
Modulation of Temoporfin Distribution in Blood by β-Cyclodextrin Nanoshuttles

 ,
, 
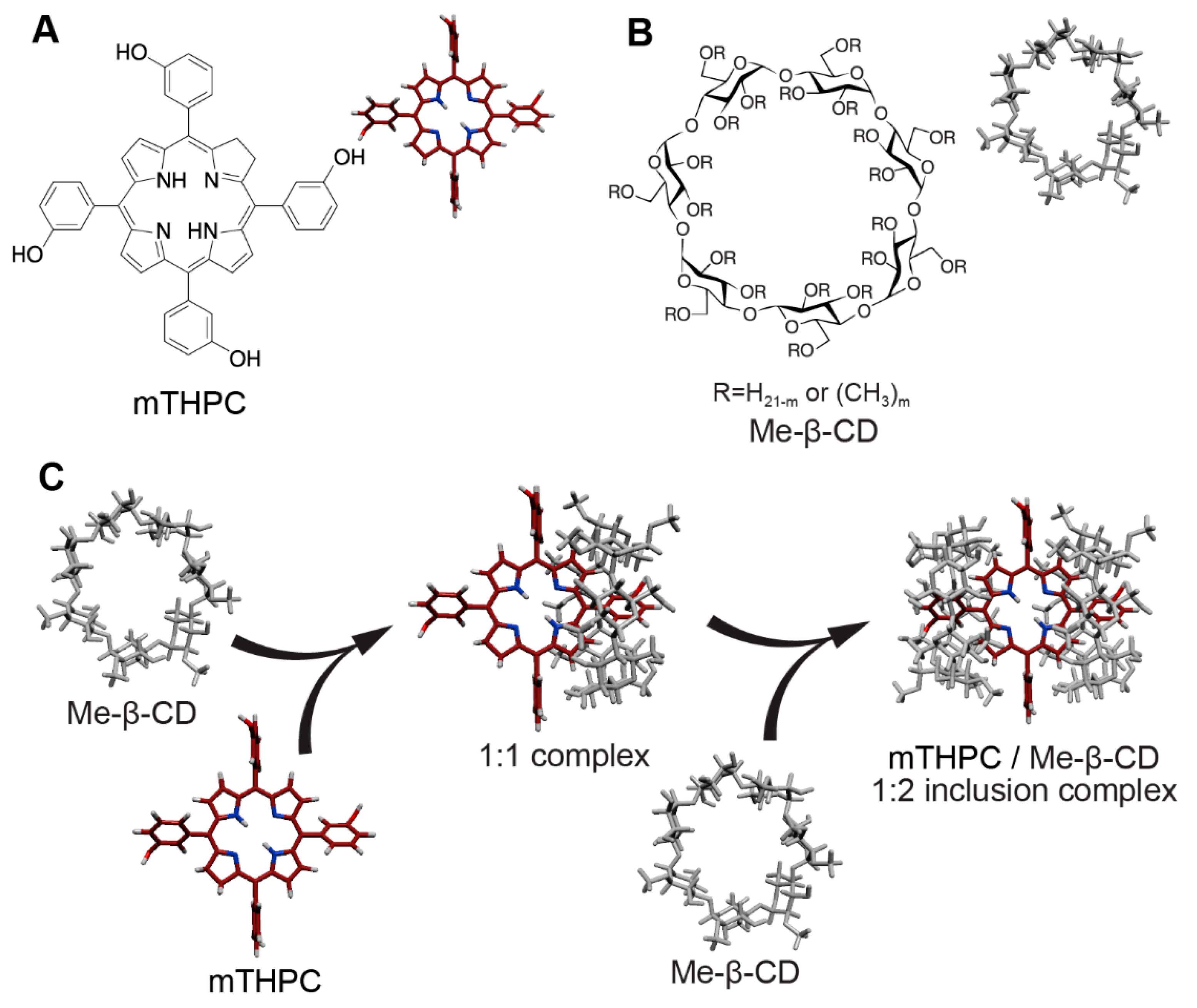{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines
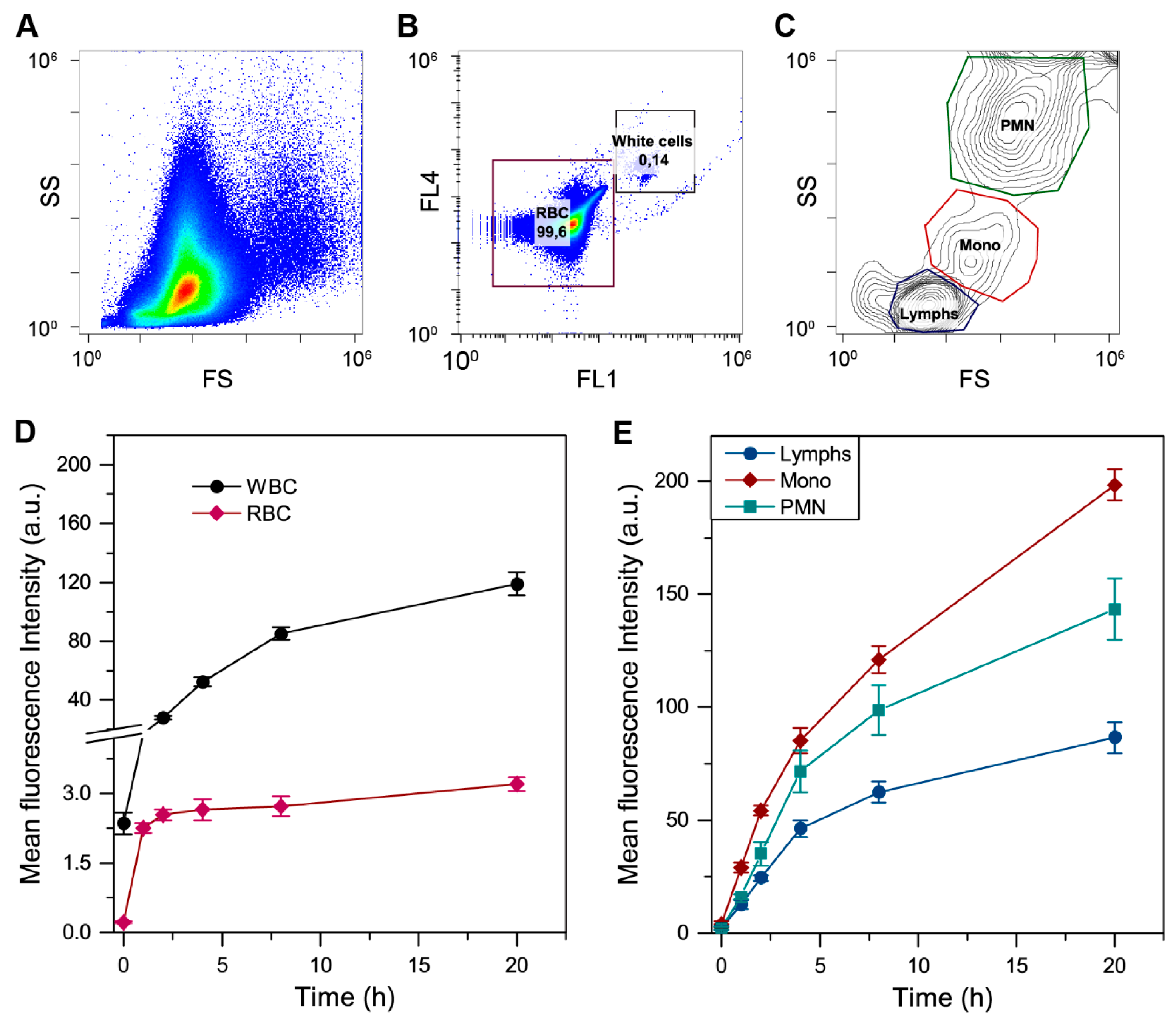
2.3. Flow Cytometry
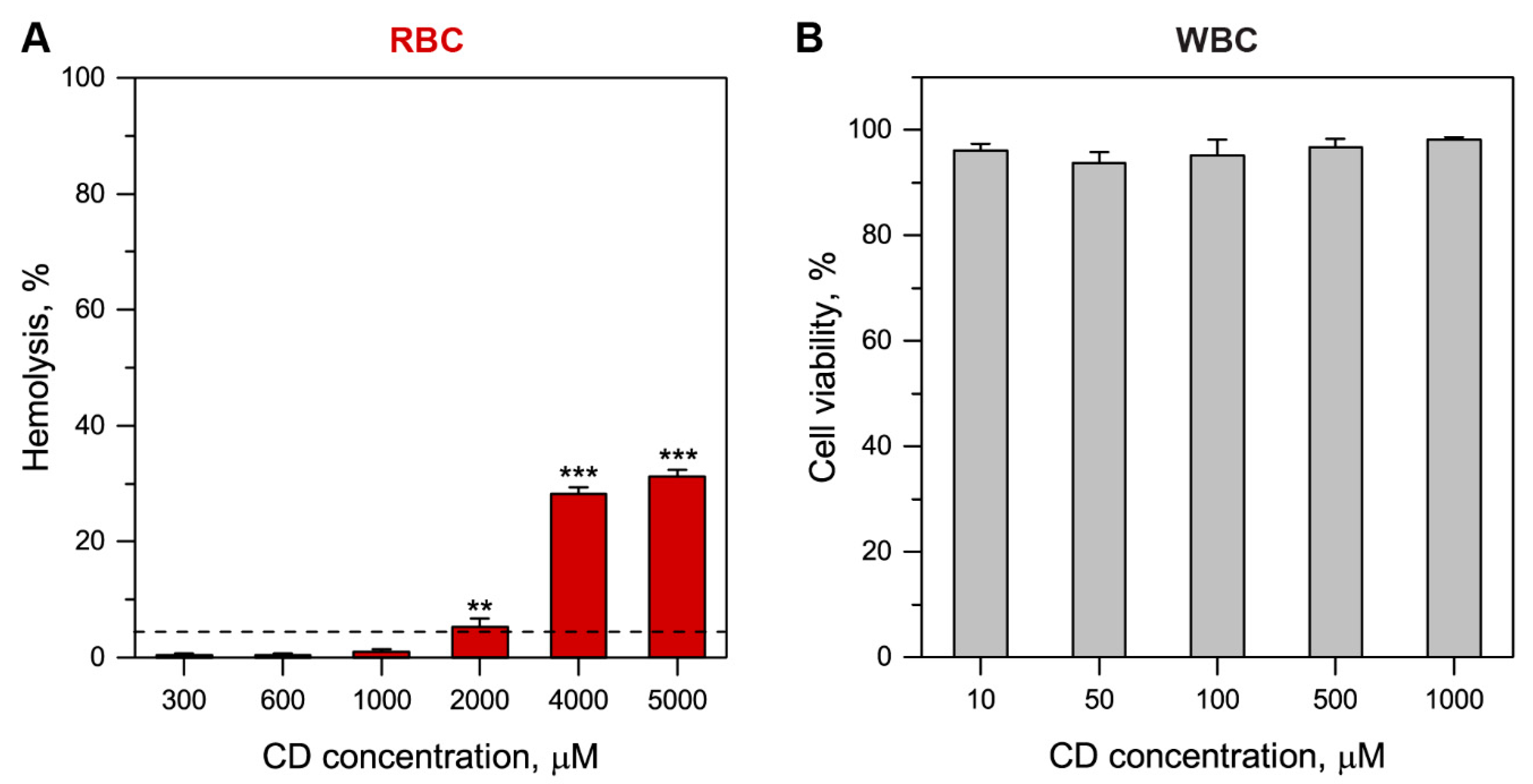
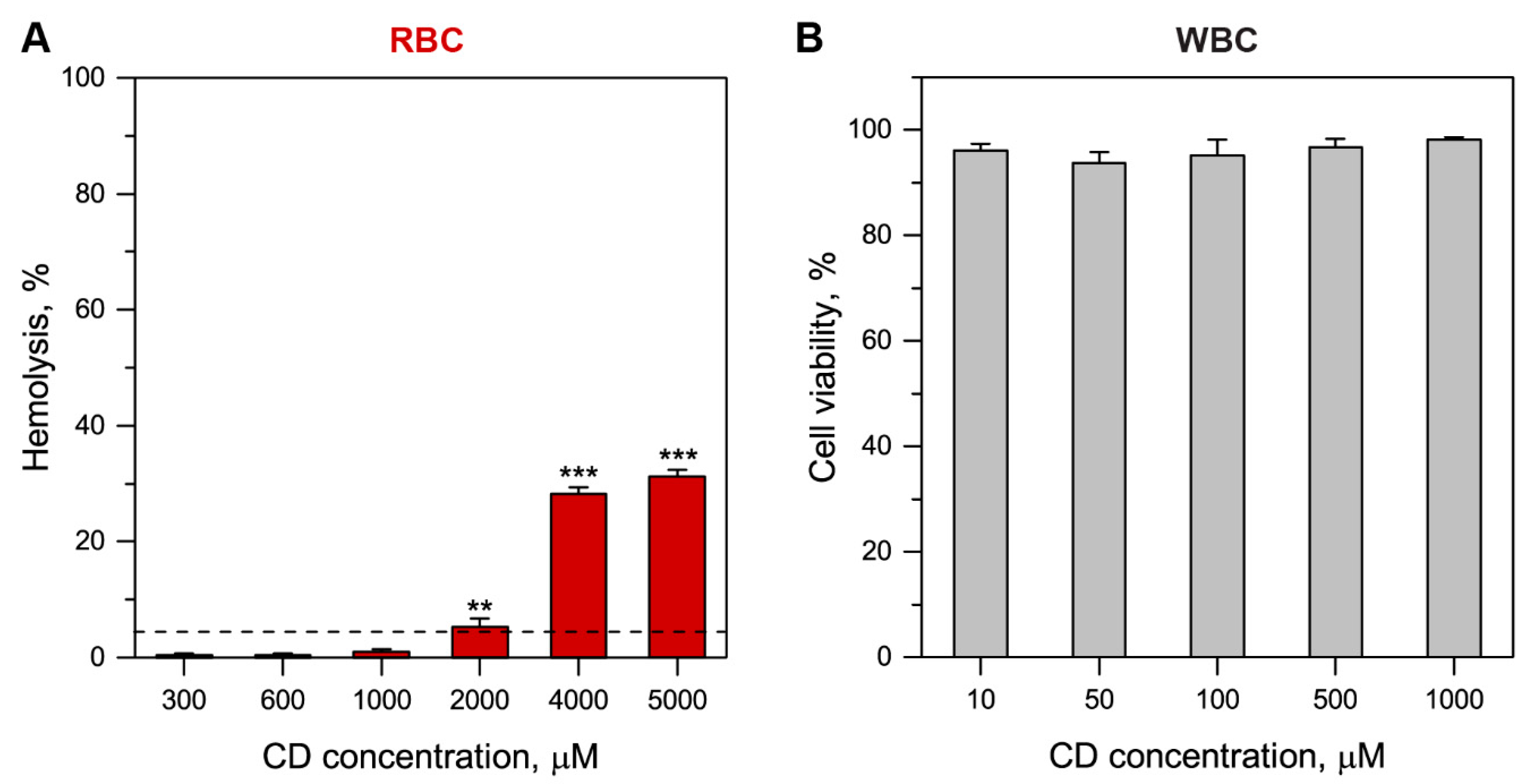
2.4. Me-β-CD Toxicity
2.5. Fluorescence Microscopy
2.6. Statistics
3. Results
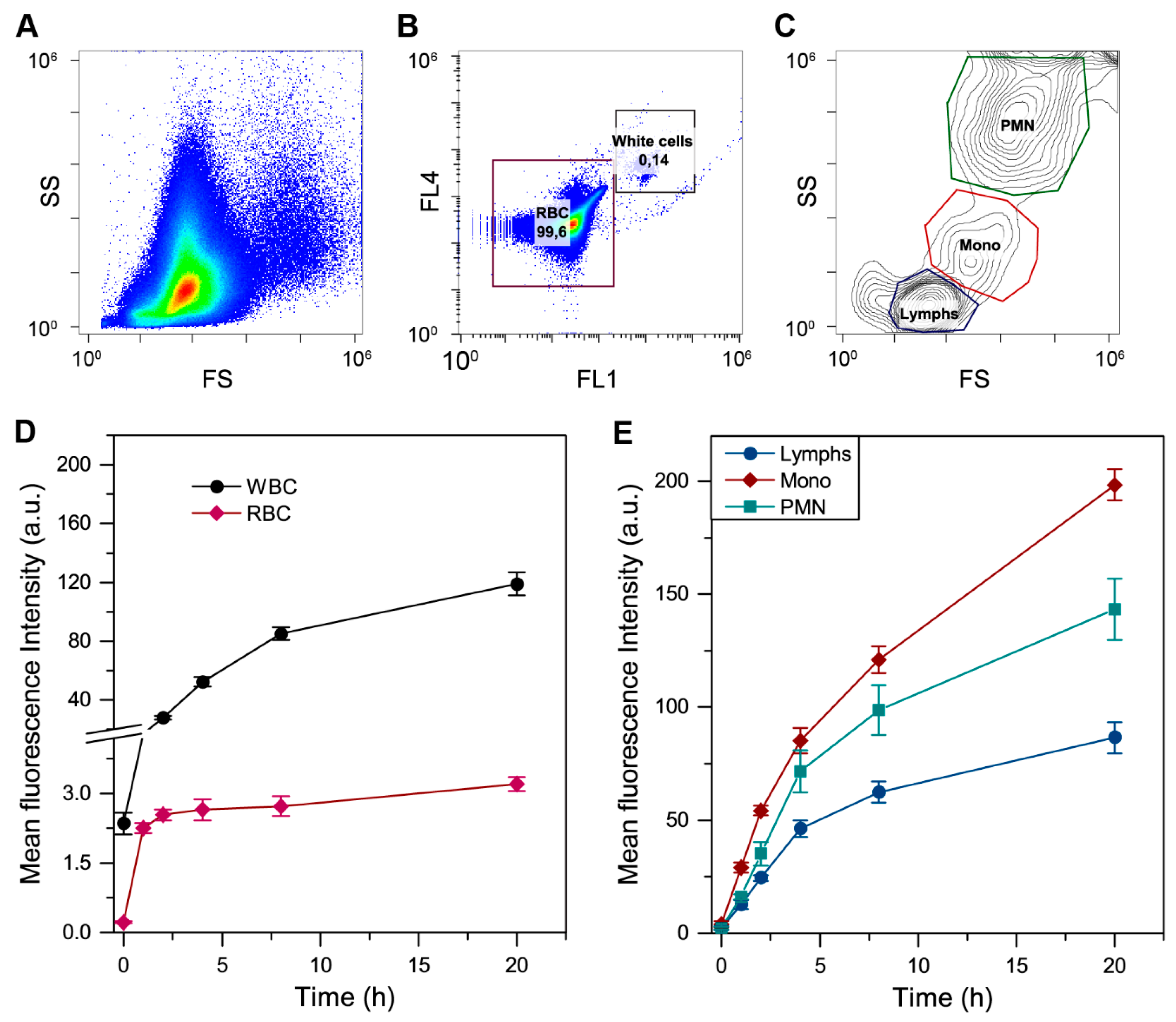
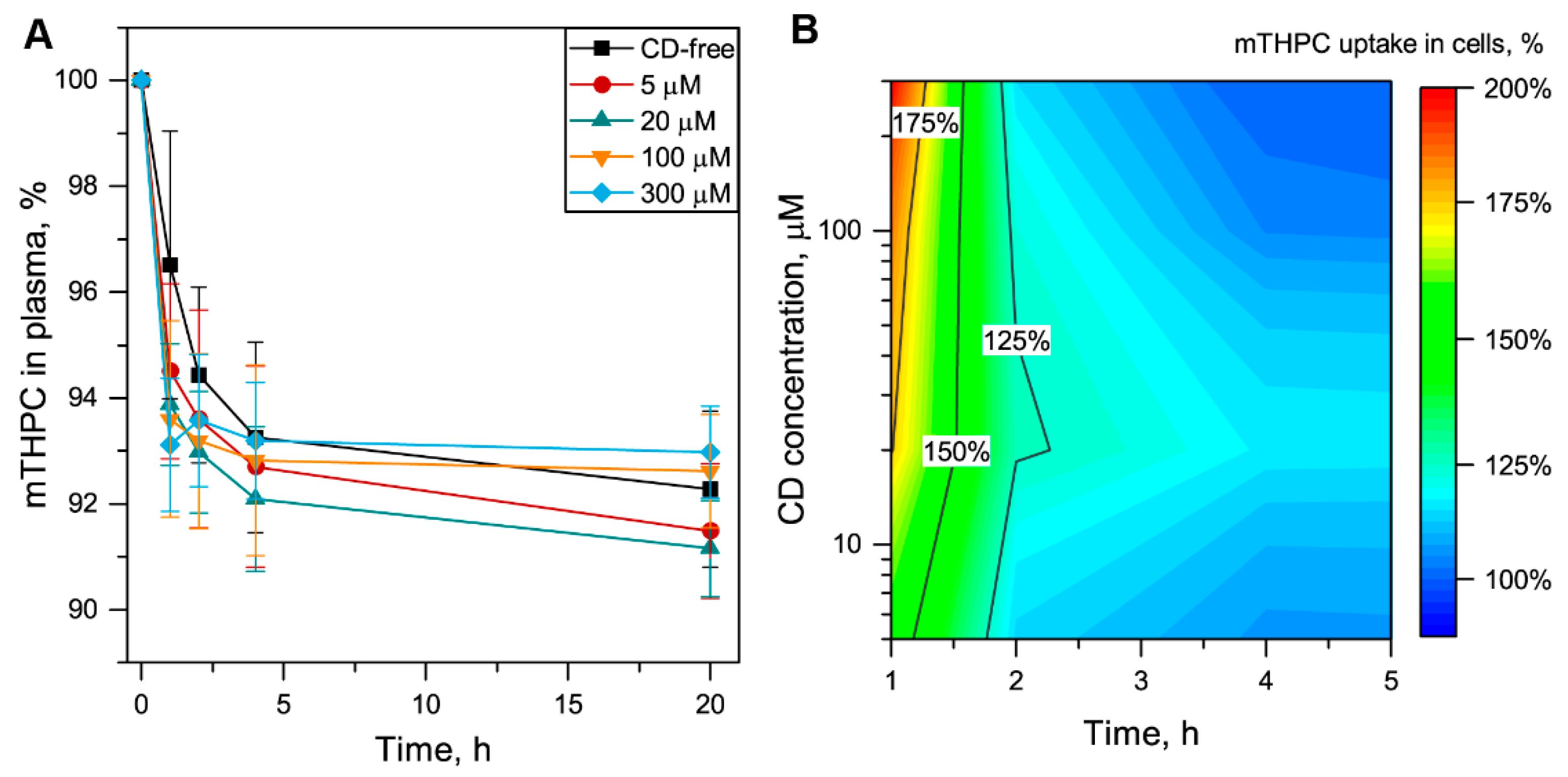
3.1. Distribution of Free mTHPC in Blood
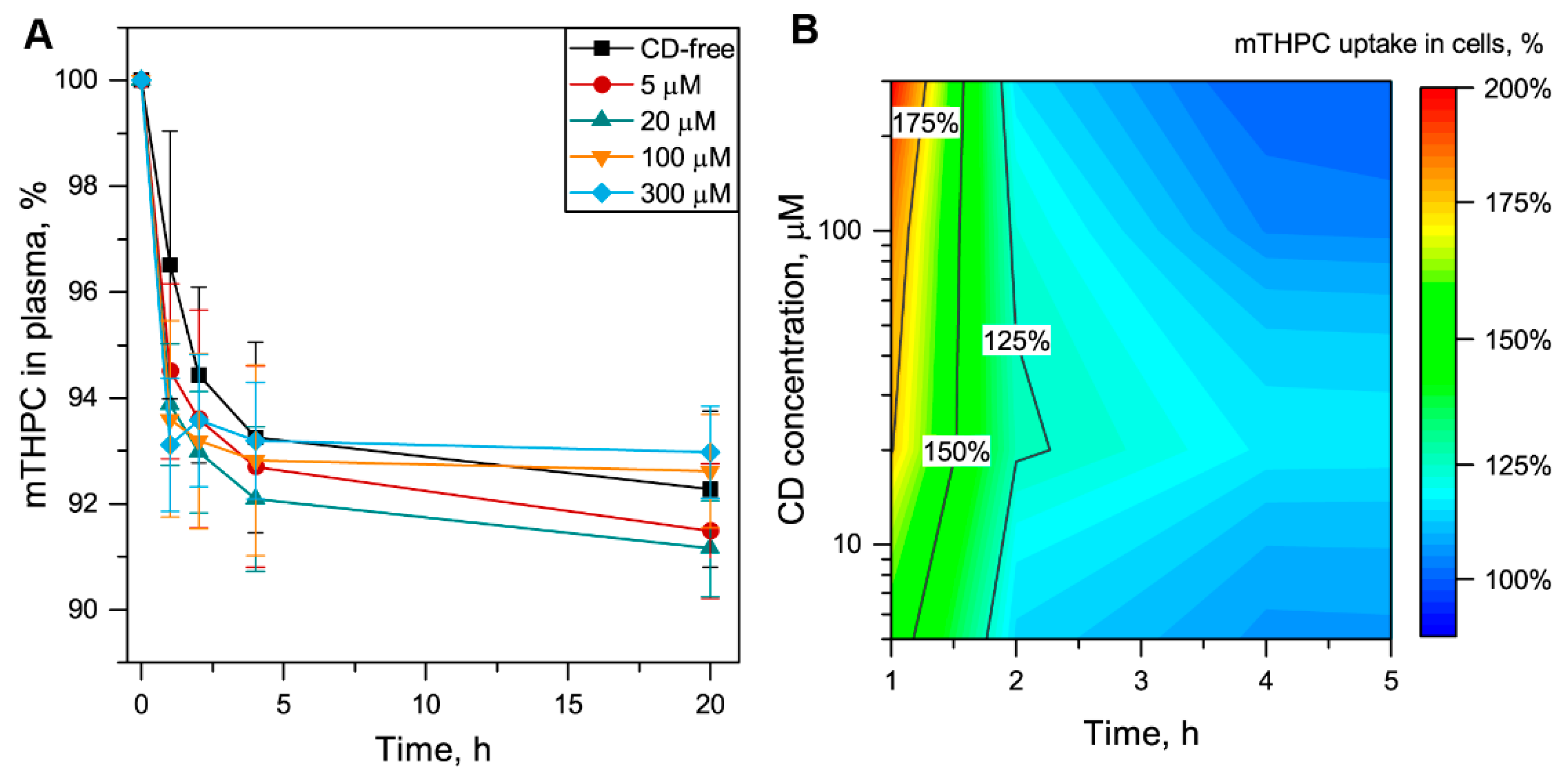
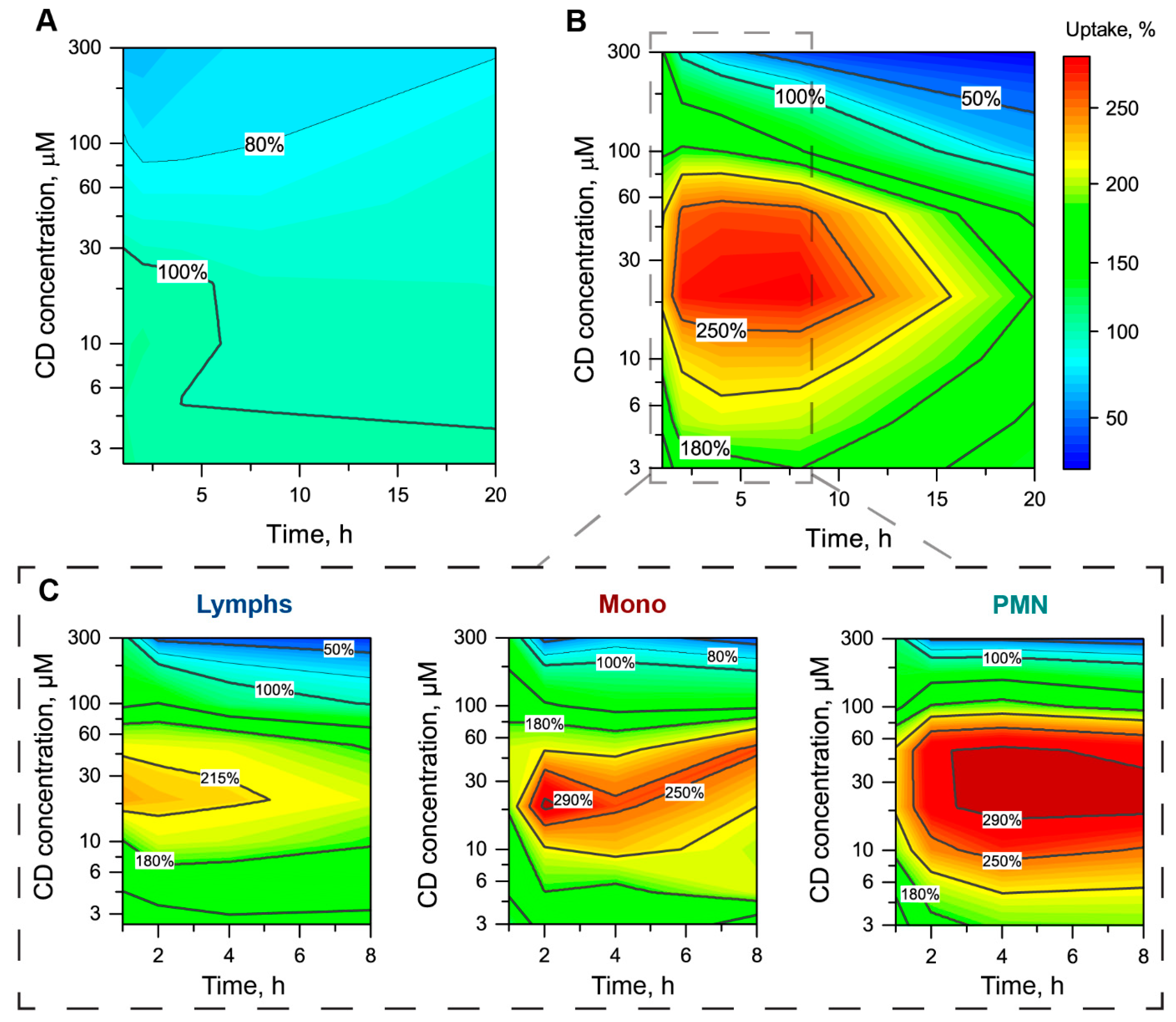
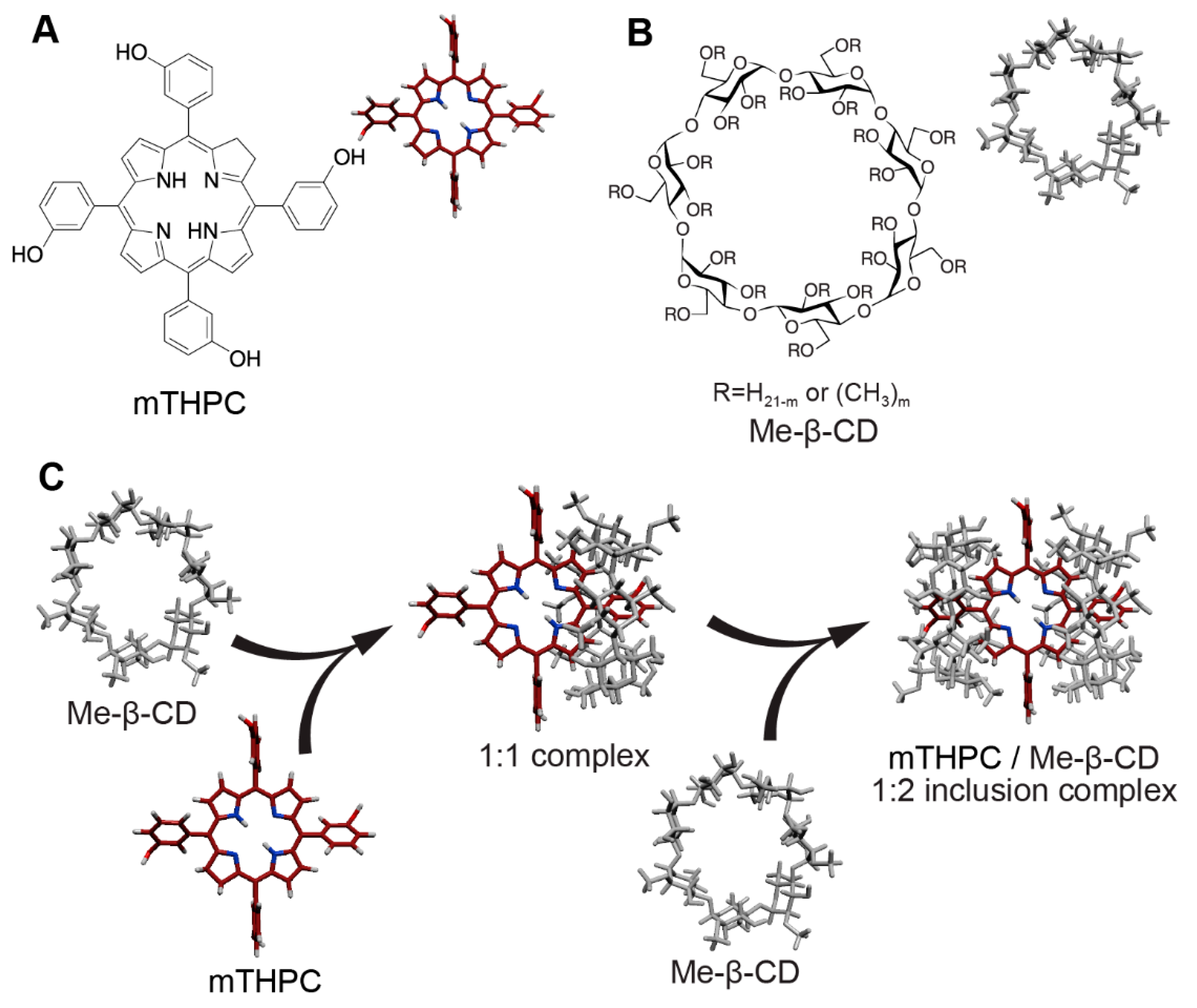
3.2. Alteration of mTHPC Distribution in Blood by Cyclodextrins
3.2.1. Me-β-CD Toxicity
3.2.2. Redistribution of mTHPC from Plasma to Blood Cells
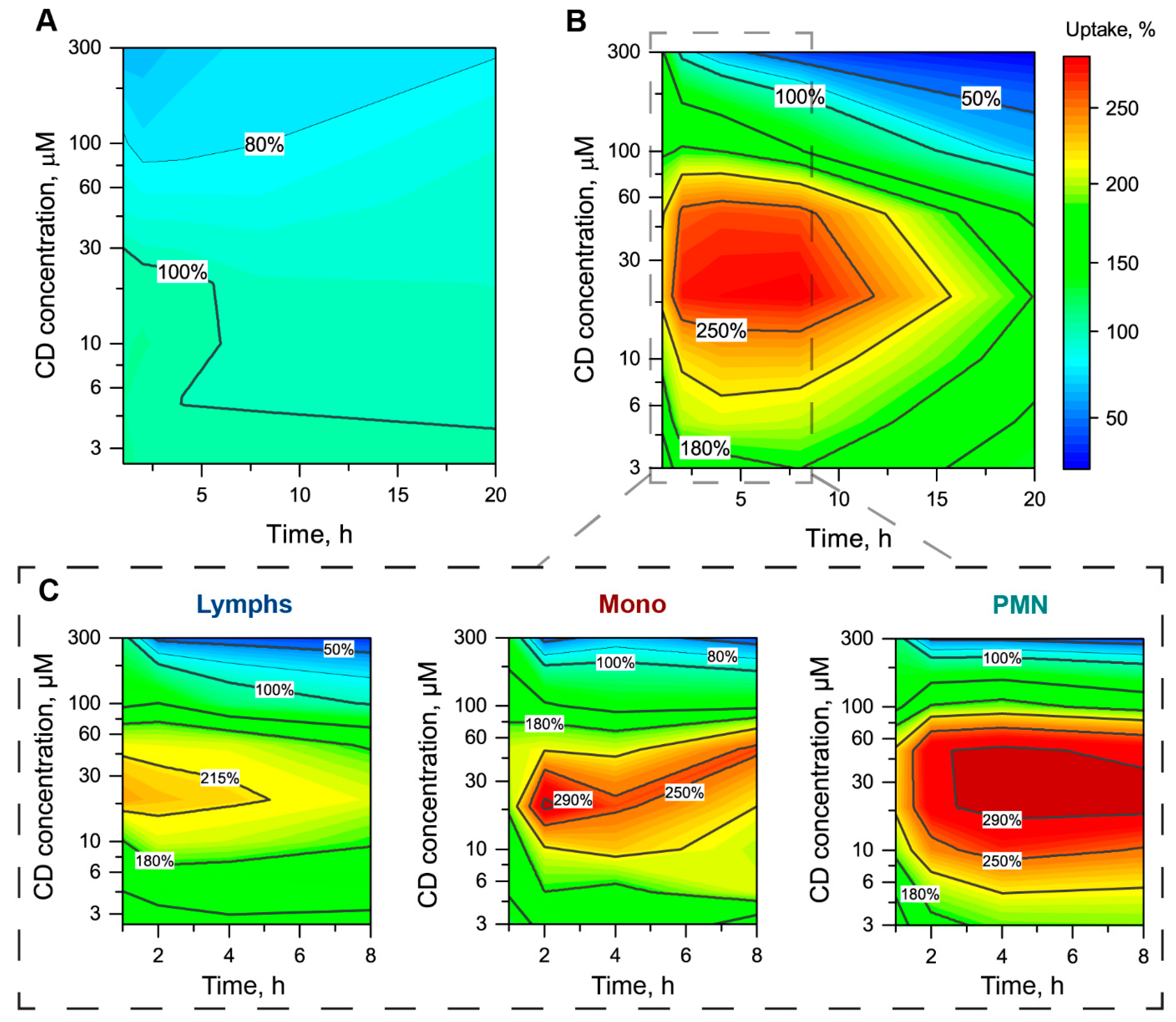
3.2.3. The mTHPC Accumulation in Subpopulations of Blood Cells
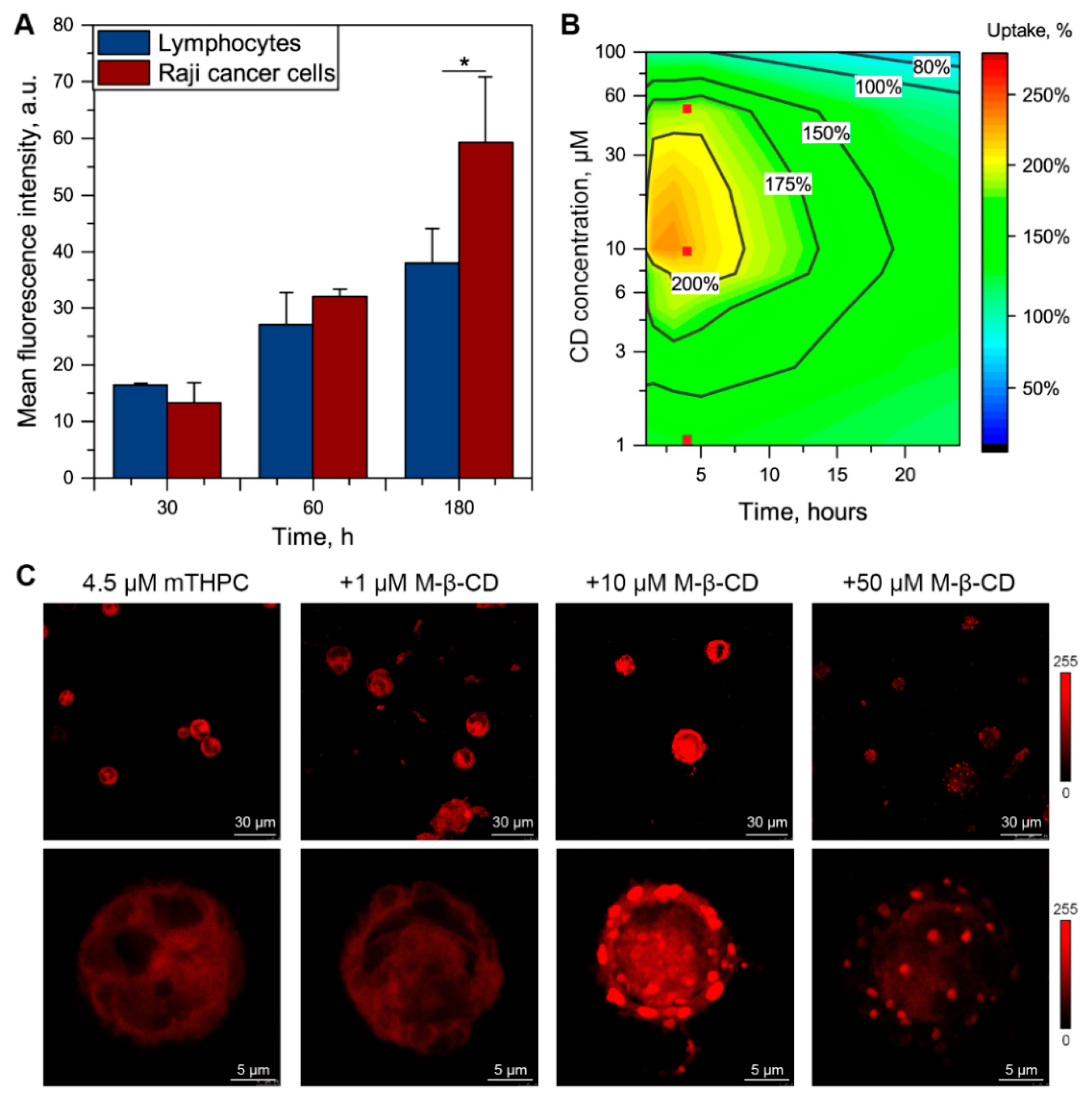
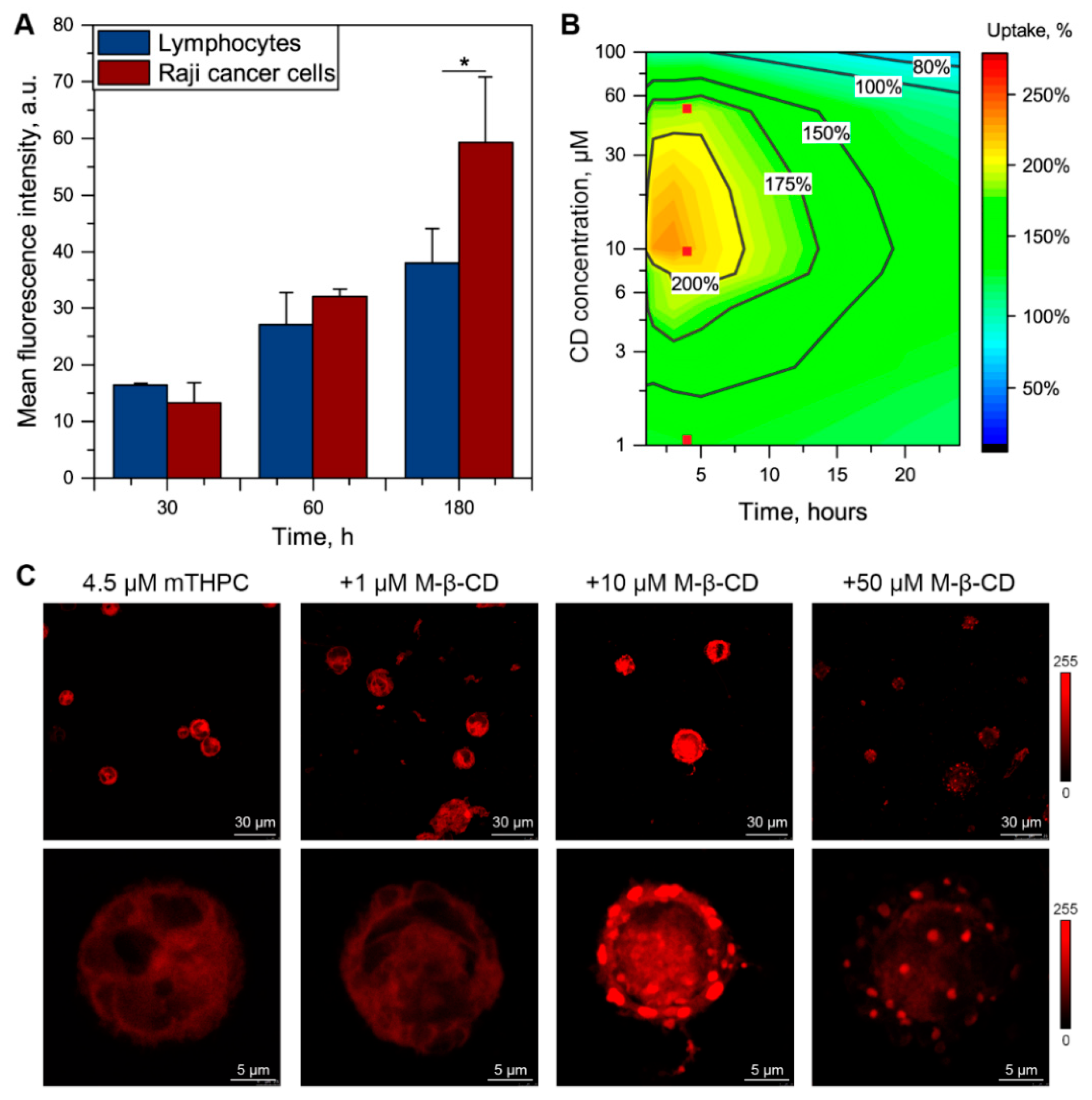
3.3. Me-β-CD-Modulated mTHPC Accumulation in Blood Cancer Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Li, X.; Lovell, J.F.; Yoon, J.; Chen, X. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one—photosensitizers, photochemistry and cellular localization. Photodiagn. Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part three—Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagn. Photodyn. Ther. 2005, 2, 91–106. [Google Scholar] [CrossRef] [Green Version]
- Senge, M.O.; Brandt, J.C. Temoporfin (Foscan®, 5,10,15,20-Tetra(m-hydroxyphenyl)chlorin)-A second-generation photosensitizer. Photochem. Photobiol. 2011, 87, 1240–1296. [Google Scholar] [CrossRef]
- Bonnett, R.; Charlesworth, P.; Djelal, B.D.; Foley, S.; McGarvey, D.J.; Truscott, T.G. Photophysical properties of 5,10,15,20-tetrakis(m-hydroxyphenyl)porphyrin (m-THPP), 5,10,15,20-tetrakis(m-hydroxyphenyl)chlorin (m-THPC) and 5,10,15,20-tetrakis(m-hydroxyphenyl)bacteriochlorin (m-THPBC): A comparative study. J. Chem. Soc. Perkin Trans. 1999, 2, 325–328. [Google Scholar] [CrossRef]
- De Visscher, S.A.H.J.; Melchers, L.J.; Dijkstra, P.U.; Karakullukcu, B.; Tan, I.B.; Hopper, C.; Roodenburg, J.L.N.; Witjes, M.J.H. mTHPC-mediated Photodynamic Therapy of Early Stage Oral Squamous Cell Carcinoma: A Comparison to Surgical Treatment. Ann. Surg. Oncol. 2013, 20, 3076–3082. [Google Scholar] [CrossRef]
- Sasnouski, S.; Zorin, V.; Khludeyev, I.; D’Hallewin, M.-A.; Guillemin, F.; Bezdetnaya, L. Investigation of Foscan® interactions with plasma proteins. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2005, 1725, 394–402. [Google Scholar] [CrossRef]
- Jablonka, L.; Ashtikar, M.; Gao, G.; Jung, F.; Thurn, M.; Preuß, A.; Scheglmann, D.; Albrecht, V.; Röder, B.; Wacker, M.G. Advanced in silico modeling explains pharmacokinetics and biodistribution of temoporfin nanocrystals in humans. J. Control. Release 2019, 308, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Ronn, A.M.; Nouri, M.; Löfgren, L.A.; Steinberg, B.M.; Westerborn, A.; Windahl, T.; Shikowitz, M.J.; Abramson, A.L. Human tissue levels and plasma pharmacokinetics of temoporfin (Foscan®, mTHPC). Lasers Med Sci. 1996, 11, 267–272. [Google Scholar] [CrossRef]
- Glanzmann, T.; Hadjur, C.; Zellweger, M.; Grosiean, P.; Forrer, M.; Ballini, J.P.; Monnier, P.; van den Bergh, H.; Lim, C.K.; Wagnières, G. Pharmacokinetics of Tetra(m-Hydroxyphenyl)Chlorin in Human Plasma and Individualized Light Dosimetry in Photodynamic Therapy. Photochem. Photobiol. 1998, 67, 596–602. [Google Scholar]
- Hong, E.J.; Choi, D.G.; Shim, M.S. Targeted and effective photodynamic therapy for cancer using functionalized nanomaterials. Acta Pharm. Sin. B 2016, 6, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Yakavets, I.; Millard, M.; Zorin, V.; Lassalle, H.-P.; Bezdetnaya, L. Current state of the nanoscale delivery systems for temoporfin-based photodynamic therapy: Advanced delivery strategies. J. Control. Release 2019, 304, 268–287. [Google Scholar] [CrossRef]
- Ben Mihoub, A.; LaRue, L.; Moussaron, A.; Youssef, Z.; Colombeau, L.; Baros, F.; Frochot, C.; Vanderesse, R.; Acherar, S. Use of Cyclodextrins in Anticancer Photodynamic Therapy Treatment. Molecules 2018, 23, 1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, V.J.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef]
- Duchene, D. Cyclodextrins and Their Inclusion Complexes. In Cyclodextrins in Pharmaceutics, Cosmetics, and Biomedicine: Current and Future Industrial Applications; Bilensoy, E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 1–18. ISBN 978-0-470-92681-9. [Google Scholar]
- Del Valle, E. Cyclodextrins and their uses: A review. Process. Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Laza-Knoerr, A.; Gref, R.; Couvreur, P. Cyclodextrins for drug delivery. J. Drug Target. 2010, 18, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Vyas, A.; Saraf, S.; Saraf, S. Cyclodextrin based novel drug delivery systems. J. Incl. Phenom. Macrocycl. Chem. 2008, 62, 23–42. [Google Scholar] [CrossRef]
- Yakavets, I.; Lassalle, H.-P.; Yankovsky, I.; Ingrosso, F.; Monari, A.; Bezdetnaya, L.; Zorin, V. Evaluation of temoporfin affinity to β-cyclodextrins assuming self-aggregation. J. Photochem. Photobiol. A: Chem. 2018, 367, 13–21. [Google Scholar] [CrossRef]
- Mosinger, J.; Kliment, V.; Sejbal, J.; Kubát, P.; Lang, K. Host-guest complexes of anionic porphyrin sensitizers with cyclodextrins. J. Porphyrins Phthalocyanines 2002, 6, 514–526. [Google Scholar] [CrossRef]
- Mosinger, J.; Slavětínská, L.; Lang, K.; Coufal, P.; Kubát, P. Cyclodextrin carriers of positively charged porphyrin sensitizers. Org. Biomol. Chem. 2009, 7, 3797–3804. [Google Scholar] [CrossRef] [PubMed]
- Yakavets, I.; Yankovsky, I.; Bezdetnaya, L.; Zorin, V. Soret band shape indicates mTHPC distribution between β-cyclodextrins and serum proteins. Dye. Pigment. 2017, 137, 299–306. [Google Scholar] [CrossRef]
- Aslanoglu, B.; Yakavets, I.; Zorin, V.; Lassalle, H.-P.; Ingrosso, F.; Monari, A.; Catak, S. Optical properties of photodynamic therapy drugs in different environments: The paradigmatic case of temoporfin. Phys. Chem. Chem. Phys. 2020, 22, 16956–16964. [Google Scholar] [CrossRef]
- Yakavets, I.V.; Yankovsky, I.V.; Khludeyev, I.I.; Lassalle, H.; Bezdetnaya, L.N.; Zorin, V.P. Optical Methods for the Analysis of the Temoprofin Photosensitizer Distribution Between Serum Proteins and Methyl-?-Cyclodextrin Nanocarriers in Blood Serum. J. Appl. Spectrosc. 2018, 84, 1–7. [Google Scholar] [CrossRef]
- Yankovsky, I.; Bastien, E.; Yakavets, I.; Khludeyev, I.; Lassalle, H.-P.; Gräfe, S.; Bezdetnaya, L.; Zorin, V. Inclusion complexation with β-cyclodextrin derivatives alters photodynamic activity and biodistribution of meta-tetra(hydroxyphenyl)chlorin. Eur. J. Pharm. Sci. 2016, 91, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Yakavets, I.; Yankovsky, I.; Millard, M.; Lamy, L.; Lassalle, H.-P.; Wiehe, A.; Zorin, V.; Bezdetnaya, L. The alteration of temoporfin distribution in multicellular tumor spheroids by β-cyclodextrins. Int. J. Pharm. 2017, 529, 568–575. [Google Scholar] [CrossRef]
- Goodhead, L.K.; Macmillan, F.M. Measuring osmosis and hemolysis of red blood cells. Adv. Physiol. Educ. 2017, 41, 298–305. [Google Scholar] [CrossRef]
- Terstappen, L.W.M.M.; Degrooth, B.G.; Visscher, K.; VanKouterik, F.A.; Greve, J. Four-Parameter white blood cell differential counting based on light scattering measurements. Cytometry 1988, 9, 39–43. [Google Scholar] [CrossRef]
- Reshetov, V.; Zorin, V.; Siupa, A.; D’Hallewin, M.-A.; Guillemin, F.; Bezdetnaya, L. Interaction of Liposomal Formulations of Meta-tetra(hydroxyphenyl)chlorin (Temoporfin) with Serum Proteins: Protein Binding and Liposome Destruction. Photochem. Photobiol. 2012, 88, 1256–1264. [Google Scholar] [CrossRef]
- Moras, M.; Lefevre, S.D.; Ostuni, M.A. From Erythroblasts to Mature Red Blood Cells: Organelle Clearance in Mammals. Front. Physiol. 2017, 8, 1076. [Google Scholar] [CrossRef]
- Kryjewski, M.; Goslinski, T.; Mielcarek, J. Functionality stored in the structures of cyclodextrin–porphyrinoid systems. Co-ord. Chem. Rev. 2015, 300, 101–120. [Google Scholar] [CrossRef]
- Mazzaglia, A. Photodynamic Tumor Therapy with Cyclodextrin Nanoassemblies. In Cyclodextrins in Pharmaceutics, Cosmetics, and Biomedicine: Current and Future Industrial Applications; Bilensoy, E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 343–361. ISBN 978-0-470-92681-9. [Google Scholar]
- Lang, K.; Mosinger, J.; Wagnerová, D. Photophysical properties of porphyrinoid sensitizers non-covalently bound to host molecules; models for photodynamic therapy. Coord. Chem. Rev. 2004, 248, 321–350. [Google Scholar] [CrossRef]
- Dentuto, P.; Catucci, L.; Cosma, P.; Fini, P.; Agostiano, A.; Hackbarth, S.; Rancan, F.; Roeder, B. Cyclodextrin/chlorophyll a complexes as supramolecular photosensitizers. Bioelectrochemistry 2007, 70, 39–43. [Google Scholar] [CrossRef]
- Kitagishi, H.; Chai, F.; Negi, S.; Sugiura, Y.; Kano, K. Supramolecular intracellular delivery of an anionic porphyrin by octaarginine-conjugated per-O-methyl-?-cyclodextrin. Chem. Commun. 2015, 51, 2421–2424. [Google Scholar] [CrossRef]
- Kolarova, H.; Macecek, J.; Nevrelova, P.; Huf, M.; Tomecka, M.; Bajgar, R.; Mosinger, J.; Strnad, M. Photodynamic therapy with zinc-tetra(p-sulfophenyl)porphyrin bound to cyclodextrin induces single strand breaks of cellular DNA in G361 melanoma cells. Toxicol. In Vitro 2005, 19, 971–974. [Google Scholar] [CrossRef]
- Yakavets, I.; Lassalle, H.-P.; Scheglmann, D.; Wiehe, A.; Zorin, V.; Bezdetnaya, L. Temoporfin-in-Cyclodextrin-in-Liposome—A New Approach for Anticancer Drug Delivery: The Optimization of Composition. Nanomaterials 2018, 8, 847. [Google Scholar] [CrossRef] [Green Version]
- Yakavets, I.; Millard, M.; Lamy, L.; Francois, A.; Scheglmann, D.; Wiehe, A.; Lassalle, H.-P.; Zorin, V.; Bezdetnaya, L. Lamy Matryoshka-Type Liposomes Offer the Improved Delivery of Temoporfin to Tumor Spheroids. Cancers 2019, 11, 1366. [Google Scholar] [CrossRef] [Green Version]
- Yakavets, I.; Francois, A.; Lamy, L.; Piffoux, M.; Gazeau, F.; Wilhelm, C.; Zorin, V.; Silva, A.K.A.; Bezdetnaya, L. Effect of stroma on the behavior of temoporfin-loaded lipid nanovesicles inside the stroma-rich head and neck carcinoma spheroids. J. Nanobiotechnol. 2021, 19, 1–18. [Google Scholar] [CrossRef]
- Yakavets, I.; Guereschi, C.; Lamy, L.; Kravchenko, I.; Lassalle, H.-P.; Zorin, V.; Bezdetnaya, L. Cyclodextrin nanosponge as a temoporfin nanocarrier: Balancing between accumulation and penetration in 3D tumor spheroids. Eur. J. Pharm. Biopharm. 2020, 154, 33–42. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yakavets, I.; Yankovsky, I.; Zorina, T.; Belevtsev, M.; Bezdetnaya, L.; Zorin, V. Modulation of Temoporfin Distribution in Blood by β-Cyclodextrin Nanoshuttles. Pharmaceutics 2021, 13, 1054. https://doi.org/10.3390/pharmaceutics13071054
Yakavets I, Yankovsky I, Zorina T, Belevtsev M, Bezdetnaya L, Zorin V. Modulation of Temoporfin Distribution in Blood by β-Cyclodextrin Nanoshuttles. Pharmaceutics. 2021; 13(7):1054. https://doi.org/10.3390/pharmaceutics13071054
Chicago/Turabian StyleYakavets, Ilya, Igor Yankovsky, Tatyana Zorina, Mikhail Belevtsev, Lina Bezdetnaya, and Vladimir Zorin. 2021. "Modulation of Temoporfin Distribution in Blood by β-Cyclodextrin Nanoshuttles" Pharmaceutics 13, no. 7: 1054. https://doi.org/10.3390/pharmaceutics13071054
APA StyleYakavets, I., Yankovsky, I., Zorina, T., Belevtsev, M., Bezdetnaya, L., & Zorin, V. (2021). Modulation of Temoporfin Distribution in Blood by β-Cyclodextrin Nanoshuttles. Pharmaceutics, 13(7), 1054. https://doi.org/10.3390/pharmaceutics13071054