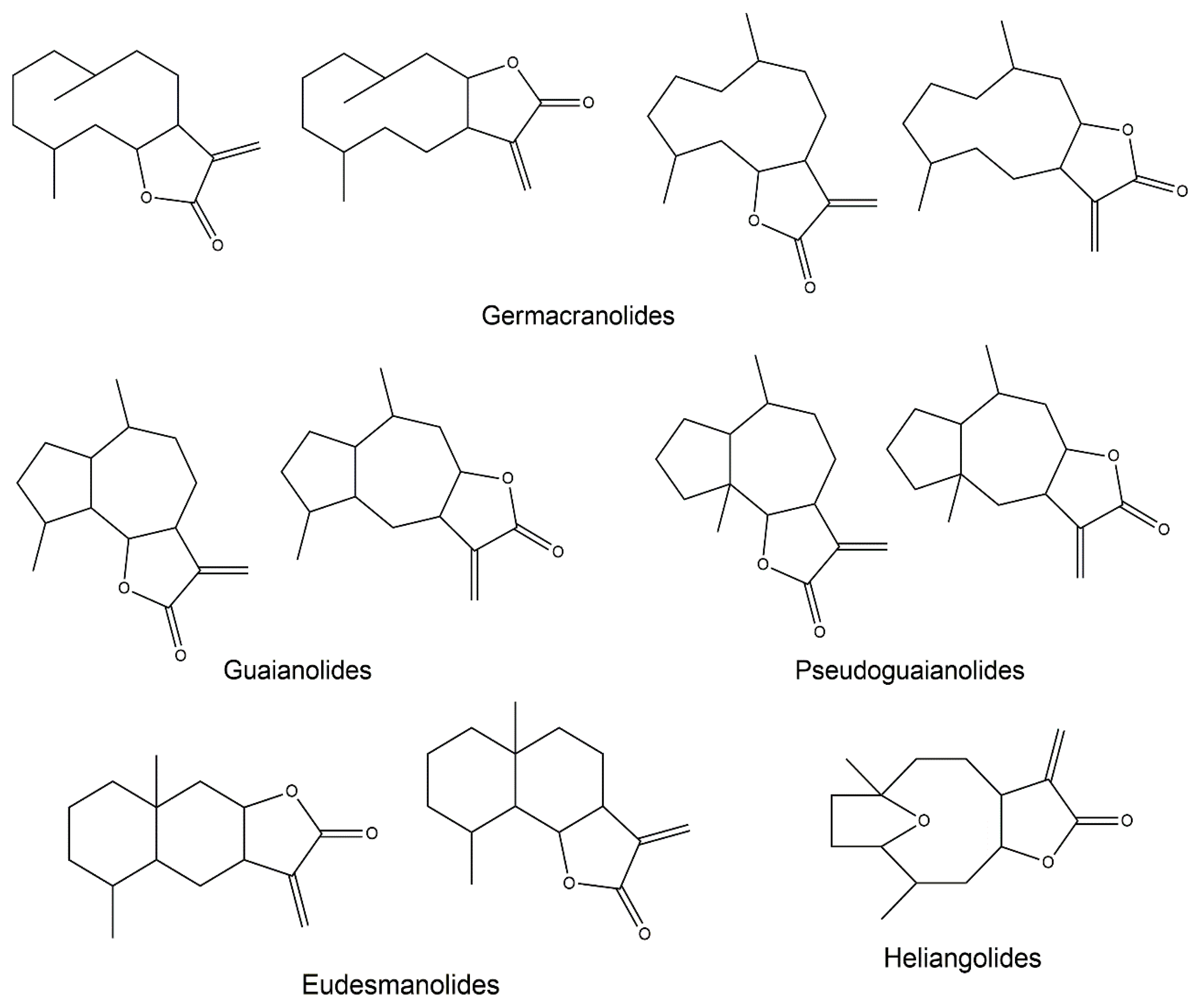
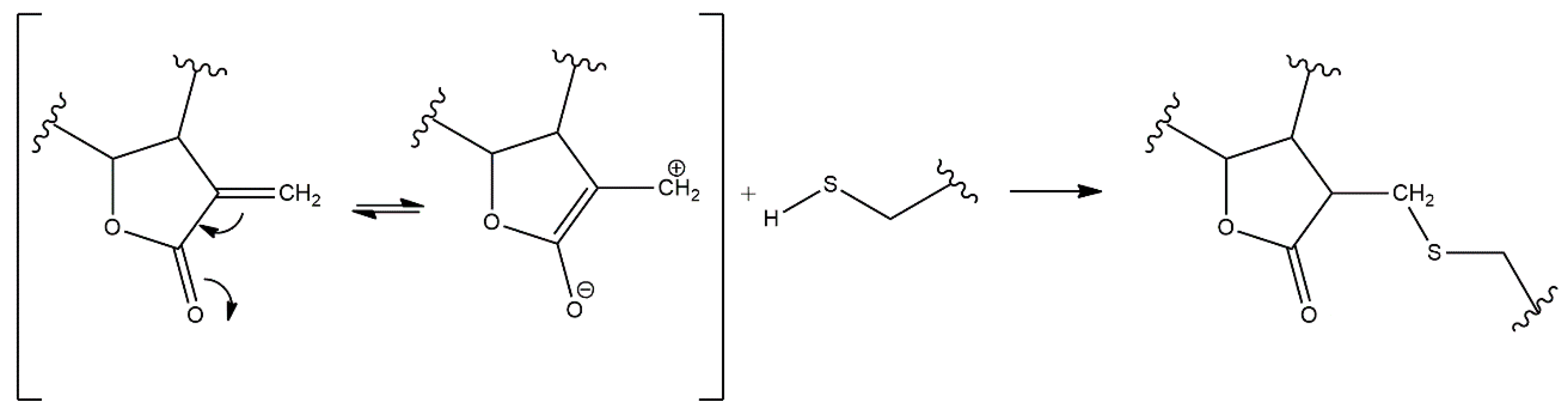
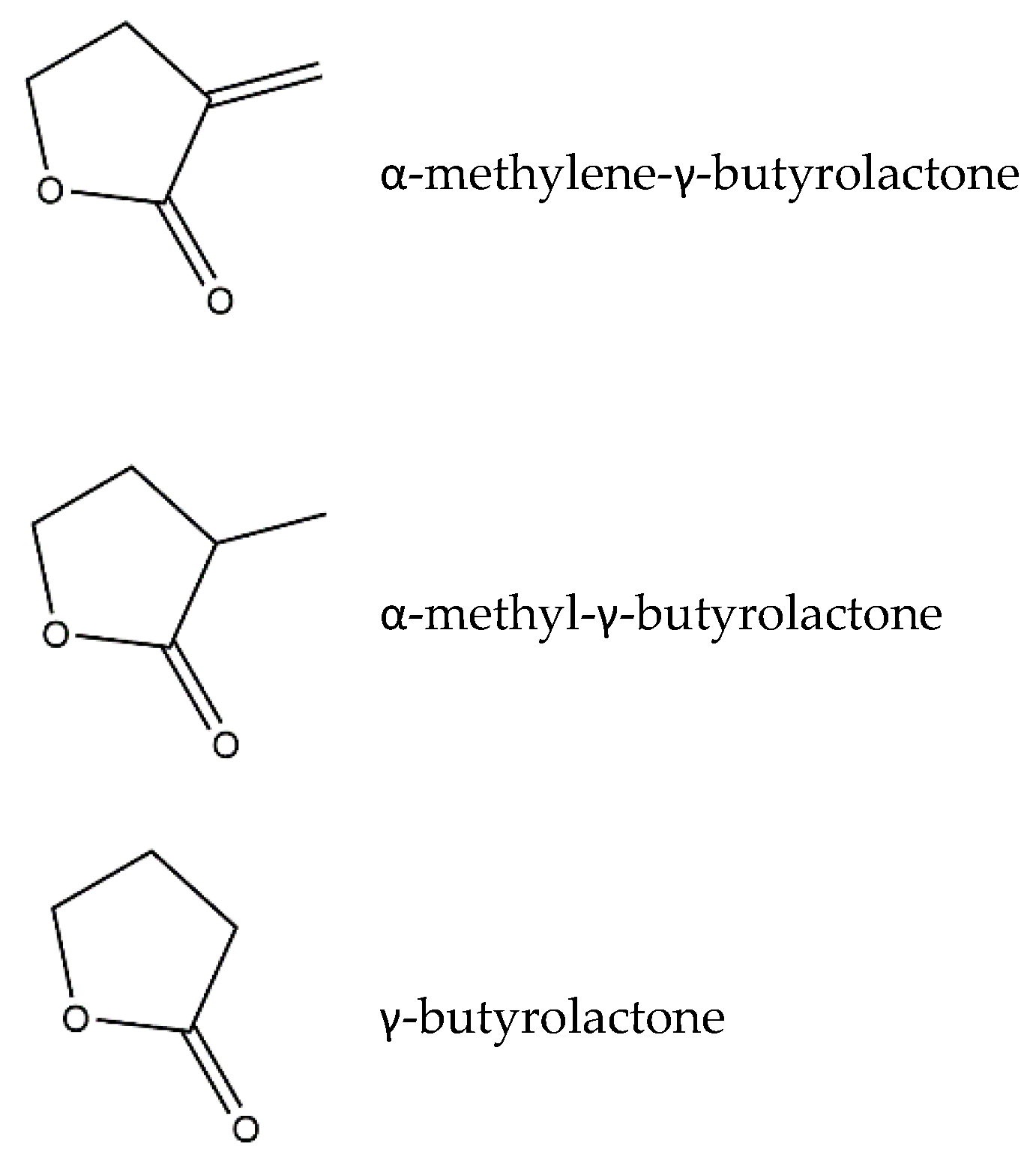
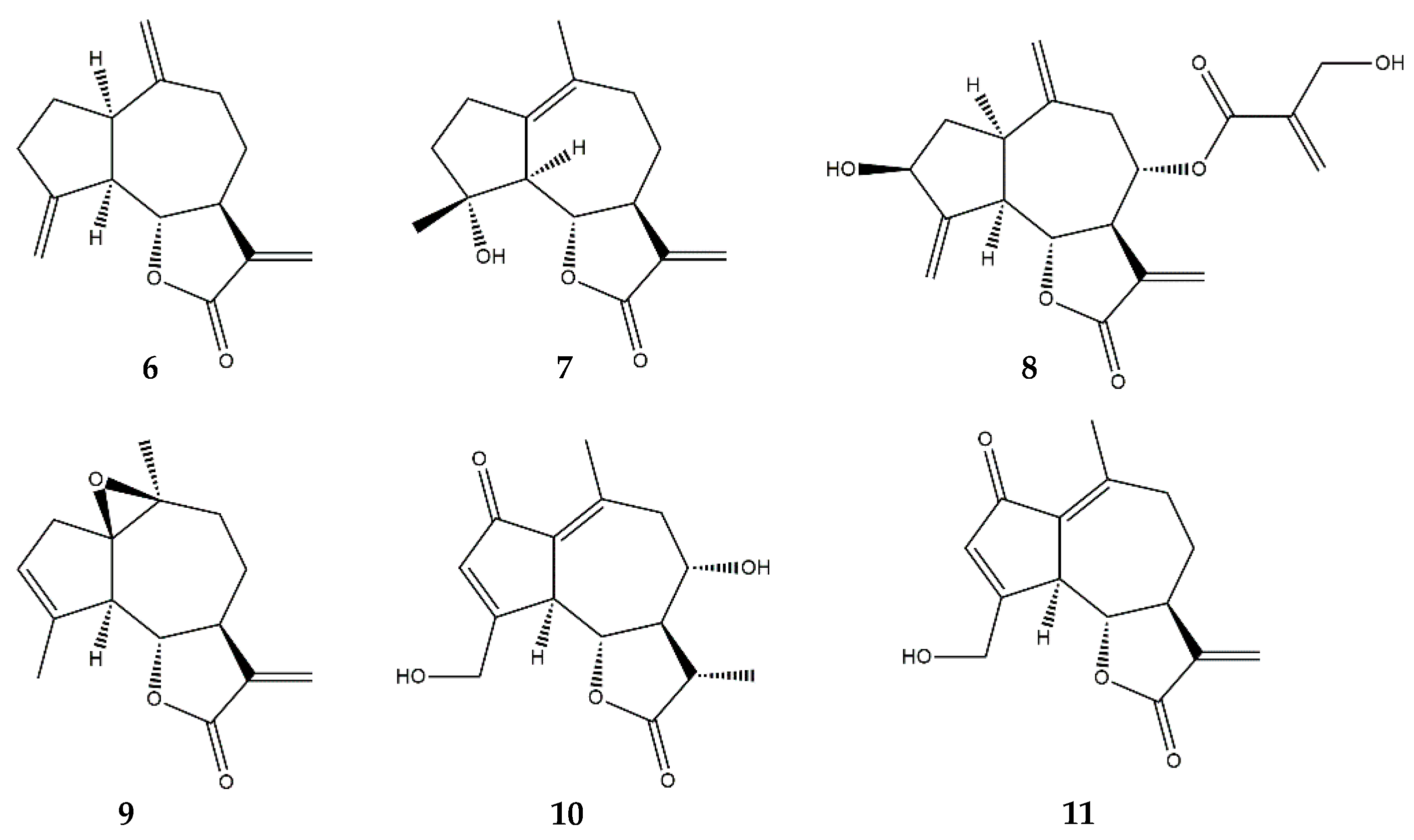
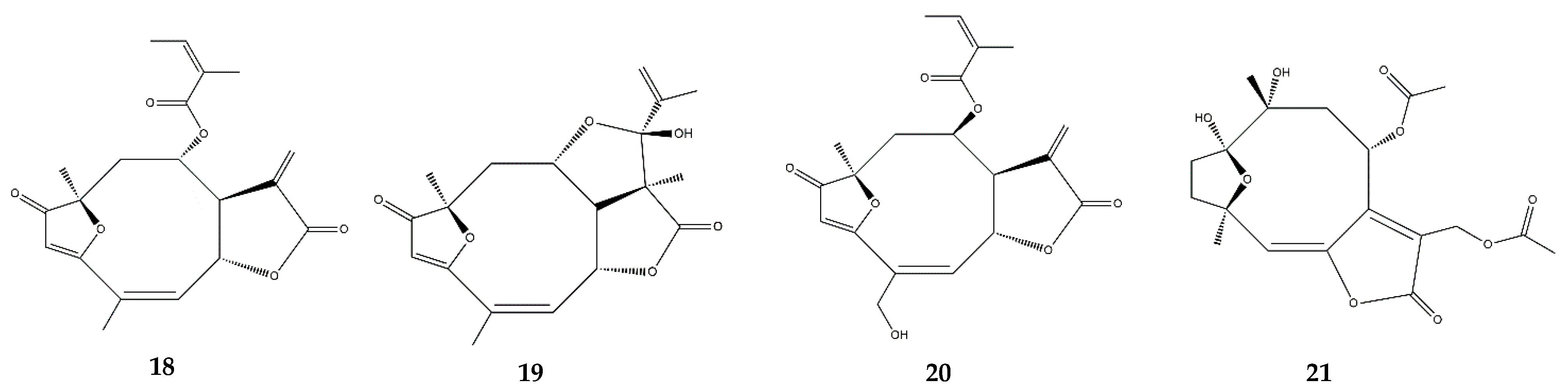
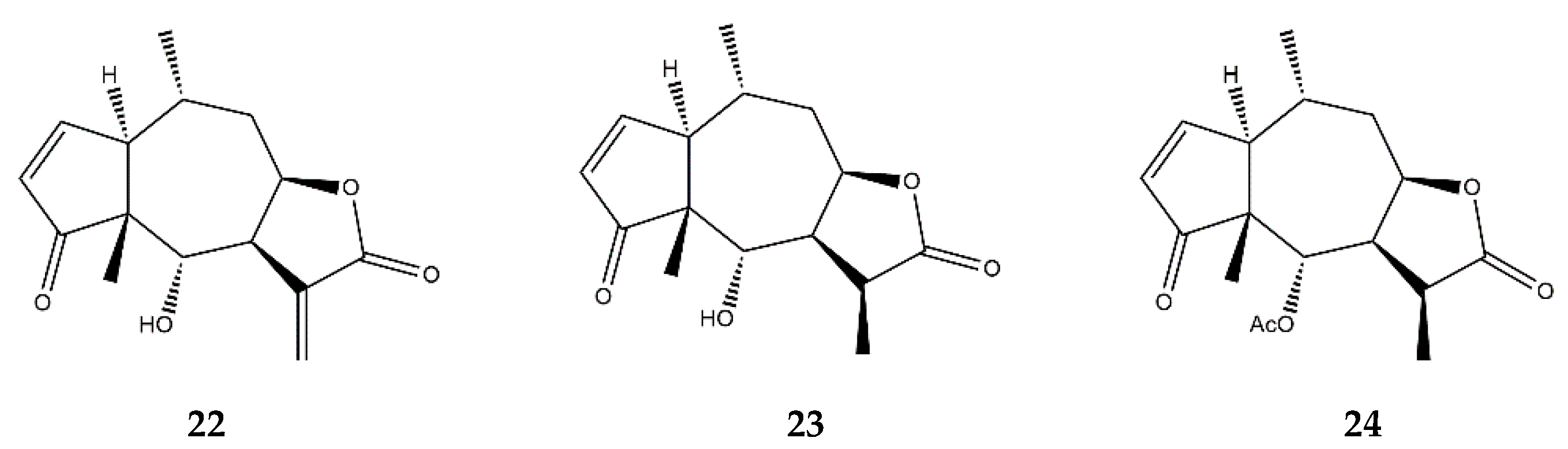
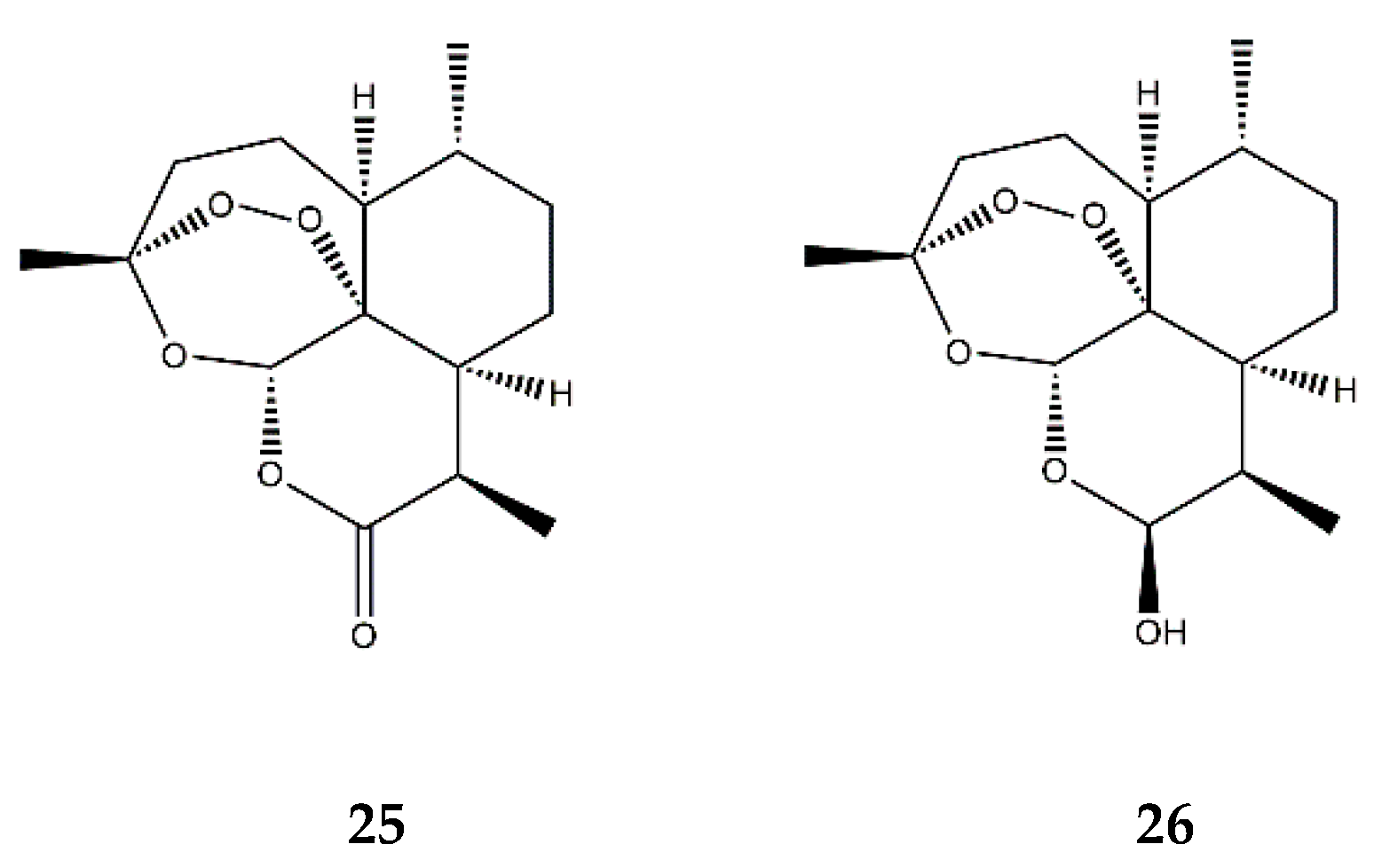
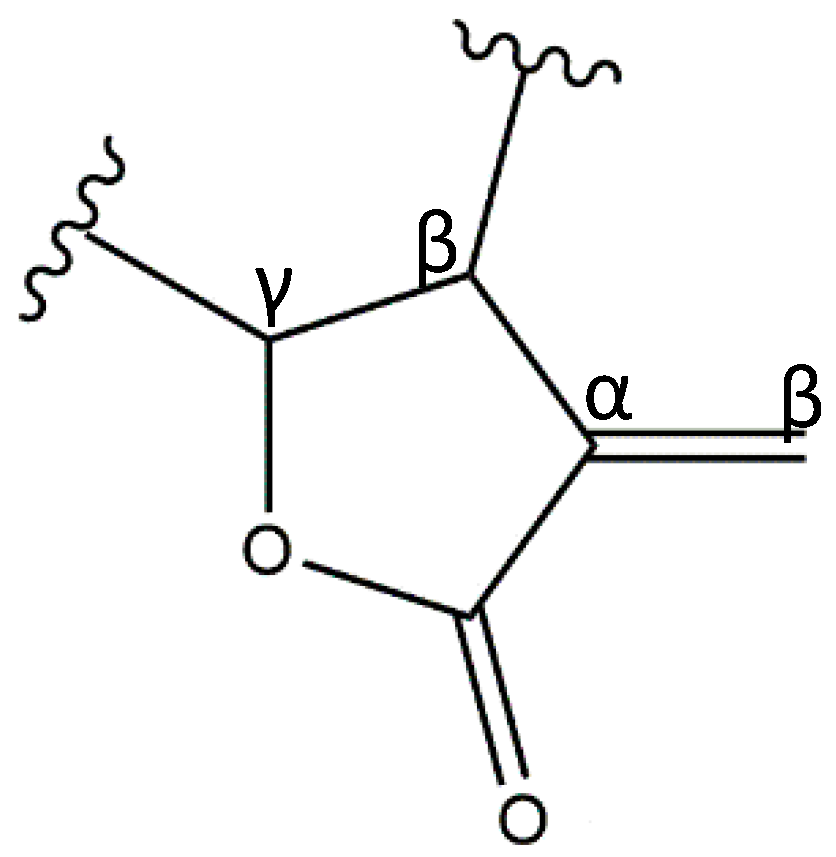
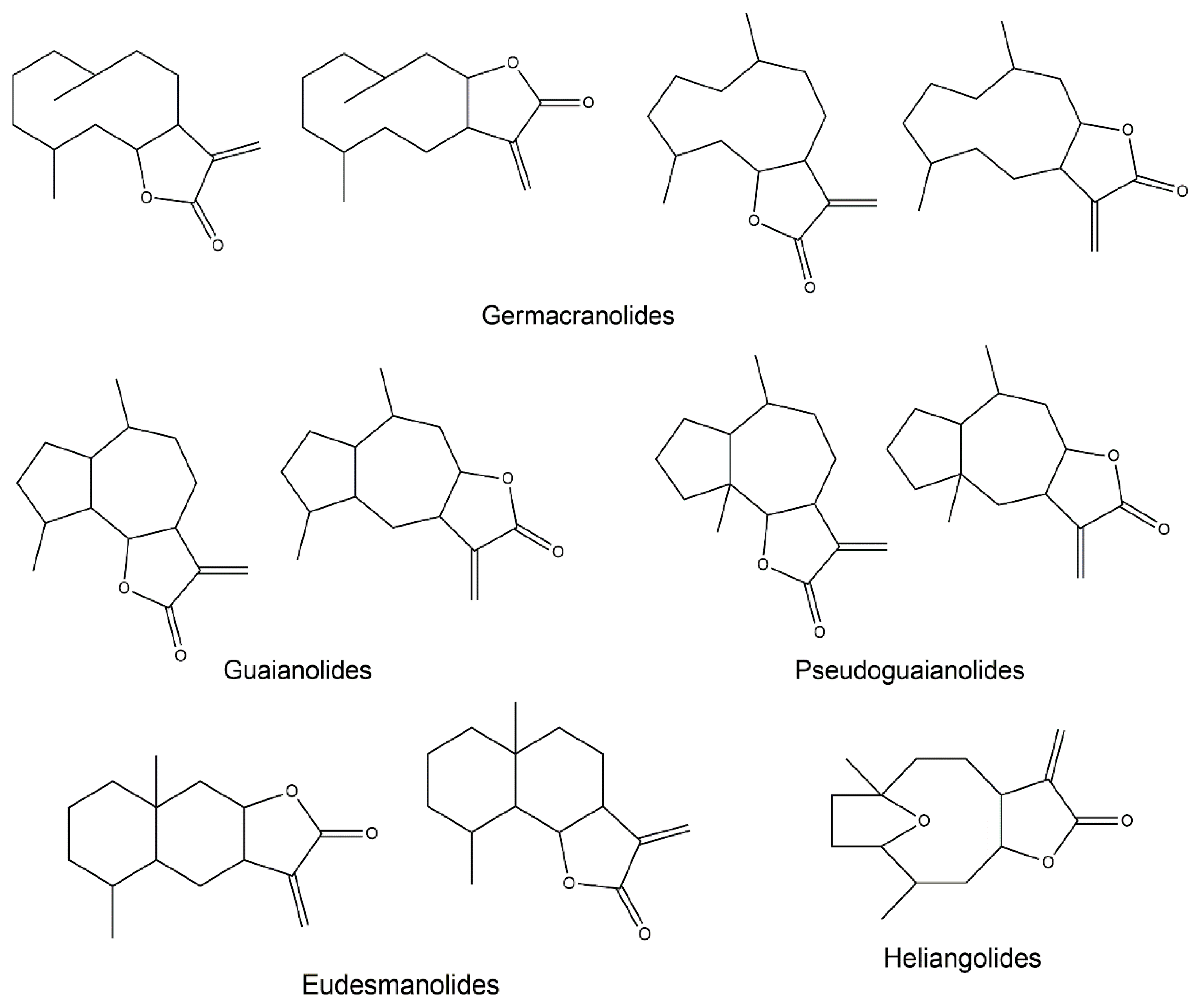
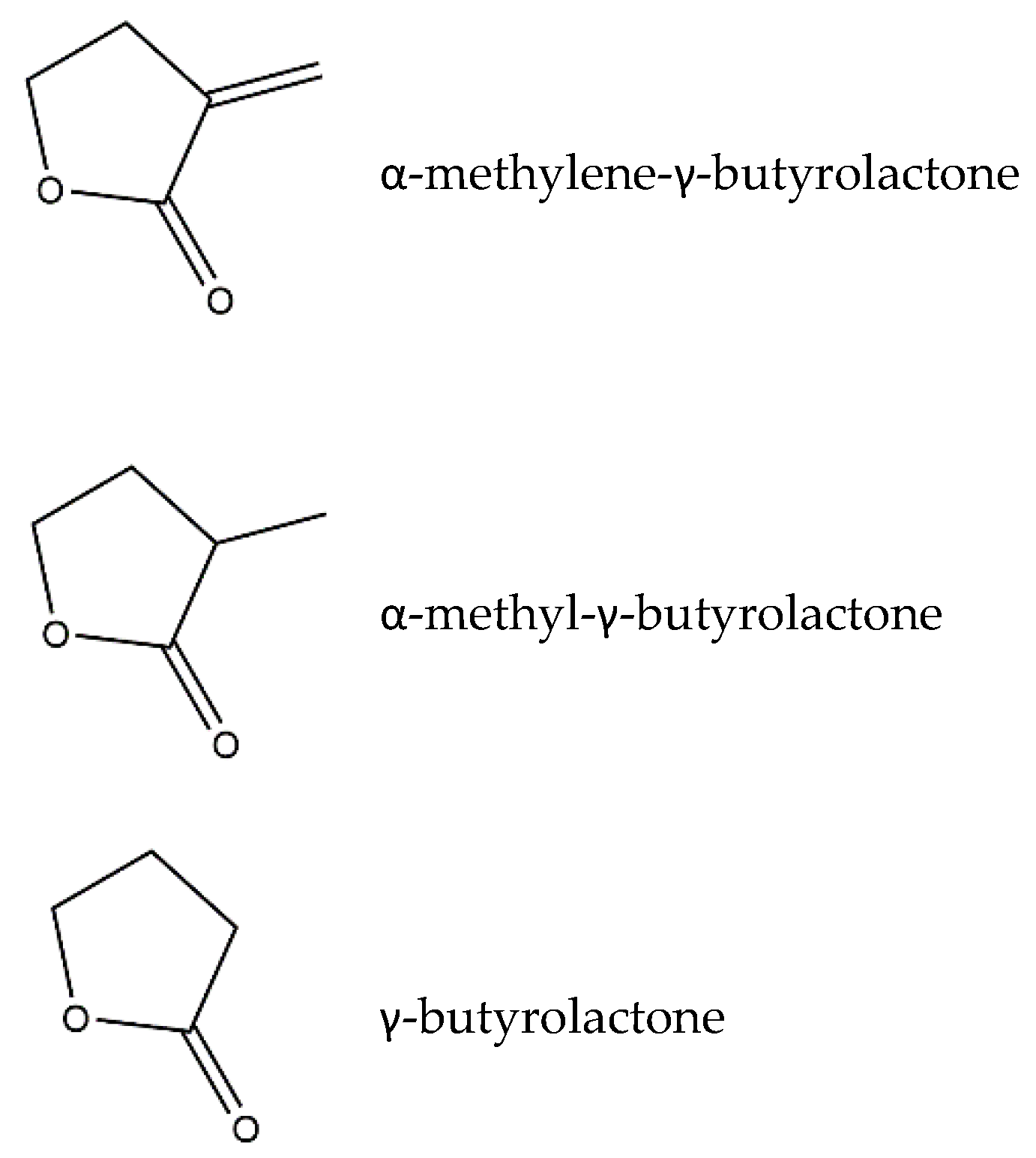
:1. Sesquiterpene Lactones
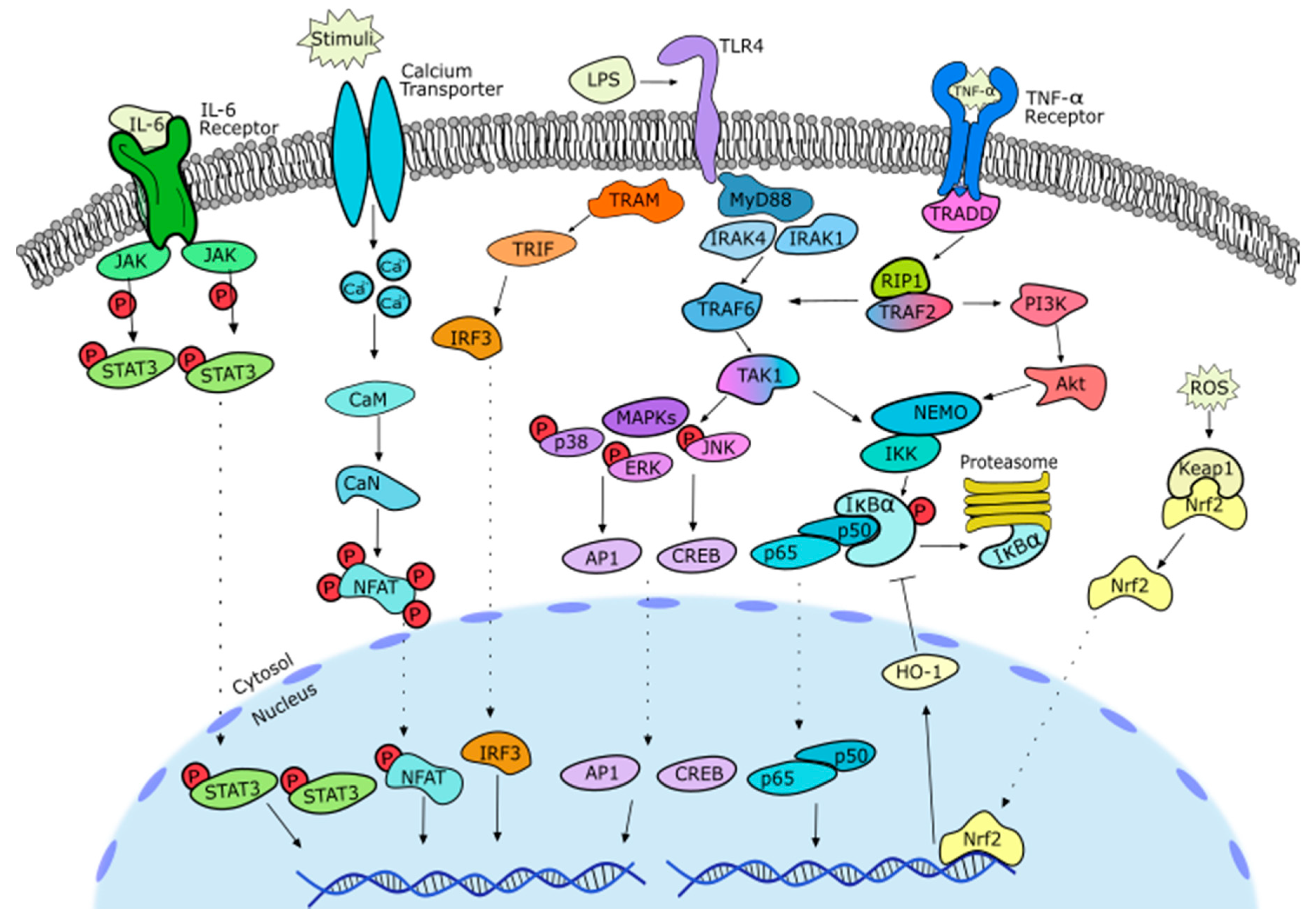
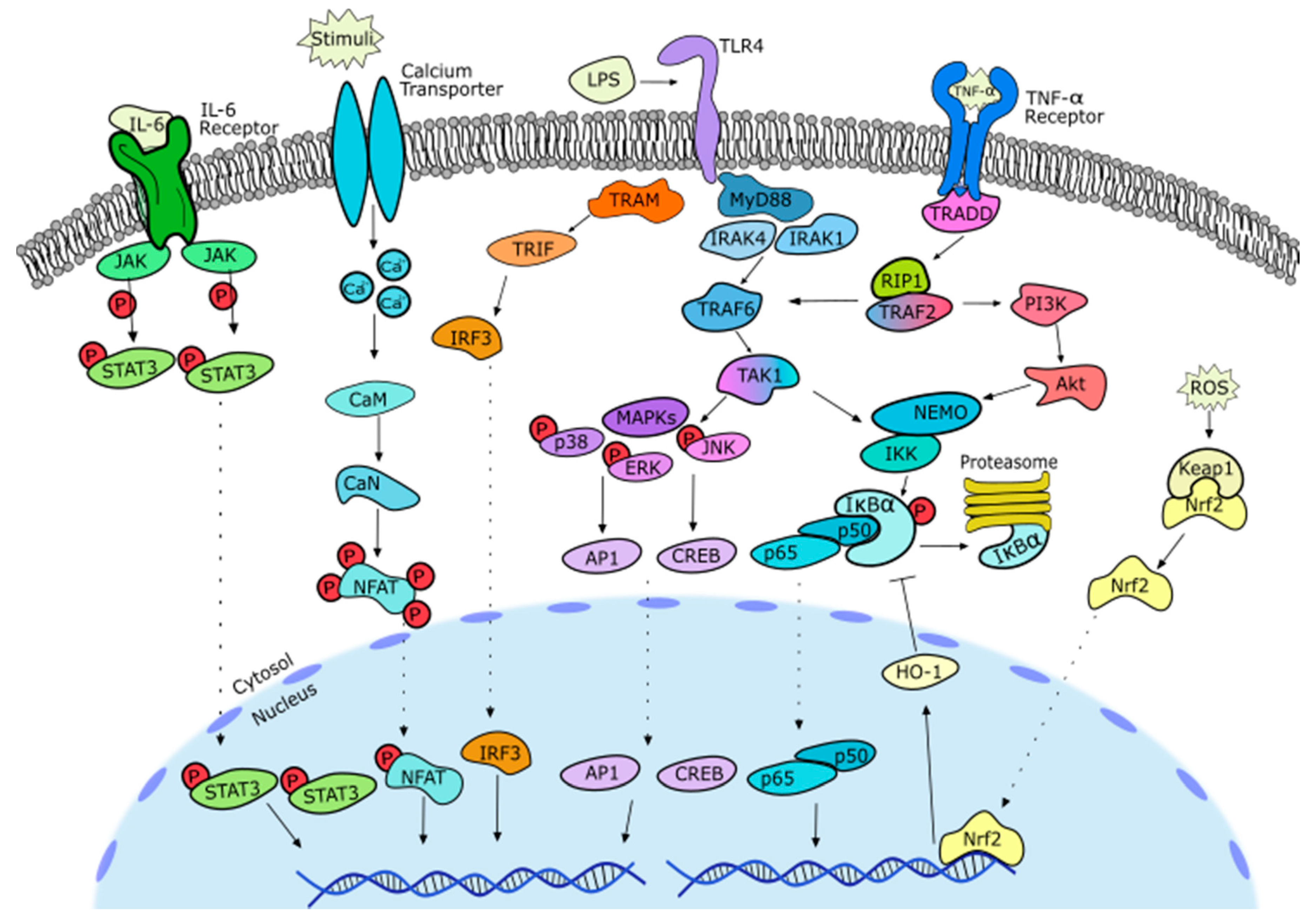
2. Anti-Inflammatory Potential of Sesquiterpene Lactones
2.1. General Notes on Inflammation
2.2. SL-Containing Extracts
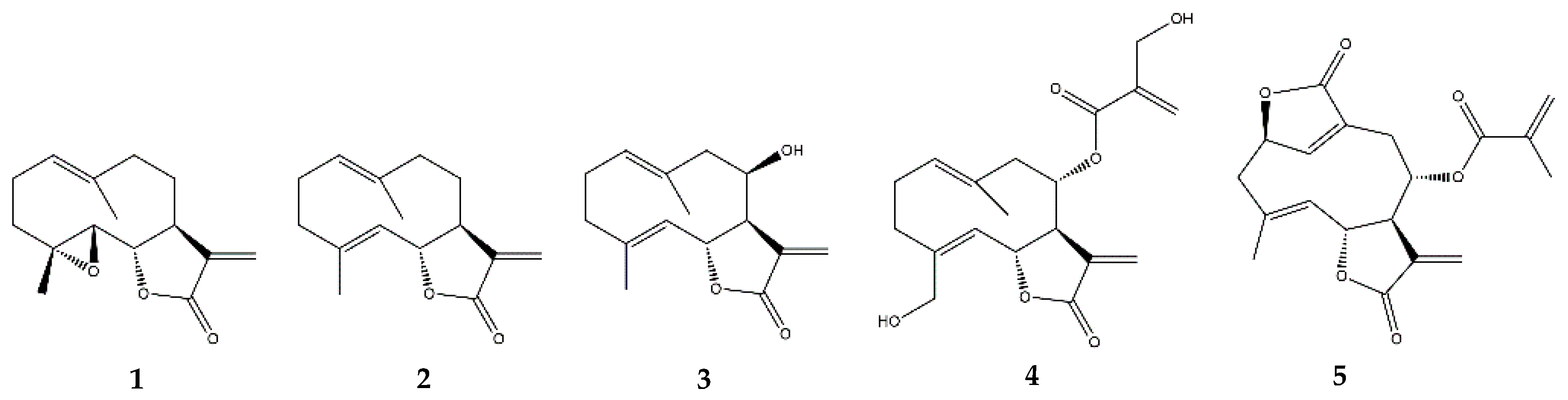
2.3. Germacranolides
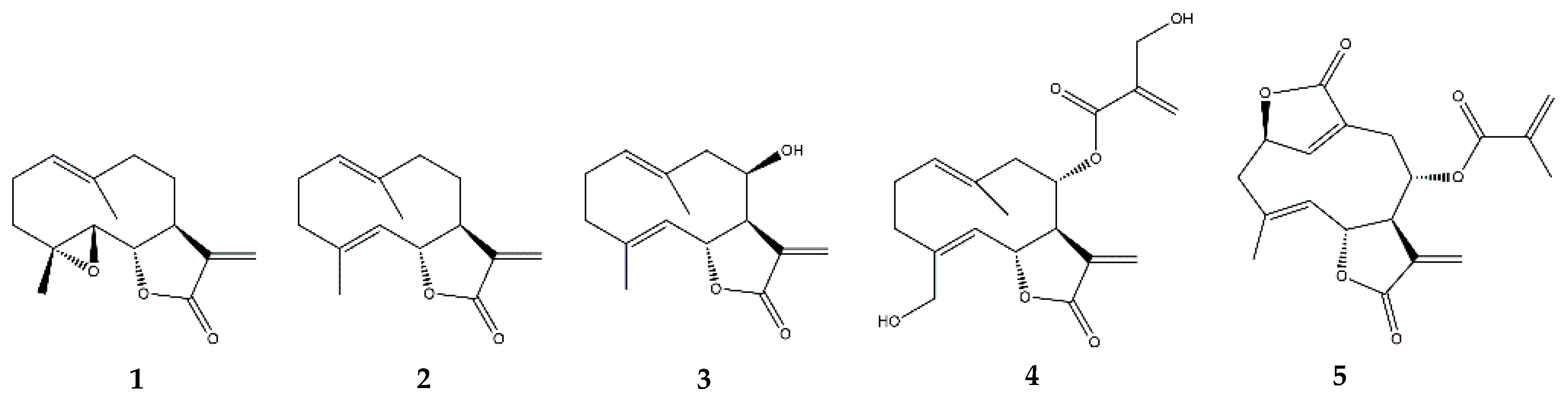
2.3.1. Parthenolide
2.3.2. Costunolide
2.3.3. Other Germacranolides
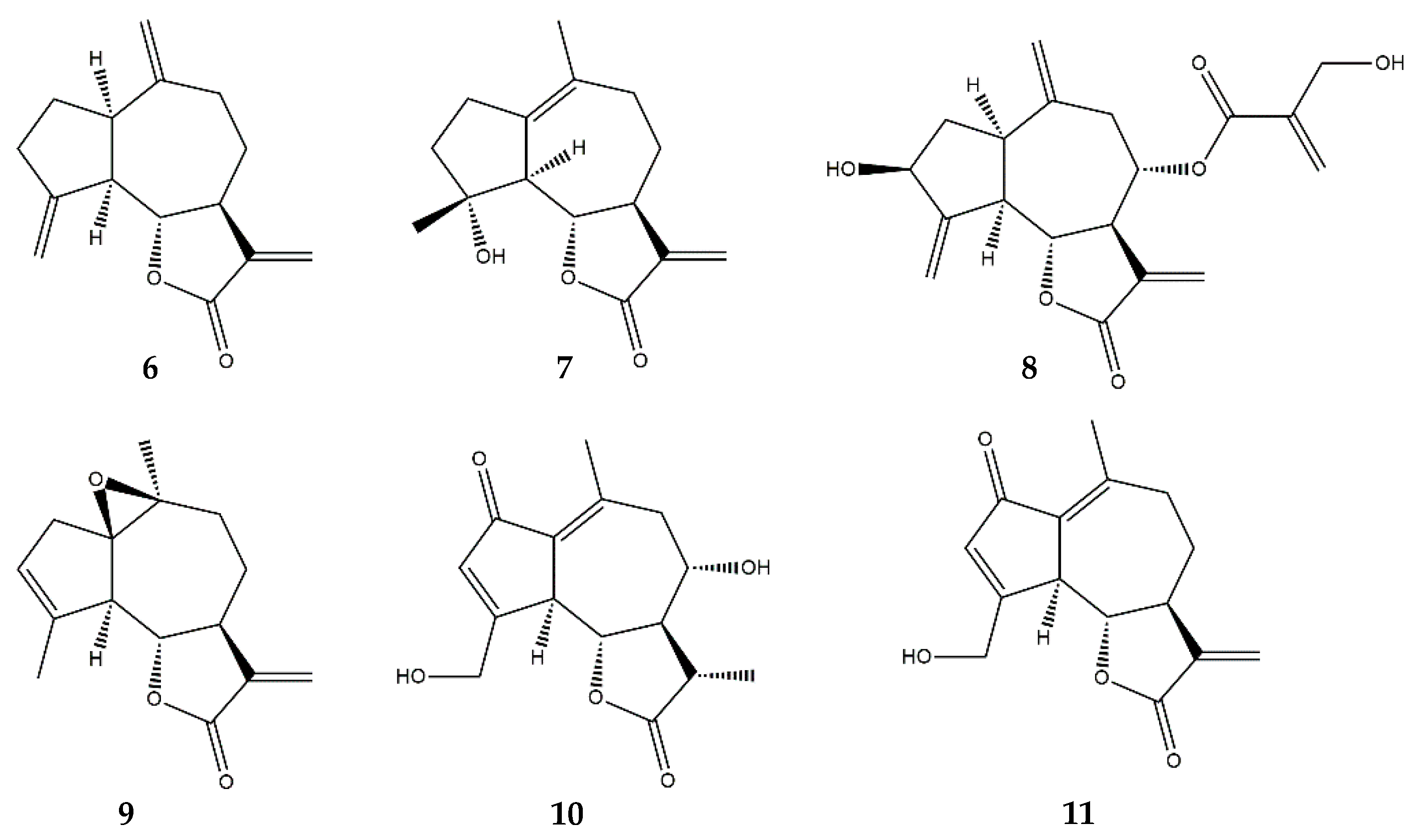
2.4. Guaianolides
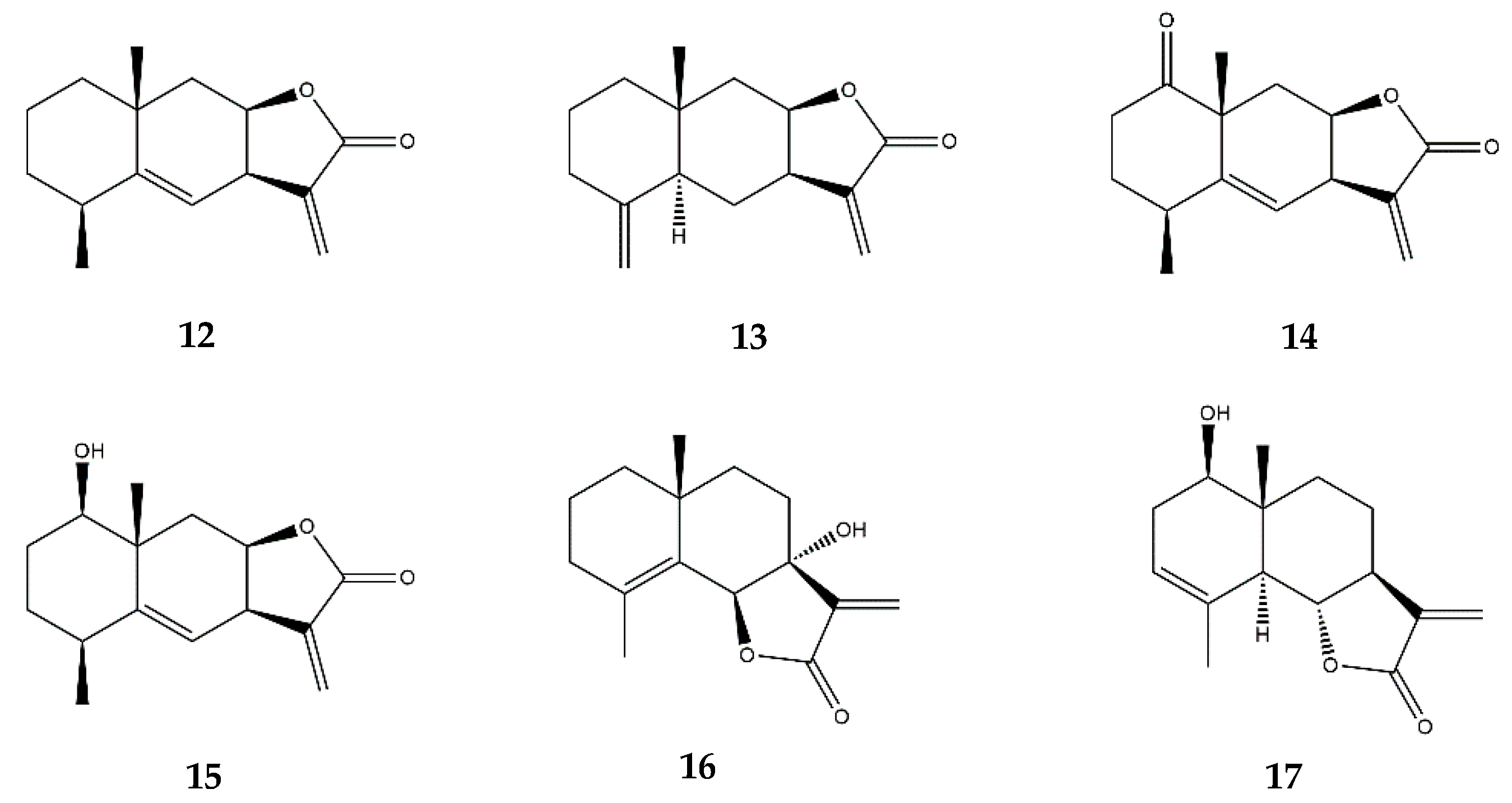
2.5. Eudesmanolides
2.6. Heliangolides
2.7. Pseudoguaianolides
2.8. Other SL Subclasses
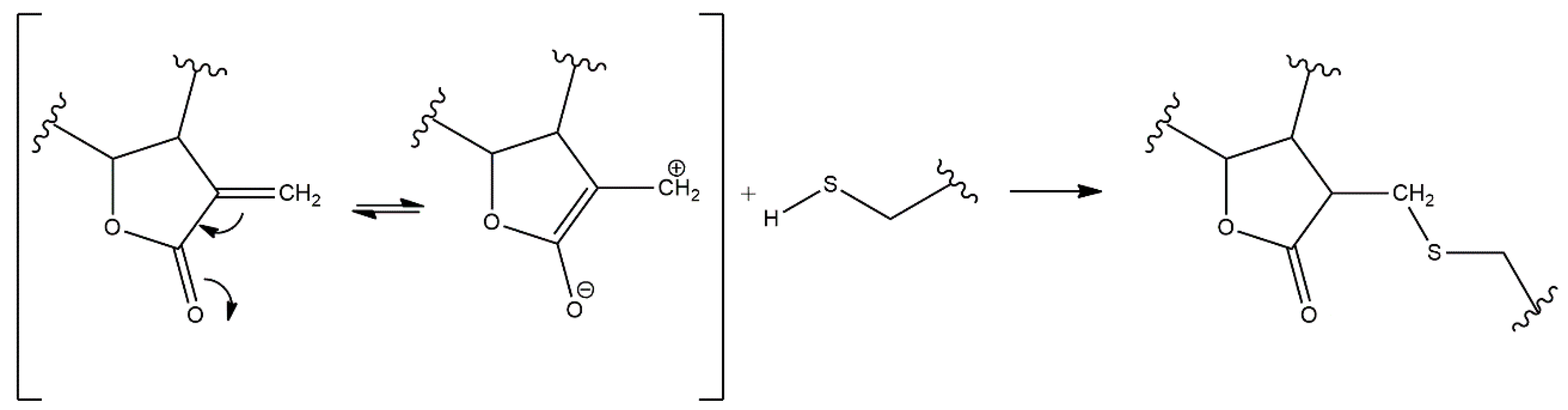
3. Structure–Activity Relationship of SLs
4. Pharmacokinetic Concerns
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| ADME | Absorption, Distribution, Metabolism, Excretion |
| Akt | Protein kinase B |
| ALT | Alanine aminotransferase |
| AP-1 | Activator protein-1 |
| apoE | Apolipoprotein E |
| BBB | Blood–brain barrier |
| BMDMs | Bone marrow-derived macrophages |
| CCL | Chemokine (C-C motif) ligand |
| CD | Cluster of differentiation |
| C/EBP | CCAAT-enhancer-binding protein |
| COX | Cyclooxygenase |
| Crz1 | Calcineurin-responsive zinc finger 1 |
| CSF | Colony-stimulating factor |
| CXCL10 | C-X-C motif chemokine ligand-10 |
| D-GalN | D-galactosamine |
| DMAPP | Dimethylallyl-diphosphate |
| DNA | Deoxyribonucleic acid |
| DSS | Dextran sulfate sodium |
| EGFR | Epidermal growth factor receptor |
| Erk | Extracellular signal-regulated kinase |
| FK506 | Tacrolimus |
| FPP | Farnesyl diphosphate |
| GLUT | Glucose transporter |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GSH | Glutathione |
| 1H-NMR | Proton Nuclear Magnetic Resonance |
| HEK | Human embryonic kidney |
| HMGB | High mobility group box |
| HO-1 | Heme oxygenase-1 |
| hPXR | Human pregnane X receptor |
| HUVECs | Human umbilical vein endothelial cells |
| IBD | Inflammatory bowel diseases |
| IC50 | Half maximal inhibitory concentration |
| ICAM | Intercellular adhesion molecule |
| IFN | Interferon |
| IκBα | NF-κB inhibitor alpha |
| IKK | IκB kinase |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| IP-10 | Interferon gamma-induced protein-10 |
| IPP | Isopentenyl diphosphate |
| IRF | Interferon regulatory factor |
| JAKs | Janus kinases |
| JNK | c-Jun N-terminal kinase |
| LDHA | Lactate dehydrogenase A |
| LOX | Lipoxygenase |
| LPS | Lipopolysaccharide |
| MAPKs | Mitogen-activated protein kinases |
| MCP | Monocyte chemoattractant protein |
| M-CSF | Macrophage colony-stimulating factor |
| MMP | Matrix metalloproteinase |
| MPO | Myeloperoxidase |
| mRNA | Messenger ribonucleic acid |
| MyD88 | Myeloid differentiation primary response 88 |
| NFAT | Nuclear factor of activated T-cells |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | Nucleotide-binding oligomeriztion domain (NOD)-like receptor family pyrin domain-containing 3 |
| NO | Nitric oxide |
| Nrf | Nuclear respiratory factor |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| Oct-1 | Octamer transcription factor-1 |
| p38 | Member of the MAPKs |
| p65 | Subunit of NF-κB |
| PBMCs | Peripheral blood mononuclear cells |
| PDK | Phosphoinositide-dependent kinase |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphatidyl inositol 3-kinase |
| PKM2 | Pyruvate kinase M2 |
| PMA | Phorbol 12-myristate 13-acetate |
| PMNLs | Polymorphonuclear leukocytes |
| ROS | Reactive oxygen species |
| SAR | Structure-activity relationship |
| 15(S)-HETE | 15-Hydroxyeicosatetraenoic acid |
| SLs | Sesquiterpene lactones |
| SOCS | Suppressor of cytokine signaling |
| SOD | Superoxide dismutase |
| SOX | SRY-box transcrition factor 9 |
| STATs | Signal transducer and activator of transcription |
| TGF-β | Transforming growth factor beta |
| Th1/Th2 | Type-1/2 helper T lymphocytes |
| TIMP | Tissue inhibitor of metalloproteinases |
| TIRAP | Toll/interleukin-1 receptor domain-containing adapter protein |
| TLR | Toll-like receptor |
| TNBS | Trinitrobenzene Sulfonic Acid |
| TNF-α | Tumor necrosis factor alpha |
| TNFR | Tumor necrosis factor receptor |
| TPA | 12-O-tetradecanoylphorbol acetate |
| TRAF | TNFR-associated factor |
| Trib1 | Tribbles pseudokinase 1 |
| TRIF | Toll-interleukin-1 receptor domain-containing adapter-inducing interferon-β |
| UbcH5 | Ubiquitin-conjugating enzyme H5 |
| VCAM | Vascular cell adhesion molecule |
| VEGF | Vascular endothelial growth factor |
References
- Chaturvedi, D.; Dwivedi, P. Recent Developments on the Antidiabetic Sesquiterpene Lactones and Their Semisynthetic Analogues. In Discovery and Development of Antidiabetic Agents from Natural Products, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 185–205. [Google Scholar]
- Vranová, E.; Coman, D.; Gruissem, W. Network Analysis of the MVA and MEP Pathways for Isoprenoid Synthesis. Annu. Rev. Plant Biol. 2013, 64, 665–700. [Google Scholar] [CrossRef]
- Lopes, A.A.; Pina, E.S.; Silva, D.B.; Pereira, A.M.S.; Silva, M.F.D.G.F.D.; Da Costa, F.B.; Lopes, N.P.; Pupo, M.T. A biosynthetic pathway of sesquiterpene lactones in Smallanthus sonchifolius and their localization in leaf tissues by MALDI imaging. Chem. Commun. 2013, 49, 9989–9991. [Google Scholar] [CrossRef]
- Ramirez, A.M.; Saillard, N.; Yang, T.; Franssen, M.C.R.; Bouwmeester, H.J.; Jongsma, M.A. Biosynthesis of Sesquiterpene Lactones in Pyrethrum (Tanacetum cinerariifolium). PLoS ONE 2013, 8, e65030. [Google Scholar] [CrossRef] [Green Version]
- Majdi, M.; Liu, Q.; Karimzadeh, G.; Malboobi, M.A.; Beekwilder, J.; Cankar, K.; De Vos, R.; Todorović, S.; Simonović, A.; Bouwmeester, H. Biosynthesis and localization of parthenolide in glandular trichomes of feverfew (Tanacetum parthenium L. Schulz Bip.). Phytochemistry 2011, 72, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Amrehn, E.; Aschenbrenner, A.-K.; Heller, A.; Spring, O. Localization of sesquiterpene lactone biosynthesis in cells of capitate glandular trichomes of Helianthus annuus (Asteraceae). Protoplasma 2016, 253, 447–455. [Google Scholar] [CrossRef]
- Hussien, T.; Mohamed, T.; Elshamy, A.; Moustafa, M.; El-Seedi, H.; Pare, P.; Hegazy, M.-E. Guaianolide Sesquiterpene Lactones from Centaurothamnus maximus. Molecules 2021, 26, 2055. [Google Scholar] [CrossRef]
- Merfort, I. Perspectives on Sesquiterpene Lactones in Inflammation and Cancer. Curr. Drug Targets 2011, 12, 1560–1573. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, M.S.; Longhi-Balbinot, D.T.; Guazelli, C.F.; Navarro, S.A.; Zarpelon, A.C.; Casagrande, R.; Arakawa, N.S.; Verri, W.A. Sesquiterpene Lactones: Structural Diversity and Perspectives as Anti-Inflammatory Molecules. In Studies in Natural Products Chemistry; Atta-ur-Rahman, F.R.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 49, pp. 243–260. [Google Scholar]
- Schmidt, T.J. Structure-activity relationships of sesquiterpene lactones. In Studies in Natural Products Chemistry; Atta-ur-Rahman, F.R.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 33, pp. 309–392. [Google Scholar]
- Wesołowska, A.; Nikiforuk, A.; Michalska, K.; Kisiel, W.; Chojnacka-Wójcik, E. Analgesic and sedative activities of lactucin and some lactucin-like guaianolides in mice. J. Ethnopharmacol. 2006, 107, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Gonzalez, G.F.; Dos Santos, F.A.; Da Costa, F.B. Sesquiterpene Lactones: More Than Protective Plant Compounds with High Toxicity. Crit. Rev. Plant Sci. 2016, 35, 18–37. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautam, R.; Jachak, S.M. Recent developments in anti-inflammatory natural products. Med. Res. Rev. 2009, 29, 767–820. [Google Scholar] [CrossRef] [PubMed]
- Seaman, F.C. Sesquiterpene lactones as taxonomic characters in the asteraceae. Bot. Rev. 1982, 48, 121–594. [Google Scholar] [CrossRef]
- Ripoll, C.; Schmidt, B.M.; Ilic, N.; Poulev, A.; Dey, M.; Kurmukov, A.G.; Raskin, I. Anti-inflammatory Effects of a Sesquiterpene Lactone Extract from Chicory (Cichorium intybus L.) Roots. Nat. Prod. Commun. 2007, 2, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.; Belolipov, I.V.; Kurmukov, A.G.; Zakirov, S.; Raskin, L. Sesquiterpene Lactone Extract from Artemisia leucodes for Reducing Inflammation and Down-Regulating Pro-Inflammatory Gene Expression. US Patent US20080145465A1, 19 June 2008. [Google Scholar]
- Emami, S.A.; Rabe, S.Z.T.; Iranshahi, M.; Ahi, A.; Mahmoudi, M. Sesquiterpene lactone fraction fromArtemisia khorassanicainhibits inducible nitric oxide synthase and cyclooxygenase-2 expression through the inactivation of NF-κB. Immunopharmacol. Immunotoxicol. 2010, 32, 688–695. [Google Scholar] [CrossRef]
- Zamani, S.; Emami, S.A.; Iranshahi, M.; Rabe, S.Z.T.; Mahmoudi, M. Sesquiterpene fractions of Artemisia plants as potent inhibitors of inducible nitric oxide synthase and cyclooxygenase-2 expression. Iran. J. Basic Med. Sci. 2019, 22, 774–780. [Google Scholar]
- Maas, M.; Deters, A.M.; Hensel, A. Anti-inflammatory activity of Eupatorium perfoliatum L. extracts, eupafolin, and dimeric guaianolide via iNOS inhibitory activity and modulation of inflammation-related cytokines and chemokines. J. Ethnopharmacol. 2011, 137, 371–381. [Google Scholar] [CrossRef]
- Bader, A.; Giner, R.M.; Martini, F.; Schinella, G.R.; Ríos, J.L.; Braca, A.; Prieto, J.M. Modulation of COX, LOX and NFκB activities by Xanthium spinosum L. root extract and ziniolide. Fitoterapia 2013, 91, 284–289. [Google Scholar] [CrossRef]
- Klaas, C.A.; Wagner, G.; Laufer, S.; Sosa, S.; Della Loggia, R.; Bomme, U.; Pahl, H.L.; Merfort, I. Studies on the Anti-Inflammatory Activity of Phytopharmaceuticals Prepared from Arnica Flowers. Planta Med. 2002, 68, 385–391. [Google Scholar] [CrossRef]
- Erel, S.B.; Demir, S.; Nalbantsoy, A.; Ballar, P.; Khan, S.; Yavasoglu, N.U.K.; Karaalp, C. Bioactivity screening of fiveCentaureaspecies andin vivoanti-inflammatory activity ofC. athoa. Pharm. Biol. 2014, 52, 775–781. [Google Scholar] [CrossRef]
- Demir, S.; Karaalp, C.; Bedir, E. Unusual sesquiterpenes from Centaurea athoa DC. Phytochem. Lett. 2016, 15, 245–250. [Google Scholar] [CrossRef]
- Gao, S.; Wang, Q.; Tian, X.-H.; Li, H.-L.; Shen, Y.-H.; Xu, X.-K.; Wu, G.-Z.; Hu, Z.-L.; Zhang, W.-D. Total sesquiterpene lactones prepared from Inula helenium L. has potentials in prevention and therapy of rheumatoid arthritis. J. Ethnopharmacol. 2017, 196, 39–46. [Google Scholar] [CrossRef]
- De Almeida, A.B.A.; Hidalgo, M.S.; Martín, A.R.; Luiz-Ferreira, A.; Trigo, J.R.; Vilegas, W.; Dos Santos, L.C.; Souza-Brito, A.R.M.; De La Lastra, C.A. Anti-inflammatory intestinal activity of Arctium lappa L. (Asteraceae) in TNBS colitis model. J. Ethnopharmacol. 2013, 146, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, H.; He, L.; Zheng, J.; Li, Q.; Liu, X.; Wang, D. Low Oral Bioavailability and Partial Gut Microbiotic and Phase II Metabolism of Brussels/Witloof Chicory Sesquiterpene Lactones in Healthy Humans. Nutrients 2020, 12, 3675. [Google Scholar] [CrossRef] [PubMed]
- Rauh, L.K.; Horinouchi, C.D.; Loddi, A.M.; Pietrovski, E.F.; Neris, R.; Souza-Fonseca-Guimaraes, F.; Buchi, D.F.; Biavatti, M.W.; Otuki, M.F.; Cabrini, D.A. Effectiveness of Vernonia scorpioides ethanolic extract against skin inflammatory processes. J. Ethnopharmacol. 2011, 138, 390–397. [Google Scholar] [CrossRef] [Green Version]
- Freund, R.R.A.; Gobrecht, P.; Fischer, D.; Arndt, H.-D. Advances in chemistry and bioactivity of parthenolide. Nat. Prod. Rep. 2020, 37, 541–565. [Google Scholar] [CrossRef]
- Wong, H.; Menendez, I.Y. Sesquiterpene Lactones Inhibit Inducible Nitric Oxide Synthase Gene Expression in Cultured Rat Aortic Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 1999, 262, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Magni, P.; Ruscica, M.; Dozio, E.; Rizzi, E.; Beretta, G.; Facino, R.M. Parthenolide Inhibits the LPS-induced Secretion of IL-6 and TNF-α and NF-κB Nuclear Translocation in BV-2 Microglia. Phytother. Res. 2012, 26, 1405–1409. [Google Scholar] [CrossRef]
- Rummel, C.; Gerstberger, R.; Roth, J.; Hübschle, T. Parthenolide attenuates LPS-induced fever, circulating cytokines and markers of brain inflammation in rats. Cytokine 2011, 56, 739–748. [Google Scholar] [CrossRef]
- Li-Weber, M.; Giaisi, M.; Treiber, M.K.; Krammer, P.H. The anti-inflammatory sesquiterpene lactone parthenolide suppresses IL-4 gene expression in peripheral blood T cells. Eur. J. Immunol. 2002, 32, 3587–3597. [Google Scholar] [CrossRef]
- Humar, M.; García-Piñeres, A.J.; Castro, V.; Merfort, I. Effect of sesquiterpene lactones on the expression of the activation marker CD69 and of IL-2 in T-lymphocytes in whole blood. Biochem. Pharmacol. 2003, 65, 1551–1563. [Google Scholar] [CrossRef]
- Li, S.; Gao, X.; Wu, X.; Cheng, L.; Zhu, L.; Shen, D.; Tong, X. Parthenolide inhibits LPS-induced inflammatory cytokines through the toll-like receptor 4 signal pathway in THP-1 cells. Acta Biochim. Biophys. Sin. 2015, 47, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Feltenstein, M.; Schühly, W.; Warnick, J.; Fischer, N.; Sufka, K. Anti-inflammatory and anti-hyperalgesic effects of sesquiterpene lactones from Magnolia and Bear’s foot. Pharmacol. Biochem. Behav. 2004, 79, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Pae, H.-O.; Jeong, G.-S.; Woo, W.H.; Rhew, H.Y.; Kim, H.S.; Sohn, D.H.; Kim, Y.-C.; Chung, H.-T. Costunolide inhibits production of tumor necrosis factor-α and interleukin-6 by inducing heme oxygenase-1 in RAW264.7 macrophages. Inflamm. Res. 2007, 56, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Sobota, R.; Szwed, M.; Kasza, A.; Bugno, M.; Kordula, T. Parthenolide Inhibits Activation of Signal Transducers and Activators of Transcription (STATs) Induced by Cytokines of the IL-6 Family. Biochem. Biophys. Res. Commun. 2000, 267, 329–333. [Google Scholar] [CrossRef]
- Butturini, E.; Cavalieri, E.; De Prati, A.C.; Darra, E.; Rigo, A.; Shoji, K.; Murayama, N.; Yamazaki, H.; Watanabe, Y.; Suzuki, H.; et al. Two Naturally Occurring Terpenes, Dehydrocostuslactone and Costunolide, Decrease Intracellular GSH Content and Inhibit STAT3 Activation. PLoS ONE 2011, 6, e20174. [Google Scholar] [CrossRef] [Green Version]
- Scarponi, C.; Butturini, E.; Sestito, R.; Madonna, S.; Cavani, A.; Mariotto, S.; Albanesi, C. Inhibition of Inflammatory and Proliferative Responses of Human Keratinocytes Exposed to the Sesquiterpene Lactones Dehydrocostuslactone and Costunolide. PLoS ONE 2014, 9, e107904. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.S.; Yoon, Y.D.; Lee, K.H.; Park, S.-K.; Kim, H.M. Costunolide inhibits interleukin-1β expression by down-regulation of AP-1 and MAPK activity in LPS-stimulated RAW 264.7 cells. Biochem. Biophys. Res. Commun. 2004, 313, 171–177. [Google Scholar] [CrossRef]
- He, Y.; Moqbel, S.A.A.; Xu, L.; Ran, J.; Ma, C.; Xu, K.; Bao, J.; Jiang, L.; Chen, W.; Xiong, Y.; et al. Costunolide inhibits matrix metalloproteinases expression and osteoarthritis via the NF-κB and Wnt/β-catenin signaling pathways. Mol. Med. Rep. 2019, 20, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraswati, S.; Alhaider, A.A.; Abdelgadir, A.M. Costunolide suppresses an inflammatory angiogenic response in a subcutaneous murine sponge model. APMIS 2018, 126, 257–266. [Google Scholar] [CrossRef]
- Lee, J.; Tae, N.; Lee, J.J.; Kim, T.; Lee, J.-H. Eupatolide inhibits lipopolysaccharide-induced COX-2 and iNOS expression in RAW264.7 cells by inducing proteasomal degradation of TRAF6. Eur. J. Pharmacol. 2010, 636, 173–180. [Google Scholar] [CrossRef]
- Formisano, C.; Sanna, C.; Ballero, M.; Chianese, G.; Sirignano, C.; Rigano, D.; Millán, E.; Muñoz, E.; Taglialatela-Scafati, O. Anti-inflammatory sesquiterpene lactones from Onopordum illyricum L. (Asteraceae), an Italian medicinal plant. Fitoterapia 2017, 116, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Hu, L.; Zhang, L.; Xu, H.; Chen, Y.; Bian, Q.; Zhu, A.; Wu, H. Deoxyelephantopin decreases the release of inflammatory cytokines in macrophage associated with attenuation of aerobic glycolysis via modulation of PKM2. Int. Immunopharmacol. 2020, 79, 106048. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-C.; Lin, K.-J.; Cheng, Y.-W.; Hsu, C.-A.; Yang, S.-S.; Shyur, L.-F. Hepatoprotective effect and mechanistic insights of deoxyelephantopin, a phyto-sesquiterpene lactone, against fulminant hepatitis. J. Nutr. Biochem. 2013, 24, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Benteldjoune, M.; Chini, M.G.; Iannuzzi, A.M.; Kabouche, A.; Kabouche, Z.; D’ambola, M.; Marzocco, S.; Autore, G.; Bifulco, G.; De Tommasi, N. Guaianolides from Ormenis mixta: Structural Insights and Evaluation of Their Anti-inflammatory Profile. Planta Med. 2019, 85, 947–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Zhang, W.-X.; He, Z.-Q.; Wu, B.-S.; Shen, Z.-F.; Shang, H.-T.; Chen, T.; Wang, Q.; Chen, Y.-G.; Han, S.-T. The Possible Anti-Inflammatory Effect of Dehydrocostus Lactone on DSS-Induced Colitis in Mice. Evid. Based Complement. Altern. Med. 2020, 2020, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Heo, S.; Kim, S.H.; Kwon, M.; Sung, N.J.; Ryu, A.-R.; Lee, M.-Y.; Park, S.-A.; Youn, H.-S. Suppressive effects of dehydrocostus lactone on the toll-like receptor signaling pathways. Int. Immunopharmacol. 2020, 78, 106075. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, G.; Tong, T.; Chen, J. Micheliolide suppresses LPS-induced neuroinflammatory responses. PLoS ONE 2017, 12, e0186592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viennois, E.; Xiao, B.; Ayyadurai, S.; Wang, L.; Wang, P.G.; Zhang, Q.; Chen, Y.; Merlin, D. Micheliolide, a new sesquiterpene lactone that inhibits intestinal inflammation and colitis-associated cancer. Lab. Investig. 2014, 94, 950–965. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Jiang, X.; Wang, Y.; Miao, Z.; He, W.; Yang, G.; Lv, Z.; Yu, Y.; Zheng, Y. Micheliolide inhibits LPS-induced inflammatory response and protects mice from LPS challenge. Sci. Rep. 2016, 6, 23240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Wang, J.; Wang, C.; Chang, G.; Lin, Y.; Zhang, H.; Zhang, H.; Li, Q.; Pang, T. Therapeutic effects of micheliolide on a murine model of rheumatoid arthritis. Mol. Med. Rep. 2014, 11, 489–493. [Google Scholar] [CrossRef]
- Cho, J.Y.; Baik, K.U.; Jung, J.H.; Park, M.H. In vitro anti-inflammatory effects of cynaropicrin, a sesquiterpene lactone, from Saussurea lappa. Eur. J. Pharmacol. 2000, 398, 399–407. [Google Scholar] [CrossRef]
- Abderrazak, A.; Couchie, D.; Mahmood, D.; El Hage, R.; Vindis, C.; Laffargue, M.; Mateo, V.; Buechele, B.; Ayala, M.R.; El Gaafary, M.; et al. Anti-Inflammatory and Antiatherogenic Effects of the NLRP3 Inflammasome Inhibitor Arglabin in ApoE2.Ki Mice Fed a High-Fat Diet. Circulation 2015, 131, 1061–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matos, M.S.; Anastácio, J.D.; Allwood, J.W.; Carregosa, D.; Marques, D.; Sungurtas, J.; McDougall, G.J.; Menezes, R.; Matias, A.A.; Stewart, D.; et al. Assessing the Intestinal Permeability and Anti-Inflammatory Potential of Sesquiterpene Lactones from Chicory. Nutrients 2020, 12, 3547. [Google Scholar] [CrossRef] [PubMed]
- Cavin, C.; Delannoy, M.; Malnoe, A.; Debefve, E.; Touché, A.; Courtois, D.; Schilter, B. Inhibition of the expression and activity of cyclooxygenase-2 by chicory extract. Biochem. Biophys. Res. Commun. 2005, 327, 742–749. [Google Scholar] [CrossRef]
- Chun, J.; Choi, R.J.; Khan, S.; Lee, D.-S.; Kim, Y.-C.; Nam, Y.-J.; Kim, Y.S. Alantolactone suppresses inducible nitric oxide synthase and cyclooxygenase-2 expression by down-regulating NF-κB, MAPK and AP-1 via the MyD88 signaling pathway in LPS-activated RAW 264.7 cells. Int. Immunopharmacol. 2012, 14, 375–383. [Google Scholar] [CrossRef]
- Ren, Y.; Yue, B.; Ren, G.; Yu, Z.; Luo, X.; Sun, A.; Zhang, J.; Han, M.; Wang, Z.; Dou, W. Activation of PXR by alantolactone ameliorates DSS-induced experimental colitis via suppressing NF-κB signaling pathway. Sci. Rep. 2019, 9, 16636. [Google Scholar] [CrossRef]
- Liu, L.; Hua, Y.; Wang, D.; Shan, L.; Zhang, Y.; Zhu, J.; Jin, H.; Li, H.; Hu, Z.; Zhang, W. A Sesquiterpene Lactone from a Medicinal Herb Inhibits Proinflammatory Activity of TNF-α by Inhibiting Ubiquitin-Conjugating Enzyme UbcH5. Chem. Biol. 2014, 21, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tang, S.-A.; Wang, R.; Qiu, Y.; Jin, M.; Kong, D. Inhibitory Effects of JEUD-38, a New Sesquiterpene Lactone from Inula japonica Thunb, on LPS-Induced iNOS Expression in RAW264.7 Cells. Inflammation 2015, 38, 941–948. [Google Scholar] [CrossRef]
- Fonseca, L.C.; Dadarkar, S.S.; Lobo, A.S.; Mishra, P.B.; Thakkar, A.D.; Chandrababu, S.; Padigaru, M. NF-κB-mediated anti-inflammatory activity of the sesquiterpene lactone 7-hydroxyfrullanolide. Eur. J. Pharmacol. 2011, 657, 41–50. [Google Scholar] [CrossRef]
- Fonseca, L.C.; Dadarkar, S.S.; Lobo, A.S.; Suthar, A.C.; Chauhan, V.S.; Chandrababu, S.; Sharma, S.D.; Dagia, N.M.; Padigaru, M. 7-hydroxyfrullanolide, a sesquiterpene lactone, inhibits pro-inflammatory cytokine production from immune cells and is orally efficacious in animal models of inflammation. Eur. J. Pharmacol. 2010, 644, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-G.; Lee, D.-S.; Li, B.; Choi, Y.H.; Lee, S.-H.; Kim, Y.-C. Santamarin, a sesquiterpene lactone isolated from Saussurea lappa, represses LPS-induced inflammatory responses via expression of heme oxygenase-1 in murine macrophage cells. Int. Immunopharmacol. 2012, 13, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.C.; Ferreira, L.C.; Souza, M.R.; Paula, C.A.; Rezende, S.A.; Grabe-Guimarães, A.; Saúde-Guimarães, D. Anti-Inflammatory Sesquiterpene Lactones from Lychnophora trichocarpha Spreng. (Brazilian Arnica). Phytother. Res. 2012, 27, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Zarpelon, A.C.; Fattori, V.; Souto, F.O.; Pinto, L.G.; Pinho-Ribeiro, F.A.; Ruiz-Miyazawa, K.W.; Turato, W.M.; Cunha, T.M.; Da Costa, F.B.; Cunha, F.Q.; et al. The Sesquiterpene Lactone, Budlein A, Inhibits Antigen-Induced Arthritis in Mice: Role of NF-κB and Cytokines. Inflammation 2017, 40, 2020–2032. [Google Scholar] [CrossRef]
- Valério, D.A.; Cunha, T.M.; Arakawa, N.S.; Lemos, H.D.P.; Da Costa, F.B.; Parada, C.A.; Ferreira, S.H.; Cunha, F.Q.; Verri, W.A. Anti-inflammatory and analgesic effects of the sesquiterpene lactone budlein A in mice: Inhibition of cytokine production-dependent mechanism. Eur. J. Pharmacol. 2007, 562, 155–163. [Google Scholar] [CrossRef]
- Fattori, V.; Zarpelon, A.C.; Staurengo-Ferrari, L.; Borghi, S.M.; Zaninelli, T.H.; Da Costa, F.B.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q.; Casagrande, R.; et al. Budlein A, a Sesquiterpene Lactone from Viguiera robusta, Alleviates Pain and Inflammation in a Model of Acute Gout Arthritis in Mice. Front. Pharmacol. 2018, 9, 1076. [Google Scholar] [CrossRef] [Green Version]
- Sabel, R.; Fronza, A.; Carrenho, L.; Maes, A.; Barros, M.; Pollo, L.A.E.; Biavatti, M.; D’Herde, K.; Vandenabeele, P.; Kreuger, M. Anti-inflammatory activity of the sesquiterpene lactone diacethylpiptocarphol in dextransulfate sodium-induced colitis in mice. J. Ethnopharmacol. 2019, 245, 112186. [Google Scholar] [CrossRef]
- Lyss, G.; Schmidt, T.J.; Merfort, I.; Pahl, H.L. Helenalin, an Anti-Inflammatory Sesquiterpene Lactone from Arnica, Selectively Inhibits Transcription Factor NF-κB. Biol. Chem. 1997, 378, 951–962. [Google Scholar] [CrossRef]
- Lyß, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The Anti-inflammatory Sesquiterpene Lactone Helenalin Inhibits the Transcription Factor NF-κB by Directly Targeting p65. J. Biol. Chem. 1998, 273, 33508–33516. [Google Scholar] [CrossRef] [Green Version]
- Berges, C.; Fuchs, D.; Opelz, G.; Daniel, V.; Naujokat, C. Helenalin suppresses essential immune functions of activated CD4+ T cells by multiple mechanisms. Mol. Immunol. 2009, 46, 2892–2901. [Google Scholar] [CrossRef]
- Zwicker, P.; Schultze, N.; Niehs, S.; Albrecht, D.; Methling, K.; Wurster, M.; Wachlin, G.; Lalk, M.; Lindequist, U.; Haertel, B. Differential effects of Helenalin, an anti-inflammatory sesquiterpene lactone, on the proteome, metabolome and the oxidative stress response in several immune cell types. Toxicol. Vitr. 2017, 40, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.B.; Fu, P.Y.; Ky, N.; Zhu, H.S.; Feng, X.; Li, J.; Srinivasan, K.G.; Hamza, M.S.; Zhao, Y. NF-κB p65 repression by the sesquiterpene lactone, Helenalin, contributes to the induction of autophagy cell death. BMC Complement. Altern. Med. 2012, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Gertsch, J.; Sticher, O.; Schmidt, T.; Heilmann, J. Influence of helenanolide-type sesquiterpene lactones on gene transcription profiles in Jurkat T cells and human peripheral blood cells: Anti-inflammatory and cytotoxic effects. Biochem. Pharmacol. 2003, 66, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Tornhamre, S.; Schmidt, T.J.; Näsman-Glaser, B.; Ericsson, I.; Lindgren, J. Åke Inhibitory effects of helenalin and related compounds on 5-lipoxygenase and leukotriene C4 synthase in human blood cells. Biochem. Pharmacol. 2001, 62, 903–911. [Google Scholar] [CrossRef]
- Wu, Z.-L.; Wang, J.-X.; Chen, L.-P.; Dong, H.-Y.; Li, H.-L.; Zhang, W.-D. Five rare C 32 sesquiterpene lactone dimers with anti-inflammation activity from Vladimiria souliei. Fitoterapia 2018, 125, 117–122. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, J.; Zhaofang, Y.; Xie, Y.; Xu, Z.; Yin, Z.; Gao, L.; Wang, C. Artemisinin inhibits monocyte adhesion to HUVECs through the NF-κB and MAPK pathways in vitro. Int. J. Mol. Med. 2016, 37, 1567–1575. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Yang, J.H.; Han, E.H.; Choi, J.H.; Khanal, T.; Jeong, M.H.; Jeong, T.C.; Jeong, H.G. Inhibitory effect of dihydroartemisinin against phorbol ester-induced cyclooxygenase-2 expression in macrophages. Food Chem. Toxicol. 2013, 56, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Siedle, B.; Cisielski, S.; Murillo, R.; Löser, B.; Castro, V.; Klaas, C.; Hucke, O.; Labahn, A.; Melzig, M.; Merfort, I. Sesquiterpene lactones as inhibitors of human neutrophil elastase. Bioorg. Med. Chem. 2002, 10, 2855–2861. [Google Scholar] [CrossRef]
- Sülsen, V.P.; Martino, V.S. Sesquiterpene Lactones—Advances in Their Chemistry and Biological Aspects, 1st ed.; Springer International Publishing: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Chadwick, M.; Trewin, H.; Gawthrop, F.; Wagstaff, C. Sesquiterpenoids Lactones: Benefits to Plants and People. Int. J. Mol. Sci. 2013, 14, 12780–12805. [Google Scholar] [CrossRef] [Green Version]
- Choodej, S.; Pudhom, K.; Mitsunaga, T. Inhibition of TNF-α-Induced Inflammation by Sesquiterpene Lactones from Saussurea lappa and Semi-Synthetic Analogues. Planta Med. 2018, 84, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Su, S.; Zhang, S.; Zhai, S.; Sheng, R.; Wu, W.; Guo, R. Structure-activity relationship and synthetic methodologies of α-santonin derivatives with diverse bioactivities: A mini-review. Eur. J. Med. Chem. 2019, 175, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Coricello, A.; El-Magboub, A.; Luna, M.; Ferrario, A.; Haworth, I.S.; Gomer, C.J.; Aiello, F.; Adams, J.D. Rational drug design and synthesis of new α-Santonin derivatives as potential COX-2 inhibitors. Bioorganic. Med. Chem. Lett. 2018, 28, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Tambewagh, U.U.; Kandhare, A.D.; Honmore, V.S.; Kadam, P.P.; Khedkar, V.M.; Bodhankar, S.L.; Rojatkar, S.R. Anti-inflammatory and antioxidant potential of Guaianolide isolated from Cyathocline purpurea: Role of COX-2 inhibition. Int. Immunopharmacol. 2017, 52, 110–118. [Google Scholar] [CrossRef]
- Tang, J.-J.; He, Q.-R.; Dong, S.; Guo, X.; Wang, Y.-G.; Lei, B.-L.; Tian, J.-M.; Gao, J.-M. Diversity Modification and Structure-Activity Relationships of Two Natural Products 1β-hydroxy Alantolactone and Ivangustin as Potent Cytotoxic Agents. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.N.A.; Choucry, M.A.; El Senousy, A.S.; Hassan, A.; El-Marasy, S.A.; El Awdan, S.A.; Omar, F.A. Ambrosin, a potent NF-κβ inhibitor, ameliorates lipopolysaccharide induced memory impairment, comparison to curcumin. PLoS ONE 2019, 14, e0219378. [Google Scholar] [CrossRef] [PubMed]
- Siedle, B.; García-Piñeres, A.J.; Murillo, R.; Schulte-Mönting, J.; Castro, V.; Rüngeler, P.; Klaas, C.A.; Da Costa, F.B.; Kisiel, W.; Merfort, I. Quantitative Structure—Activity Relationship of Sesquiterpene Lactones as Inhibitors of the Transcription Factor NF-κB. J. Med. Chem. 2004, 47, 6042–6054. [Google Scholar] [CrossRef] [PubMed]
- Talhouk, R.S.; Nasr, B.; Fares, M.-B.; Ajeeb, B.; Nahhas, R.; Al Aaraj, L.; Talhouk, S.N.; Ghaddar, T.H.; Saliba, N.A. Anti-Inflammatory and Cytostatic Activities of a Parthenolide-Like Sesquiterpene Lactone fromCota palaestinasubsp.syriaca. Evid. Based Complement. Altern. Med. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Heilmann, J.; Wasescha, M.R.; Schmidt, T.J. The influence of glutathione and cysteine levels on the cytotoxicity of helenanolide type sesquiterpene lactones against KB cells. Bioorganic. Med. Chem. 2001, 9, 2189–2194. [Google Scholar] [CrossRef]
- Guzman, M.L.; Rossi, R.M.; Neelakantan, S.; Li, X.; Corbett, C.A.; Hassane, D.; Becker, M.W.; Bennett, J.M.; Sullivan, E.; Lachowicz, J.L.; et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 2007, 110, 4427–4435. [Google Scholar] [CrossRef]
- Li, X.; Payne, D.T.; Ampolu, B.; Bland, N.; Brown, J.T.; Dutton, M.J.; Fitton, C.A.; Gulliver, A.; Hale, L.; Hamza, D.; et al. Derivatisation of parthenolide to address chemoresistant chronic lymphocytic leukaemia. MedChemComm 2019, 10, 1379–1390. [Google Scholar] [CrossRef] [Green Version]
- Adekenov, S. Chemical modification of arglabin and biological activity of its new derivatives. Fitoterapia 2016, 110, 196–205. [Google Scholar] [CrossRef]
- Andreasen, M.F.; Kroon, P.A.; Williamson, G.; Conesa, M.T.G. Esterase Activity Able to Hydrolyze Dietary Antioxidant Hydroxycinnamates is Distributed along the Intestine of Mammals. J. Agric. Food Chem. 2001, 49, 5679–5684. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Kratz, F.; Merfort, I. In vitro Behaviour of Sesquiterpene Lactones and Sesquiterpene Lactone-Containing Plant Preparations in Human Blood, Plasma and Human Serum Albumin Solutions. Planta Med. 2004, 70, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Koukoulitsa, C.; Geromichalos, G.D.; Skaltsa, H. VolSurf analysis of pharmacokinetic properties for several antifungal sesquiterpene lactones isolated from Greek Centaurea sp. J. Comput. Mol. Des. 2005, 19, 617–623. [Google Scholar] [CrossRef] [PubMed]
- García, C.J.; Beltrán, D.; Tomás-Barberán, F.A. Human Gut Microbiota Metabolism of Dietary Sesquiterpene Lactones: Untargeted Metabolomics Study of Lactucopicrin and Lactucin Conversion In Vitro and In Vivo. Mol. Nutr. Food Res. 2020, 64. [Google Scholar] [CrossRef]
- Xu, R.; Zhou, G.; Peng, Y.; Wang, M.; Li, X. Pharmacokinetics, Tissue Distribution and Excretion of Isoalantolactone and Alantolactone in Rats after Oral Administration of Radix Inulae Extract. Molecules 2015, 20, 7719–7736. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Main SLs | Model | Extract Concentration Range | Inflammatory Pathways | Consequences | References | |
|---|---|---|---|---|---|---|---|
| Cichorium intybus L. | Dihydrolactucin, lactucin, deoxylactucin, jacquinelin and dihydrolactucopicrin | In vitro | RAW 264.7 murine macrophages + LPS | IC50 (μg/mL): 117 for COX-2; 39 for iNOS; 48 for TNF-α; 22 for IL-1β; 21 for NO | - | ↓ COX-2, iNOS, TNF-α, IL-1β, NO | [17] |
| In vivo | Paw edema model: Wistar rats + carrageenan (subcutaneous) | 50–100 mg/kg (oral administration) | - | ↓ paw volume (edema) | |||
| Arthritis model: Wistar rats + collagen (intravenous) | 200 mg/kg (oral administration) | - | |||||
| Artemisia leucodes L. | Leukomisin and austricin | In vitro | RAW264.7 murine macrophages + LPS | 2–100 μg/mL | - | ↓ COX-2, iNOS, IL-1β, NO | [18] |
| COX-1 and -2 enzymatic assay | 45–225 μg/mL | ↓ COX-2 | |||||
| In vivo | Paw edema model: Wistar rats + carrageenan (subcutaneous) | 50–200 mg/kg (oral administration) | ↓ paw edema | ||||
| Chronic inflammation model: Wistar rats + cotton implant granuloma test | 50 mg/kg (oral administration) | ↓ granuloma and inflammatory cell infiltrate | |||||
| Artemisia khorassanica L. | Unspecified | In vitro | J774A.1 murine macrophages + LPS | 10–100 μg/mL | ↓ NF-κB | ↓ COX-2, PGE2, iNOS, NO, TNF-α and IL-1β | [19] |
| Artemisia sps (A. kopetdaghensis, A. santolina, A. Sieberi, A. Fragrans, A. Absinthium, A. ciniformis) | Saturated, unsaturated and unusual SLs | In vitro | J774A.1 murine macrophages + LPS | 10–100 μg/mL | - | ↓ COX-2, PGE2, iNOS and NO | [20] |
| Eupatorium perfoliatum L. | Diguaiaperfolin (dimeric guaianolide) and Eupafolin (flavonoid) | In vitro | RAW264.7 murine macrophages + LPS | 1–100 μg/mL | - | ↓ NO, CSF-3, IL-6, IL-1α, IL-1β, TNF, Chemokine (C-C motif) ligand (CCL)-2, CCL22 and CXCL10 | [21] |
| Xanthium spinosum L. | Ziniolide | In vitro | Rat polymorphonuclear leukocytes (PMNLs) + ionophore A23187 and Ca2+ | 0–100 μg/mL | ↓ NF-κB and arachidonic acid | ↓ 5-LOX | [22] |
| Human platelets + ionophore A23187 | 25–200 μg/mL | ↓ COX-1 and 12-LOX; ↑ 15(S)-HETE | |||||
| HeLa cells + Phorbol 12-myristate 13-acetate (PMA) | 12.5–100 μg/mL | ↓ NF-κB activation | |||||
| Arnica montana L. | Helenalin and dihydrohelenalin ester derivatives | In vitro | Jurkat T cells + TNF-α or PMA | 0.5–10 μL/mL | ↓ NF-κB and NFAT | ↓ NF-κB and NFAT DNA-binding | [23] |
| Human PBMCs from healthy donors + LPS | 0.001–10 μL/mL | ↓ TNF-α and IL-1β | |||||
| Centaurea L. species (C. aphrodisea, C. athoa, C. hyalolepis, C. iberica, C. polyclada) | SL fraction (athoin, 14-O-acetylathoin and methyl-14-O-acetylathoin-12-oate in C. athoa) | In vitro | SW1353 human chondrosarcoma cells + PMA | 0–100 μg/mL | ↓ NF-κB | ↓ NF-κB activity | [24,25] |
| RAW264.7 murine macrophages + LPS | ↓ NO | ||||||
| In vivo | Paw edema model: Wistar rats + carrageenan (subcutaneous) | 6.75–50 mg/kg (oral administration) | ↓ edema | ||||
| Inula helenium L | Alantolactone and isoalantolactone | In vitro | bEnd.3 mouse endothelial cells + TNF-α | 0.6–2.4 μg/mL | ↓ NF-κB and MAPKs | ↓NF-κB inhibitor (IκB)-α, NF-κB p65, p38 and c-Jun N-terminal kinase (JNK) phosphorylation | [26] |
| RAW264.7 murine macrophages + LPS | ↓ IL-1, IL-6 and iNOS | ||||||
| Primary synovial fibroblasts from rheumatoid arthritis patients + TNF-α | ↓ IL-1, MCP-1 and MMP-3 | ||||||
| In vivo | Adjuvant-induced mice arthritis model | 12.5–50 mg/kg (oral administration) | ↓ paw swelling | ||||
| Collagen-induced mice arthritis model | |||||||
| Arctium lappa L. | Onopordopicrin | In vivo | Colitis model: Wistar rats + Trinitrobenzene Sulfonic Acid (TNBS) (enteral instillation) | 25–50 mg/kg (oral administration) | - | ↓ TNF-α and COX-2; ↓ histological damage; ↓ mucin layer loss; ↓ neutrophil infiltration | [27] |
| Vernonia scorpioides L. | Diacethylpiptocarphol and related hirsutinolides | In vivo | Acute ear edema model: Swiss mice + 12-O-tetradecanoylphorbol acetate (TPA) (topical) | 0.003–1 mg (topical) | ↓ NF-κB | ↓ neutrophil infiltration, edema and epidermal proliferation | [29] |
| Chronic ear edema model: Swiss mice + arachidonic acid (topical) or croton oil (topical) | 1 mg (topical) | ||||||
| SL Subclass | Compound Name (ID Number) | Model | Compound Concentration Ranges Tested | Inflammatory Pathways | Consequences | References | |
|---|---|---|---|---|---|---|---|
| Germacranolides | Parthenolide (1) | In vitro | Rat aortic smooth muscle cells + LPS/IFN-γ | 3–30 μM | ↓ NF-κB | ↓ iNOS and NO release | [31] |
| In vitro | BV2 mouse microglia + LPS | 5 μM | ↓ NF-κB | ↓ IL-6 and TNF-α | [32] | ||
| In vitro | Rat primary neural-glial cells + LPS | 403 μM | ↓ NF-κB, NF-IL6, Nrf-1 and PGC1α | ↓ IL-6 and TNF-α | [33] | ||
| In vivo | Wistar rats + LPS (intraperitoneal injection) | 1 mg/kg (intraperitoneal injection) | ↓ IL-6 and TNF-α in plasma; ↓ COX-2, NF-IL6, SOCS3, IκBα and Tribbles pseudokinase 1 (Trib1) in hypothalamus; ↓ fever | ||||
| In vitro | Jurkat T cells and primary peripheral human T cells + PMA/ionomycin or anti-CD3/CD28 | 1.25–5 μM | ↓ NF-κB and AP-1 | ↓ IL-4, IL-2 and IFN-γ | [34] | ||
| Primary peripheral human T cells + PMA/ionomycin or anti-CD3/CD28 | |||||||
| In vitro | Blood from healthy donors + PMA/ionomycin | 10–500 μM | - | ↓ IL-2; ↓ T-lymphocyte activation | [35] | ||
| In vitro | Human THP-1 monocytes + LPS | 0.75–12 μM | ↓ NF-κB and MAPKs | ↓ IL-6, TNF-α, IL-1β, IL-8, IL-18 and NO; ↓ iNOS, TLR4 and TRAF6 | [36] | ||
| Human primary monocytes + LPS | |||||||
| In vivo | Hindpaw edema model: Holtzman rats + carrageenan (subcutaneous injection) | 5–20 mg/kg (intraperitoneal injection) | - | ↓ Hyperalgesia and edema | [37] | ||
| In vitro | HepG2 human hepatocytes + IL-6, oncostatin M or leukemia inhibitory factor | 5 μM | ↓ STAT3 and JAKs | ↓ STAT3 phosphorylation, dimerization and activity | [39] | ||
| Costunolide (2) | In vitro | Human THP-1 monocytes + IL-6 | 6–25 μM | ↓ IL-6/STAT3 and JAKs | ↓ MCP-1, CXCL10, ICAM-1; ↓ STAT3 phosphorylation and DNA-binding activity; ↓ Intracellular GSH | [40] | |
| In vitro | RAW264.7 murine macrophages + LPS | 0.1–1 μM | ↑ Nrf-2; ↓ NF-κB | ↑ HO-1; ↓ IL-6 and TNF-α | [38] | ||
| In vitro | Human keratinocytes from healthy donors + IL-22, IFN-γ or TNF-α | 12.5 μM | ↓ STAT3 and STAT1 | ↓ Intracellular GSH; ↓ CCL2, CXCL10, ICAM-1 and SOCS3; ↑ Epidermal growth factor receptor (EGFR) and Erk1/2 | [41] | ||
| In vitro | RAW264.7 murine macrophages + LPS | 0.1–3 μM | ↓ AP-1 and MAPKs | ↓ IL-1β | [42] | ||
| In vitro | Primary rat chondrocytes + IL-1β | 2–6 μM | ↓ NF-κB and Wnt/β-catenin; ↑ SOX-9 | ↓ MMP-3, MMP-9, MMP-13, iNOS, COX-2 and IL-6; ↑ collagen II | [43] | ||
| In vivo | Sprague-Dawley rats (surgically induced osteoarthritis model) | 6 μM (intra-articular injection) | attenuation of cartilage degeneration | ||||
| In vivo | Angiogenesis model: Swiss albino mice + polyester-polyurethane sponge implants | 5–20 mg/kg (cannula) | - | ↓ Angiogenesis, macrophage and neutrophil accumulation, and collagen deposition; ↓ IL-1β, IL-6, IL-17, TNF-α, TGF-β; ↑ IL-10 | [44] | ||
| Eupatolide (3) | In vitro | RAW264.7 murine macrophages + LPS | 0.1–10 μM | ↓ NF-κB, AP-1, MAPKs, Akt | ↓ COX-2, PGE2, iNOS, NO and TRAF6 | [45] | |
| Human embryonic kidney (HEK)-293 cells + LPS | ↑ proteossomal degradation of TRAF6 | ||||||
| Onopordopicrin (4) | In vitro | NIH-3T3 cell line + TNF-α | IC50 (μM): 8.6 for NF-κB; 15.3 for STAT3; EC50 (μM): 2.2 for Nrf-2 | ↓ NF-κB and STAT3; ↑ Nrf-2 | ↓ NF-κB activity | [46] | |
| HeLa cell line + IFN-γ | ↓ STAT3 activity | ||||||
| HaCaT keratinocytes | ↑ Nrf-2 activity | ||||||
| Deoxyelephantopin (5) | In vitro | RAW264.7 murine macrophages + LPS | 2.5–10 μM | - | ↓ high mobility group box (HMGB) 1, pyruvate kinase M2 (PKM2), glucose transporter 1 (GLUT1), lactate dehydrogenase A (LDHA) and phosphoinositide-dependent kinase 1 (PDK1) and IL-1β | [47] | |
| In vivo | C57BL/6J mice + LPS (intraperitoneal injection) | 10 mg/kg (intraperitoneal injection) | - | ↓ endotoxic shock and sepsis | |||
| Guaianolides | Dehydrocostuslactone (6) | In vitro | THP-1 human cells + IL-6 | 6–25 μM | ↓ IL-6/STAT3 and JAKs | ↓ MCP-1, CXCL10, ICAM-1; ↓ STAT3 phosphorylation and DNA-binding activity; ↓ Intracellular GSH | [40] |
| In vitro | Human keratinocytes from healthy donors + IL-22, IFN-γ or TNF-α | 12.5 μM | ↓ STAT3 and STAT1 | ↓ Intracellular GSH; ↓ CCL2, CXCL10, ICAM-1 and SOCS3; ↑ EGFR and Erk1/2 | [41] | ||
| In vivo | Colitis model: BALB/c mice + Dextran sulfate sodium (DSS) (oral administration) | 10–20 mg/kg | ↓ IL-6/STAT3 | ↓ TNF-α, IL-1β, MPO, SOD, IL-6, IL-17, IL-23, COX-2, iNOS | [50] | ||
| In vitro | RAW 264.7 macrophages + LPS | 10–20 μM | ↓ MyD88/TRIF; ↓ NF-κB; ↓ IRF-3 | ↓ COX-2, INF-β, IP-10 | [51] | ||
| Micheliolide (7) | In vitro | BV2 microglia cells + LPS | 1–10 μM | ↓ NF-κB; ↓ PI3K/Akt ↓ MAPKs | ↓ TNF-α, IL-6, IL-1β, COX-2, iNOS | [52] | |
| In vitro | RAW 264.7 macrophages + LPS | 1–10 μM | ↓ NF-κB | ↓ IL-6, TNF-α, IL-1β | [53] | ||
| In vitro | RAW264.7 macrophages + LPS | 0–10 μM | ↓ NF-κB; ↓ PI3K/Akt | ↓ IL-6, TNF-α, MCP-1, INF-β and IL-1β | [54] | ||
| Human dendritic cells and monocytes + LPS | ↓ IL-6, TNF-α, MCP-1, INF-β | ||||||
| In vivo | Arthritis model: DBA/1 mice + collagen (intradermal injection) | 30 mg/kg (intraperitoneal injection) | - | ↓ TIMP-1, M-CSF, ICAM-1, INF-γ | [55] | ||
| Cynaropicrin (8) | In vitro | RAW 264.7 macrophages + LPS | 0–35 μM | - | ↓ TNF-α and NO | [56] | |
| Human macrophages U937 + LPS | |||||||
| Primary splenocytes from mice + concanavalin A, phytohemagglutinin and LPS | ↓ lymphocyte proliferation | ||||||
| Arglabin (9) | In vitro | Peritoneal macrophages from ApoE2.Ki mice + LPS and cholesterol crystals | 50 nM | ↓ NF-κB; ↓ NLRP3 | ↓ IL-1α, IL-1β, IL-18 | [57] | |
| 11β,13-dihydrolactucin (10) | In vitro | Yeast S. cerevisiae + MnCl2 | 0.36–18 μM | ↓ Calcineurin-Crz1 (NFAT) | ↓ NFAT nuclear translocation and transcriptional activity | [58] | |
| 8-deoxylactucin (11) | In vitro | Human colon-cancer cells HT29 + TNF-α | 115 μM | ↓ NF-κB | ↓ PGE2 | [59] | |
| Eudesmanolides | Alantolactone (12) | In vitro | bEnd.3 mouse endothelial cells + TNF-α | 2.6–10.3 μM | ↓ NF-κB and MAPKs | ↓ IκBα, NF-κB p65, p38 and JNK phosphorylation | [26] |
| RAW264.7 murine macrophages + LPS; | ↓ IL-1, IL-6 and iNOS | ||||||
| Primary synovial fibroblasts from rheumatoid arthritis patients + TNF-α | ↓ IL-1, MCP-1 and MMP-3 | ||||||
| In vitro | RAW 264.7 macrophages + LPS | 1.25–10 μM | ↓ NF-κB; ↓ MyD88 | ↓ iNOS, COX-2, TNF-α | [60] | ||
| In vivo | Colitis model: C57BL/6 mice + DSS (oral administration) | 50 mg/kg (oral administration) | ↓ NF-κB; ↑ hPXR | ↓ iNOS, ICAM-1, COX-2, TNF-α, IFN-γ, IL-6 | [61] | ||
| Isoalantolactone (13) | In vitro | bEnd.3 mouse endothelial cells + TNF-α | 2.6–10.3 μM | ↓ NF-κB and MAPKs | ↓ IκBα, NF-κB p65, p38 and JNK phosphorylation | [26] | |
| RAW264.7 murine macrophages + LPS | ↓ IL-1, IL-6 and iNOS | ||||||
| Primary synovial fibroblasts from rheumatoid arthritis patients + TNF-α | ↓ IL-1, MCP-1 and MMP-3 | ||||||
| In vitro | 293T cells + TNF-α | 2.5–10 μM | ↓ NF-κB and MAPKs | ↓ UbcH5 | [62] | ||
| In vivo | Hepatitis model: BALB/c mice + TNF-α and D-galactosamine (D-GalN) (intraperitoneal injection) | 10 mg/kg (intraperitoneal injection) | ↓ serum alanine aminotransferase (ALT); ↓ hepatocyte damage; ↓ IL-6, MCP-1, ICAM-1 and VCAM-1 | ||||
| JEUD-38 (14) | In vitro | RAW 264.7 macrophages + LPS | 2.5–10 μM | ↓ NF-κB and MAPKs | ↓ iNOS | [63] | |
| 7-hydroxyfrullanolide (16) | In vitro | THP-1 cell + LPS | 0.3–100 μM | ↓ NF-κB | ↓ NF-κB activation and nuclear translocation | [64] | |
| HUVECs + LPS | ↓ ICAM-1, VCAM-1, E-selectin ↓ Monocyte adhesion | ||||||
| PBMCs + LPS | ↓ NF-κB-related gene expression | ||||||
| In vitro | PBMCs + LPS | 0.3–100 μM | - | ↓ IL-6 and TNF-α | [65] | ||
| Primary human synovial tissue cells | |||||||
| In vivo | Colitis model: BALB/c mice + DSS (oral administration) | 75 mg/kg (oral administration) | - | ↓ TNF-α and IL-6; ↓ Colonic edema; ↓ Shortening of the colon; ↓ hemoglobin and rectal bleeding; ↓ neutrophil infiltration | |||
| Paw edema model: Wistar rats + carrageenan (subcutaneous injection) | 100 mg/kg | - | ↓ paw edema | ||||
| Arthritis model: DBA/1J mice + collagen (intradermal injection) | 25–75 mg/kg (oral administration) | - | ↓ joint deformities and bone destruction | ||||
| Santamarin (17) | In vitro | RAW264.7 macrophages + LPS | 5–40 μM | ↓ NF-κB; ↑ Nfr2 | ↓ COX-2 and iNOS; ↓ TNF-α, IL-1β ↑ HO-1 | [66] | |
| Murine peritoneal macrophages + LPS | ↓ COX-2 and iNOS; ↓ TNF-α, IL-1β | ||||||
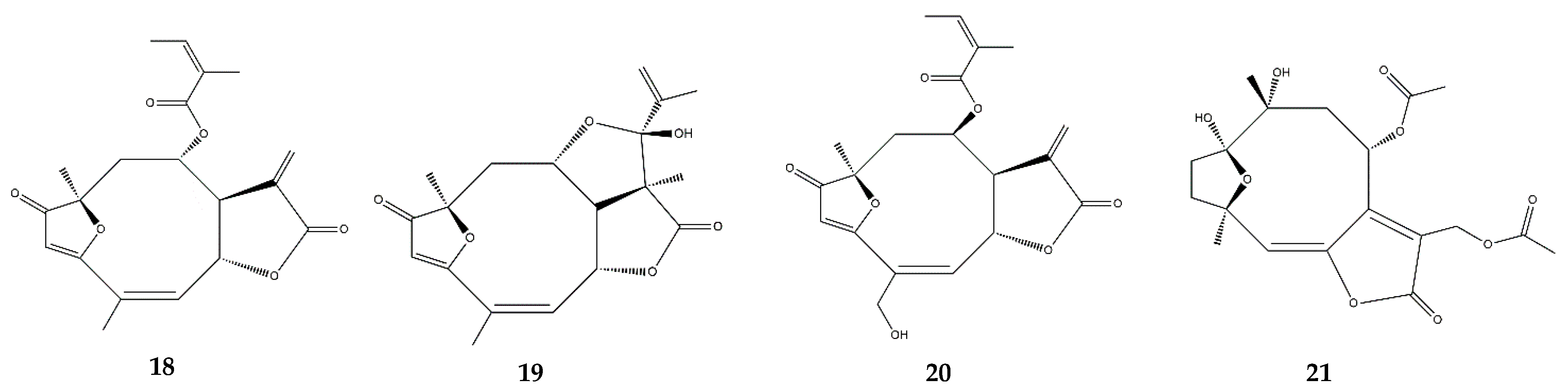
| Heliangolides | Lychnopholide (18) | In vitro | J774A.1 macrophages + INF-γ and LPS | 0.0125–0.2 μM | - | ↑ IL-10; ↓ NO | [67] |
| Eremantholide (19) | J774A.1 macrophages + INF-γ and LPS | 0.625–10 μM | - | ↑ IL-10; ↓ TNF-α | |||
| Budlein A (20) | In vitro | RAW264.7 + TNF-α or IL-1β | 2.7 × 104–26.7 μM | ↓ NF-κB | ↓ NF-κB activity | [68] | |
| In vivo | Arthritis model: C57BL/6 mice + methylated bovine serum albumin (intra-articular injection) | 1–10 mg/kg (oral administration) | ↓ edema; ↓ neutrophil and leukocyte infiltration; ↓ proteoglycan degradation; ↓ IL-33, TNF-α, IL-1β, COX-2 | ||||
| In vivo | Paw edema model: Swiss mice + carrageenan (subcutaneous injection) | 1–10 mg/kg | - | ↓ TNF-α, IL-1β; ↓ edema, and neutrophil infiltration; ↓ mechanical hypernocecipetion | [69] | ||
| In vivo | Gout arthritis model: Swiss mice + monosodium urate crystals (intra-articular injection) | 1–10 mg/kg (oral administration) | ↓ NF-κB; ↓ NLRP3 inflammasome | ↓ TNF-α and IL-1β; ↓ neutrophil recruitment; ↓ edema and mechanical hypersensitivity | [70] | ||
| In vitro | Bone marrow derived macrophages (BMDMs) + LPS and monosodium urate crystals | 2.7–26.7 mM | ↓ TNF-α and IL-1β | ||||
| Diacethylpiptocarphol (21) | In vivo | Colitis model: BALB/c mice + DSS (oral administration) | 5 mg/kg (oral administration) | - | ↓ TNF-α; ↑ TGF-β; ↓ immune cell infiltration and tissue damage | [71] | |
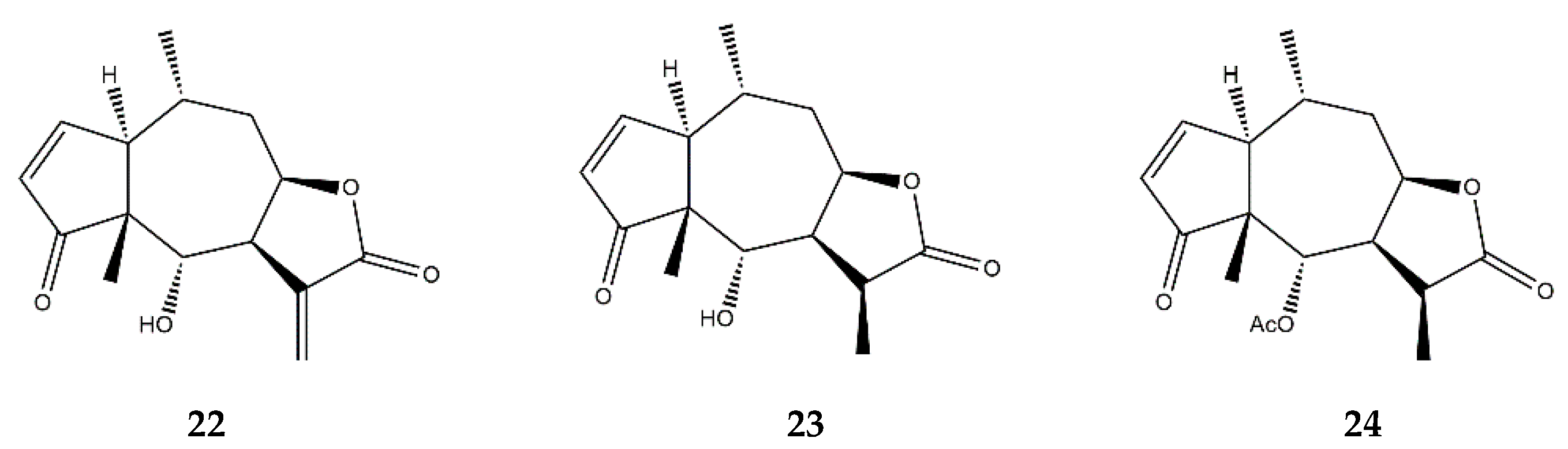
| Pseudoguaianolides | Helenalin (22) | In vitro | Jurkat T cells + TNF-α | 5–200 μM | ↓ NF-κB | ↓ NF-κB DNA-binding | [72] |
| In vitro | Jurkat T cells + TNF-α | 10 μM | ↓ NF-κB | ↓ NF-κB DNA-binding and nuclear translocation | [73] | ||
| In vitro | Jurkat CD4+ T-cells | 0.5–5 μM | ↓ NFAT ↓ NF-κB | ↓ IL-2 ↓ proliferation of CD4+ cells | [23,74] | ||
| In vitro | THP-1 cells + LPS | 0.52–1.08 μM | ↓ NF-κB | ↓ IL-1α, IL-19, MCP-3, GM-CSF | [75] | ||
| In vitro | A2780 human ovarian cancer cell line | 0.5–2 μM | ↓ NF-κB | ↓ NF-κB p65 expression | [76] | ||
| 11α,13-dihydrohelenalin (23) | In vitro | PBMCs + LPS | 2–20 μM | ↓ NF-κB and NFAT | ↓ IL-2, IL-6, GM-CSF, TNF-α, INF-γ, iNOS | [77] | |
| Jurkat T-cells + LPS | ↓ NF-κB and NFAT levels | ||||||
| 11α,13-dihydrohelenalin–acetate (24) | In vitro | PBMCs + LPS | 2–20 μM | ↓ NF-κB and NFAT | ↓ IL-2, IL-6, GM-CSF, TNF-α, INF-γ, iNOS | ||
| Jurkat T-cells + LPS | ↓ NF-κB and NFAT levels | ||||||
| In vitro | Human granulocytes + Ionophore A23187 | 1–600 μM | ↓ Arachidonic Acid | ↓ Leukotriene C4 synthase; ↓ 5-lipooxygenase | [78] | ||
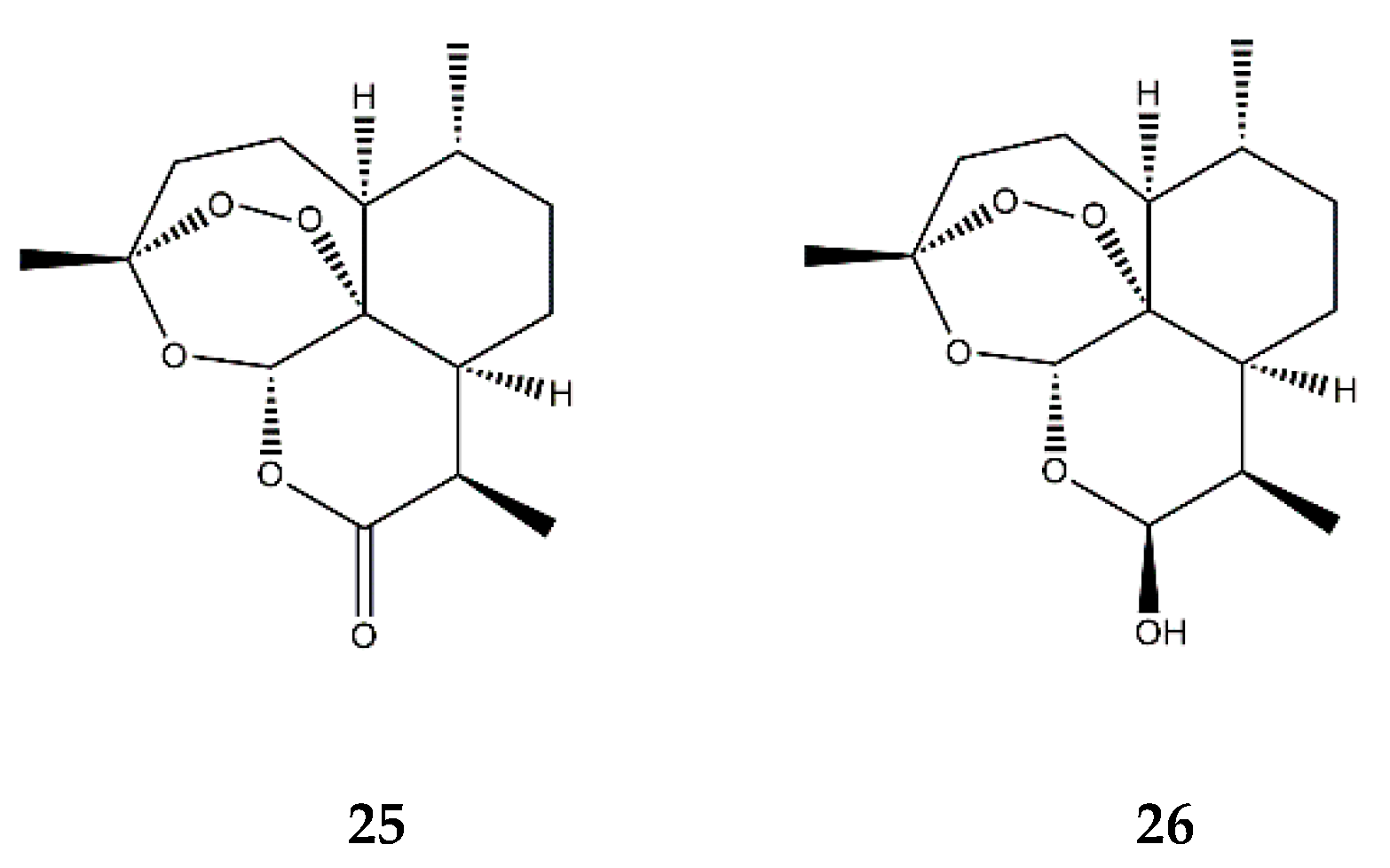
| Endoperoxide SL | Artemisinin (25) | In vitro | HUVECs + TNF-α | 50–200 μM | ↓ NF-κB; ↓ MAPKs | ↓ ICAM-1, VCAM-1; ↓ adhesion of monocytes | [80] |
| Dihydroartemisinin (26) | In vitro | RAW 264.7 macrophages + PMA | 5–25 μM | ↓ NF-κB, AP-1 and MAPKs | ↓ COX-2 | [81] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matos, M.S.; Anastácio, J.D.; Nunes dos Santos, C. Sesquiterpene Lactones: Promising Natural Compounds to Fight Inflammation. Pharmaceutics 2021, 13, 991. https://doi.org/10.3390/pharmaceutics13070991
Matos MS, Anastácio JD, Nunes dos Santos C. Sesquiterpene Lactones: Promising Natural Compounds to Fight Inflammation. Pharmaceutics. 2021; 13(7):991. https://doi.org/10.3390/pharmaceutics13070991
Chicago/Turabian StyleMatos, Melanie S., José D. Anastácio, and Cláudia Nunes dos Santos. 2021. "Sesquiterpene Lactones: Promising Natural Compounds to Fight Inflammation" Pharmaceutics 13, no. 7: 991. https://doi.org/10.3390/pharmaceutics13070991
APA StyleMatos, M. S., Anastácio, J. D., & Nunes dos Santos, C. (2021). Sesquiterpene Lactones: Promising Natural Compounds to Fight Inflammation. Pharmaceutics, 13(7), 991. https://doi.org/10.3390/pharmaceutics13070991