
Alteration of Anticancer and Protein-Binding Properties of Gold(I) Alkynyl by Phenolic Schiff Bases Moieties

 ,
,  ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods and Instrumentation
2.3. Synthesis and Characterizations
2.4. Crystal Structure Determination
2.5. HSA Binding Studies
2.6. Molecular Docking Studies
2.7. Anticancer Studies
3. Results and Discussion
3.1. Synthesis and Characterization
3.2. Crystallographic Studies
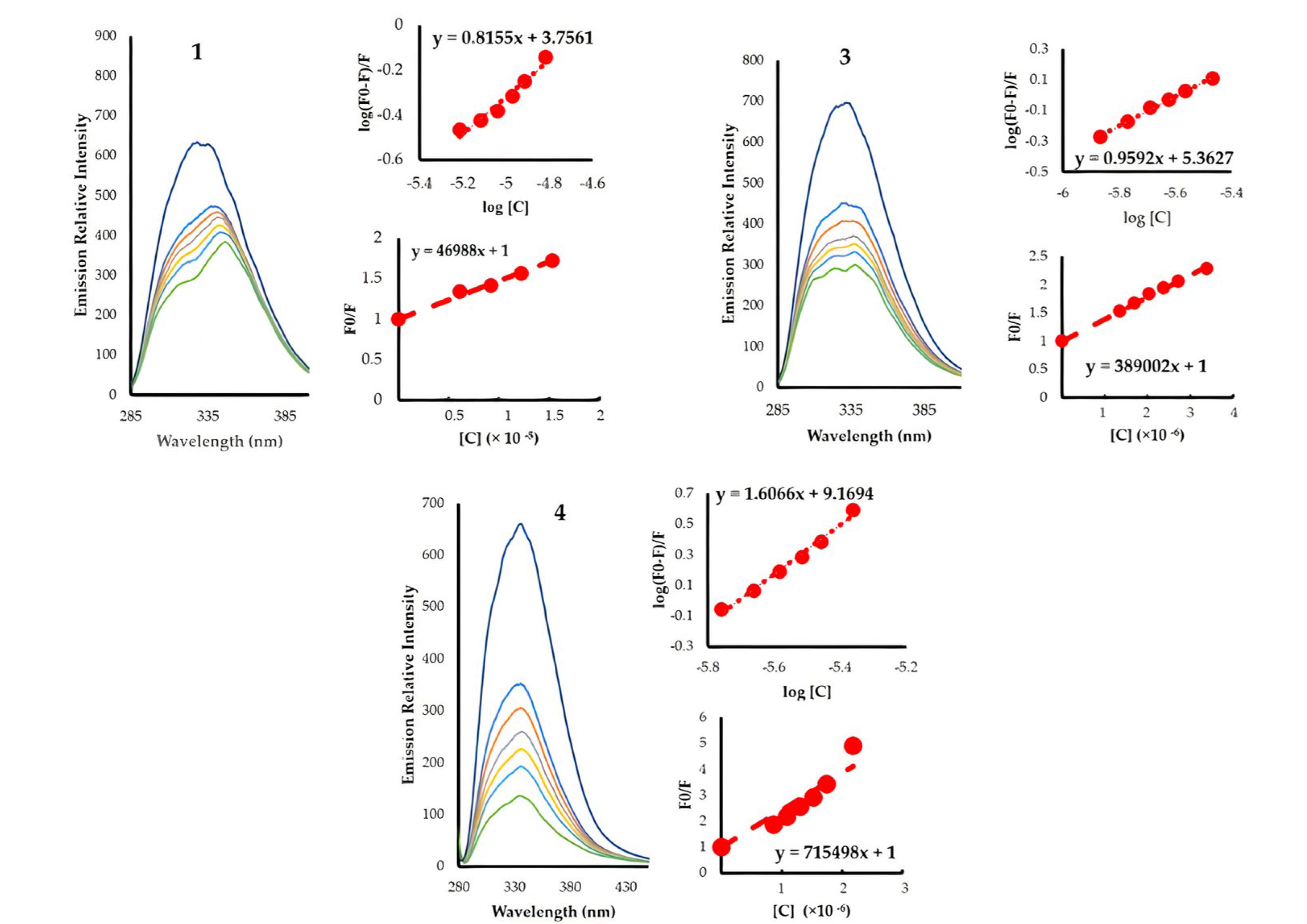
3.3. HSA Binding
3.4. Anticancer Studies
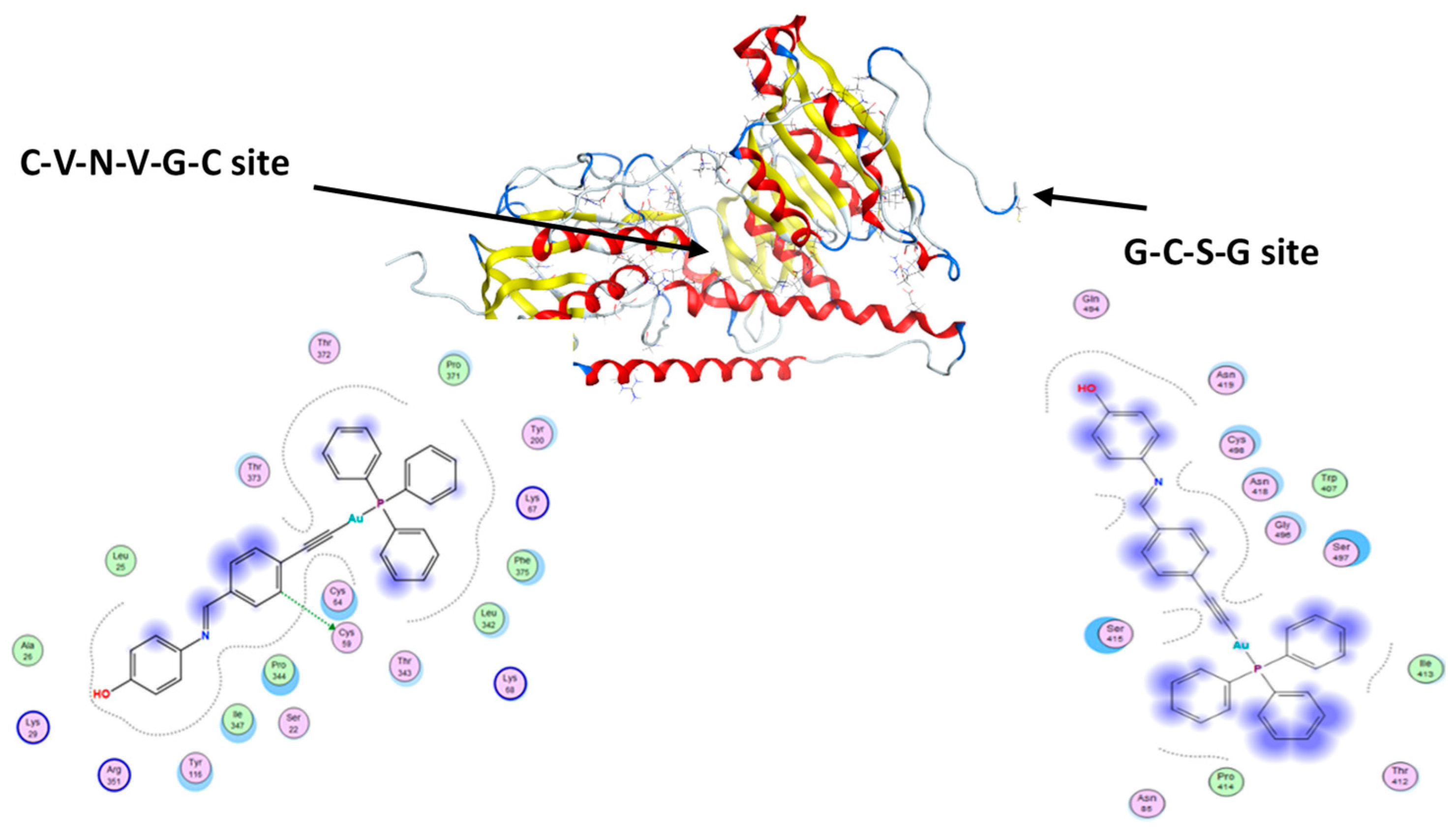
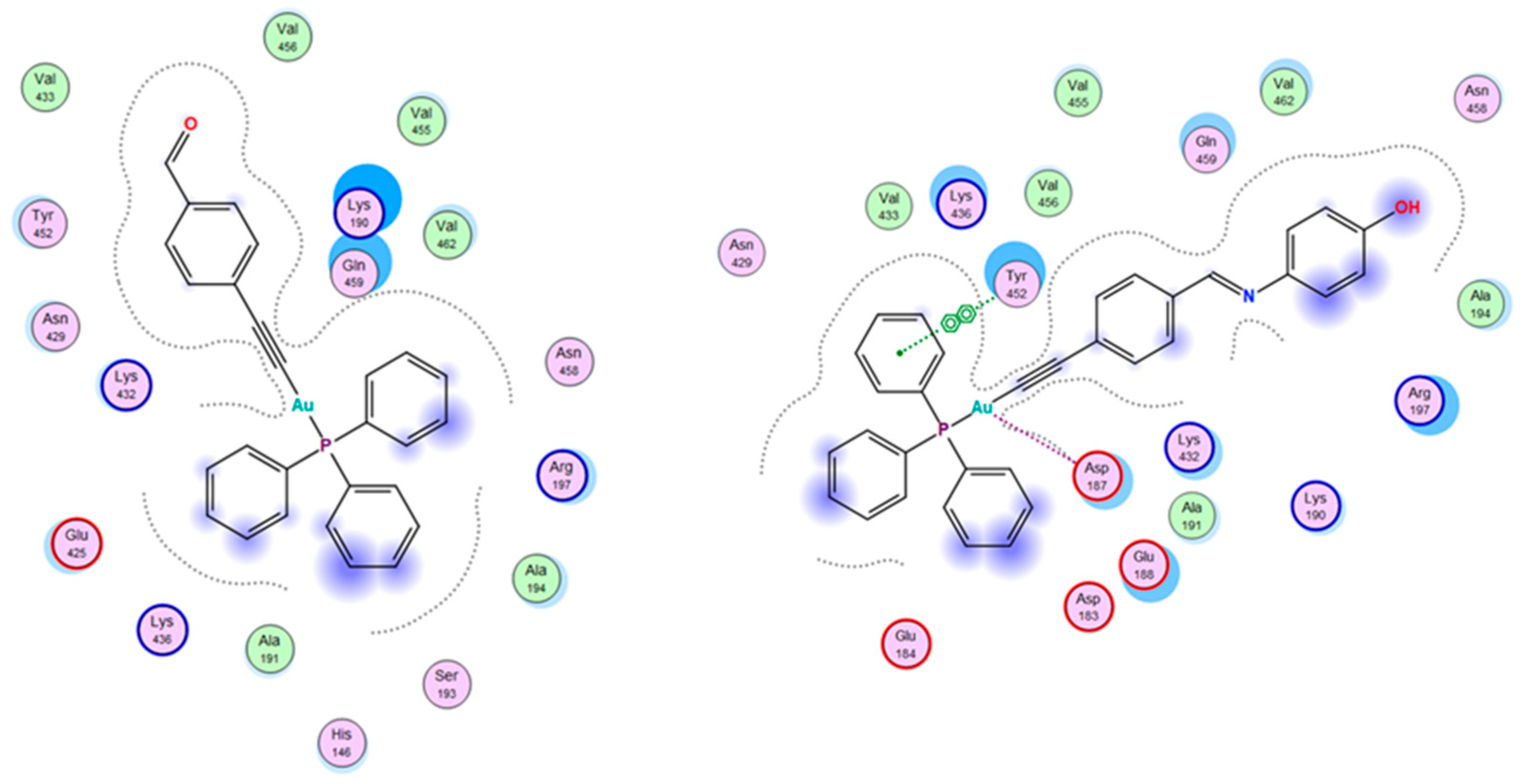
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- Lazarević, T.; Rilak, A.; Bugarčić, Ž.D. Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives. Eur. J. Med. Chem. 2017, 142, 8–31. [Google Scholar] [CrossRef]
- Deo, K.M.; Ang, D.L.; McGhie, B.; Rajamanickam, A.; Dhiman, A.; Khoury, A.; Holland, J.; Bjelosevic, A.; Pages, B.; Gordon, C.; et al. Platinum coordination compounds with potent anticancer activity. Coord. Chem. Rev. 2018, 375, 148–163. [Google Scholar] [CrossRef]
- Trudu, F.; Amato, F.; Vanhara, P.; Pivetta, T.; Pena-Mendez, E.M.; Havel, J. Coordination compounds in cancer: Past, present and perspectives. J. Appl. Biomed. 2015, 13, 79–103. [Google Scholar] [CrossRef]
- De Gramont, A.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G.; et al. Leucovorin and Fluorouracil with or without Oxaliplatin as First-Line Treatment in Advanced Colorectal Cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.L. Efficacy of oxaliplatin in the treatment of colorectal cancer. Oncology 2000, 14, 9–14. [Google Scholar]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Łos, M.J. Autophagy, Apoptosis, Mitoptosis and Necrosis: Interdependence Between Those Pathways and Effects on Cancer. Arch. Immunol. Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Sánchez-de-Diego, C.; Mármol, I.; Pérez, R.; Gascón, S.; Rodriguez-Yoldi, M.J.; Cerrada, E. The anticancer effect related to disturbances in redox balance on Caco-2 cells caused by an alkynyl gold(I) complex. J. Inorg. Biochem. 2017, 166, 108–121. [Google Scholar] [CrossRef]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef]
- Shaw, C.F. Gold-Based Therapeutic Agents. Chem. Rev. 1999, 99, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.F. Metal Compounds in Cancer Therapy; Fricker, S.P., Ed.; Chapman & Hall: London, UK, 1994; pp. 46–64. [Google Scholar]
- Tiekink, E.R.T. Gold derivatives for the treatment of cancer. Crit. Rev. Oncol. Hematol. 2002, 42, 225–248. [Google Scholar] [CrossRef]
- Ott, I. On the medicinal chemistry of gold complexes as anticancer drugs. Coord. Chem. Rev. 2009, 253, 1670–1681. [Google Scholar] [CrossRef]
- Andermark, V.; Göke, K.; Kokoschka, M.; Abu El Maaty, M.A.; Lum, C.T.; Zou, T.; Sun, R.W.-Y.; Aguiló, E.; Oehninger, L.; Rodríguez, L.; et al. Alkynyl gold(I) phosphane complexes: Evaluation of structure-activity-relationships for the phosphane ligands, effects on key signaling proteins and preliminary in-vivo studies with a nanoformulated complex. J. Inorg. Biochem. 2016, 160, 140–148. [Google Scholar] [CrossRef]
- Mármol, I.; Virumbrales-Muñoz, M.; Quero, J.; Sánchez-de-Diego, C.; Fernández, L.; Ochoa, I.; Cerrada, E.; Yoldi, M.J.R. Alkynyl gold(I) complex triggers necroptosis via ROS generation in colorectal carcinoma cells. J. Inorg. Biochem. 2017, 176, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Bagowski, C.P.; Kokoschka, M.; Stefanopoulou, M.; Alborzinia, H.; Can, S.; Vlecken, D.H.; Sheldrick, W.S.; Wölfl, S.; Ott, I. On the Biological Properties of Alkynyl Phosphine Gold(I) Complexes. Angew. Chem. Int. Ed. 2012, 51, 8895–8899. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. Methods Mol. Biol. 2008, 443, 365–382. [Google Scholar] [PubMed]
- Austin, W.B.; Bilow, N.; Kelleghan, W.J.; Lau, K.S.Y. Facile synthesis of ethynylated benzoic acid derivatives and aromatic compounds via ethynyltrimethylsilane. J. Org. Chem. 1981, 46, 2280–2286. [Google Scholar] [CrossRef]
- Isab, A.A.; Fettouhi, M.; Ahmad, S.; Ouahab, L. Mixed ligand gold(I) complexes of phosphines and thiourea and X-ray structure of (thiourea-κS)(tricyclohexylphosphine)gold(I)chloride. Polyhedron 2003, 22, 1349–1354. [Google Scholar] [CrossRef]
- McAuliffe, C.A.; Parish, R.V.; Randall, P.D. Gold(I) complexes of unidentate and bidentate phosphorus-, arsenic-, antimony-, and sulphur-donor ligands. J. Chem. Soc. Dalton Trans. 1979, 1730–1735. [Google Scholar] [CrossRef]
- Hurst, S.K.; Lucas, N.T.; Cifuentes, M.P.; Humphrey, M.G.; Samoc, M.; Luther-Davies, B.; Asselberghs, I.; Van Boxel, R.; Persoons, A. Organometallic complexes for nonlinear optics: Part 23. Quadratic and cubic hyperpolarizabilities of acetylide and vinylidene complexes derived from protected and free formylphenylacetylenes. J. Organomet. Chem. 2001, 633, 114–124. [Google Scholar] [CrossRef]
- APEX3-Software Suite for Crystallographic Programs; Bruker AXS Inc.: Madison, WI, USA, 2016.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Villarreal, W.; Colina-Vegas, L.; Visbal, G.; Corona, O.; Corrêa, R.S.; Ellena, J.; Cominetti, M.R.; Batista, A.A.; Navarro, M. Copper(I)–Phosphine Polypyridyl Complexes: Synthesis, Characterization, DNA/HSA Binding Study, and Antiproliferative Activity. Inorg. Chem. 2017, 56, 3781–3793. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Jankeje, K.; Albani, J.R. Origin of Fluorescence Lifetimes in Human Serum Albumin. Studies on Native and Denatured Protein. J. Fluoresc. 2010, 20, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Jankeje, K.; Albani, J.R. Characterization of human serum albumin forms with pH. Fluorescence lifetime studies. J. Pharm. Biomed. Anal. 2010, 51, 1097–1102. [Google Scholar] [CrossRef]
- Lineweaver, H.; Burk, D. The Determination of Enzyme Dissociation Constants. J. Am. Chem. Soc. 1934, 56, 658–666. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Bakheit, A.H.; Almutairi, F.M.; AlRabiah, H.; Kadi, A.A. Biophysical and In Silico Studies of the Interaction between the Anti-Viral Agents Acyclovir and Penciclovir, and Human Serum Albumin. Molecules 2017, 22, 1906. [Google Scholar] [CrossRef]
- Urig, S.; Becker, K. On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin. Cancer Biol. 2006, 16, 452–465. [Google Scholar] [CrossRef]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018, 130, 73–89. [Google Scholar] [CrossRef]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal Structure Analysis of Warfarin Binding to Human Serum Albumin: ANATOMY OF DRUG SITE I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef]
- Fritz-Wolf, K.; Kehr, S.; Stumpf, M.; Rahlfs, S.; Becker, K. Crystal structure of the human thioredoxin reductase-thioredoxin complex. Nat. Commun. 2011, 2, 383. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), Version 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2019.
- Abdel-Rhman, M.H.; Hussien, M.A.; Mahmoud, H.M.; Hosny, N.M. Synthesis, characterization, molecular docking and cytotoxicity studies on N-benzyl-2-isonicotinoylhydrazine-1-carbothioamide and its metal complexes. J. Mol. Struct. 2019, 1196, 417–428. [Google Scholar] [CrossRef]
- Cole, J.C.; Murray, C.W.; Nissink, J.W.M.; Taylor, R.D.; Taylor, R. Comparing protein–ligand docking programs is difficult. Proteins Struct. Funct. Bioinform. 2005, 60, 325–332. [Google Scholar] [CrossRef]
- Jain, A.N. Bias, reporting, and sharing: Computational evaluations of docking methods. J. Comput.-Aided Mol. Des. 2008, 22, 201–212. [Google Scholar] [CrossRef]
- Muanza, D.N.; Kim, B.W.; Euler, K.L.; Williams, L. Antibacterial and Antifungal Activities of Nine Medicinal Plants from Zaire. Int. J. Pharmacogn. 1994, 32, 337–345. [Google Scholar] [CrossRef]
- Pezzuto, J.M.; Che, C.-T.; McPherson, D.D.; Zhu, J.-P.; Topcu, G.; Erdelmeier, C.A.J.; Cordell, G.A. DNA as an Affinity Probe Useful in the Detection and Isolation of Biologically Active Natural Products. J. Nat. Prod. 1991, 54, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. JNCI J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Alsaeedi, M.S.; Babgi, B.A.; Hussien, M.A.; Abdellattif, M.H.; Humphrey, M.G. DNA-Binding and Anticancer Activity of Binuclear Gold(I) Alkynyl Complexes with a Phenanthrenyl Bridging Ligand. Molecules 2020, 25, 1033. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, D.; Vitone, F.; Schiavone, P.; Ponti, C.; La Placa, M.; Re, M.C. Quantitative detection of human immunodeficiency virus type 1 (HIV-1) proviral DNA in peripheral blood mononuclear cells by SYBR green real-time PCR technique. J. Clin. Virol. 2004, 29, 282–289. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Ware, W.R. Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process. J. Phys. Chem. 1962, 66, 455–458. [Google Scholar] [CrossRef]
- Thangavel, S.; Rajamanikandan, R.; Friedrich, H.B.; Ilanchelian, M.; Omondi, B. Binding interaction, conformational change, and molecular docking study of N-(pyridin-2-ylmethylene)aniline derivatives and carbazole Ru(II) complexes with human serum albumins. Polyhedron 2016, 107, 124–135. [Google Scholar] [CrossRef]
- Al-Harthi, S.; Lachowicz, J.I.; Nowakowski, M.E.; Jaremko, M.; Jaremko, Ł. Towards the functional high-resolution coordination chemistry of blood plasma human serum albumin. J. Inorg. Biochem. 2019, 198, 110716. [Google Scholar] [CrossRef]
- Lissi, E.; Calderón, C.; Campos, A. Evaluation of the Number of Binding Sites in Proteins from their Intrinsic Fluorescence: Limitations and Pitfalls. Photochem. Photobiol. 2013, 89, 1413–1416. [Google Scholar] [CrossRef]
- Gromer, S.; Arscott, L.D.; Williams, C.H., Jr.; Schirmer, R.H.; Becker, K. Human Placenta Thioredoxin Reductase: Isolation of the selenoenzyme, steady state kinetics, and inhabitation by therapeutic gold compounds. J. Biol. Chem. 1998, 273, 20096–20101. [Google Scholar] [CrossRef]
- Becker, K.; Gromer, S.; Schirmer, R.H.; Müller, S. Thioredoxin reductase as a pathophysiological factor and drug target. Eur. J. Biochem. 2000, 267, 6118–6125. [Google Scholar] [CrossRef] [PubMed]
- Seliman, A.A.A.; Altaf, M.; Onawole, A.T.; Ahmad, S.; Ahmed, Y.; Al-Saadi, A.A.; Altuwaijri, S.; Bahita, G.; Singh, J.; Isab, A.A. Synthesis, X-ray structures and anticancer activity of gold(I)-carbene complexes with selenones as co-ligands and their molecular docking studies with thioredoxin reductase. J. Organomet. Chem. 2017, 848, 175–183. [Google Scholar] [CrossRef]
- Scalcon, V.; Bindoli, A.; Rigobello, M.P. Significance of the mitochondrial thioredoxin reductase in cancer cells: An update on role, targets and inhibitors. Free Radic. Biol. Med. 2018, 127, 62–79. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, B.; Li, X.; Han, X.; Liu, R.; Fang, J. Small molecule inhibitors of mammalian thioredoxin reductase as potential anticancer agents: An update. Med. Res. Rev. 2019, 39, 5–39. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Nakka, S.; Guruprasad, L.; Samanta, A. Interaction of Bovine Serum Albumin with Dipolar Molecules: Fluorescence and Molecular Docking Studies. J. Phys. Chem. B 2009, 113, 2143–2150. [Google Scholar] [CrossRef] [PubMed]









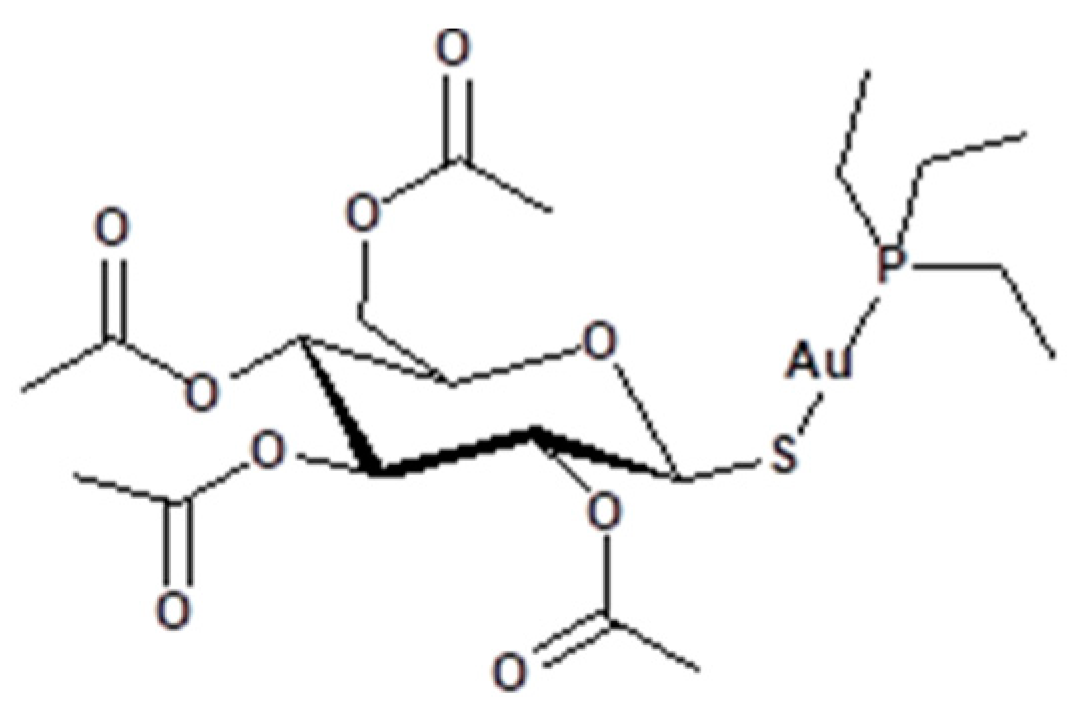{kind=link}
{kind=link}
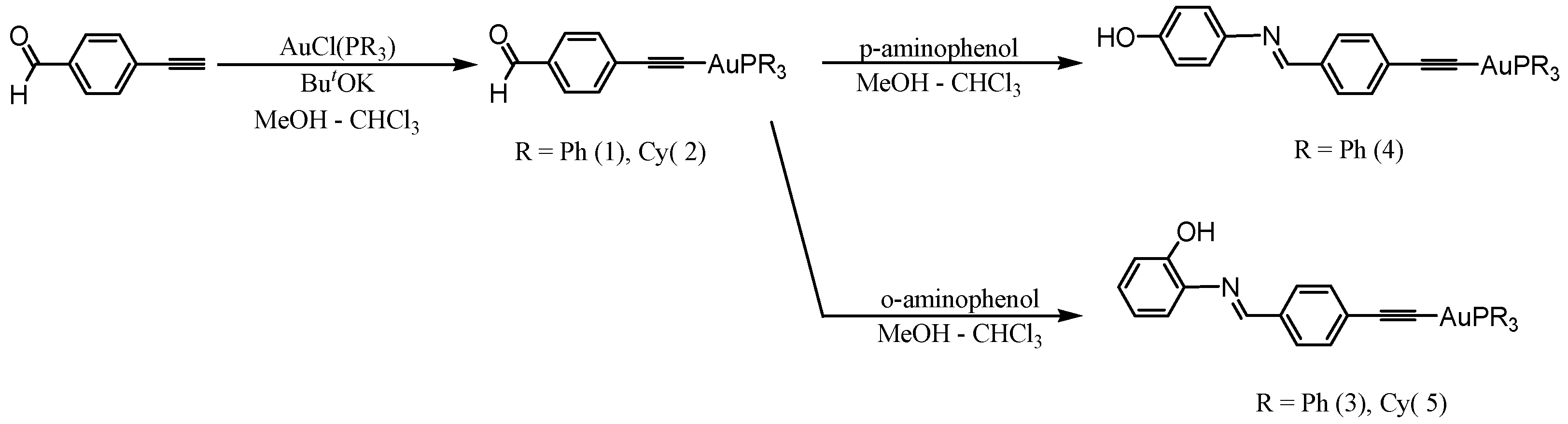{kind=link}
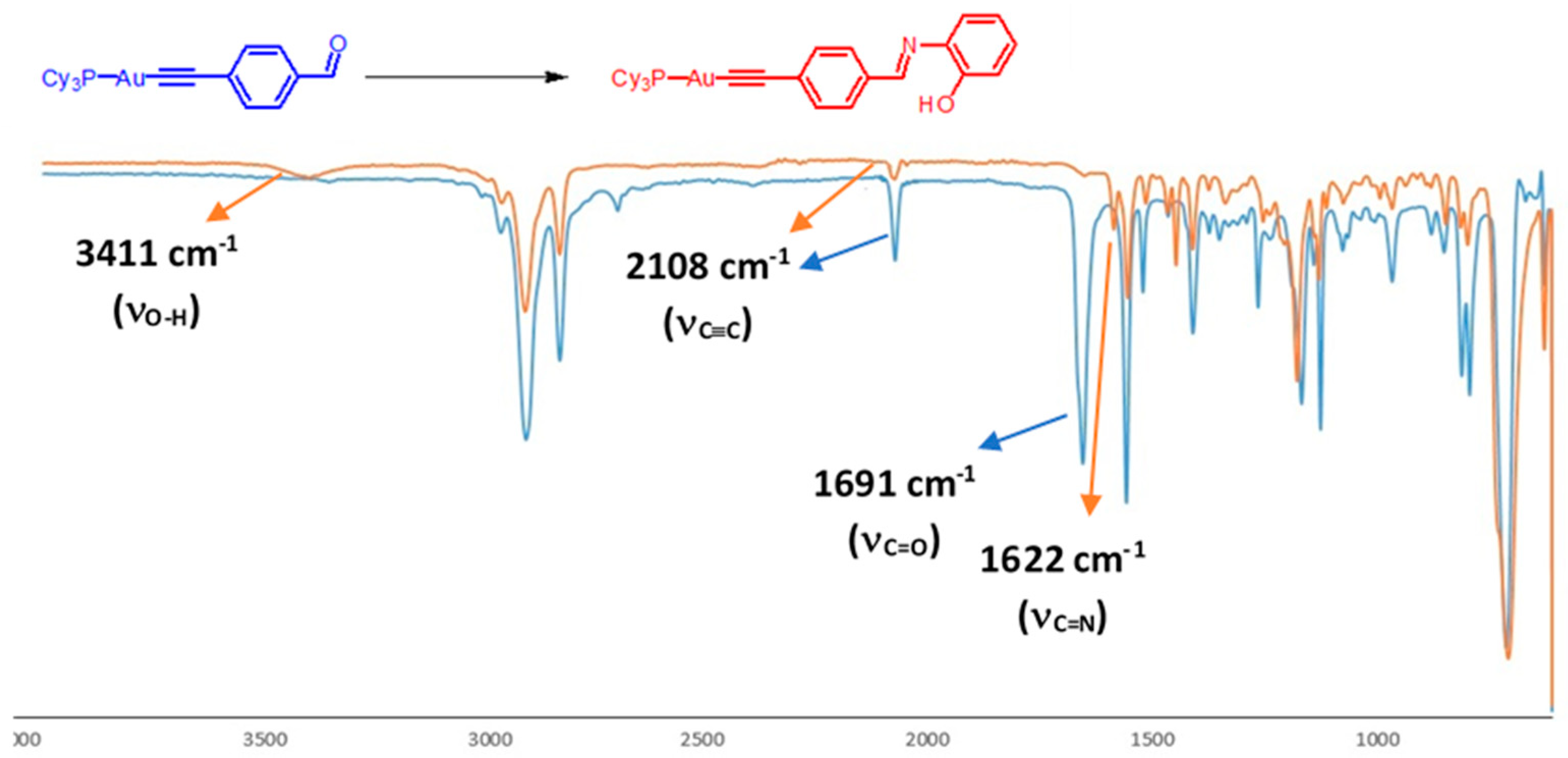{kind=link}
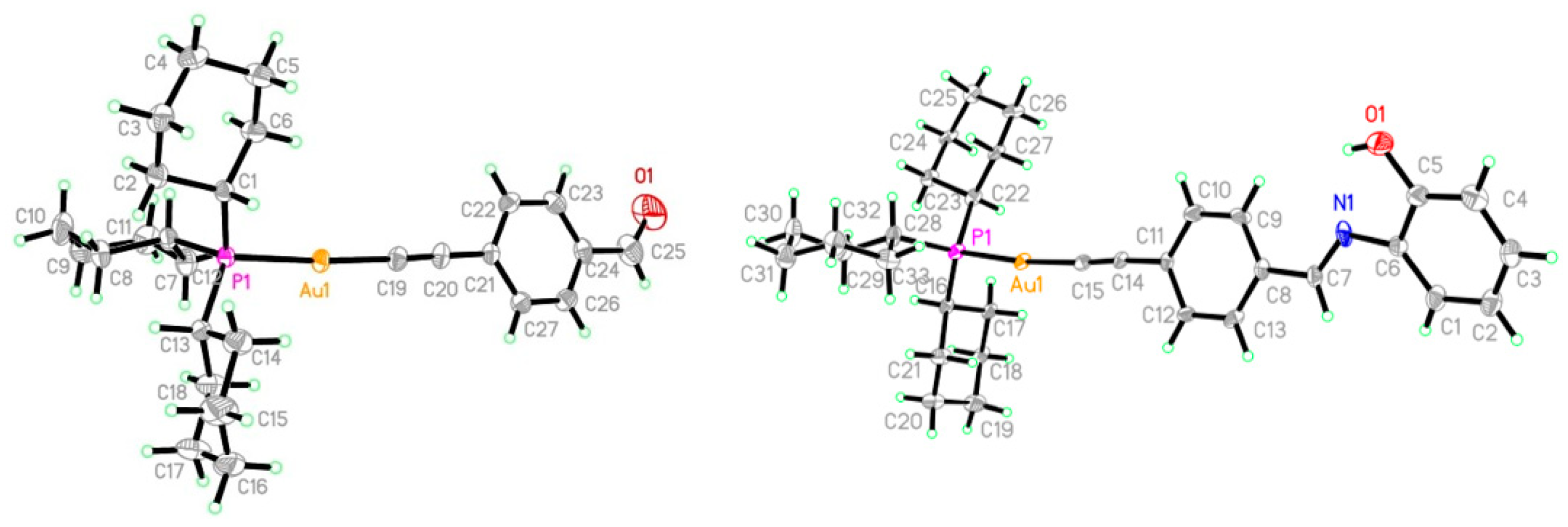{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex 2 | Complex 5 | ||
|---|---|---|---|
| Au1-C8 | 2.006(4) | Au1-C15 | 2.001(9)–2.007(9) |
| P1-C22 | 1.842(4) | P1-C22 | 1.832(9)–1.834(9) |
| O1-C9 | 1.175(7) | N1=C7 | 1.237(12)–1.267(12) |
| Au1-P1 | 2.2931(10) | Au1-P1 | 2.293(2) |
| Compound | Ksv (×104) | Kq (×1012) | Kb | n | ∆G0 (kJ mole−1) |
|---|---|---|---|---|---|
| 1 | 4.7 | 6.71 | 5.70 × 103 | 0.82 | −21.2 |
| 2 | 2.8 | 4.00 | 1.69 × 105 | 1.16 | −29.9 |
| 3 | 38.9 | 55.57 | 2.31 × 105 | 0.96 | −30.29 |
| 4 | 71.5 | 102.21 | 1.48 × 109 | 1.61 | −51.8 |
| 5 | 37.1 | 52.96 | 4.03 × 107 | 1.37 | −42.95 |
| Compound | OVCAR-3 (Ovarian Carcinoma Cancer Cell Line) (IC50% in µM) | HOP-62 (Non-Small-Cell Lung Cancer Cell Line) (IC50% in µM) |
|---|---|---|
| 1 | 13.65 ± 0.53 | 12.45 ± 0.43 |
| 2 | 15.86 ± 0.53 | 14.82 ± 0.13 |
| 3 | 09.40 ± 0.17 | 07.25 ± 0.21 |
| 4 | 05.27 ± 0.11 | 08.16 ± 0.43 |
| 5 | 09.11 ± 0.12 | 07.55 ± 0.43 |
| cisplatin | 05.89 ± 0.12 | 03.91 ± 0.20 |
| Complexes | Docking Scores against Human Thioredoxin Reductase (TrxR) (3QFB) | Docking Scores against HSA (1H9Z) | |
|---|---|---|---|
| Site 1 | Site 2 | ||
| 1 | −8.54 | −8.69 | −7.22 |
| 2 | −7.79 | −9.03 | −6.98 |
| 3 | −8.09 | −8.13 | −7.99 |
| 4 | −8.30 | −7.92 | −8.21 |
| 5 | −8.43 | −7.65 | −8.21 |
| auranofin | −8.14 | −7.65 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babgi, B.A.; Alsayari, J.; Alenezi, H.M.; Abdellatif, M.H.; Eltayeb, N.E.; Emwas, A.-H.M.; Jaremko, M.; Hussien, M.A. Alteration of Anticancer and Protein-Binding Properties of Gold(I) Alkynyl by Phenolic Schiff Bases Moieties. Pharmaceutics 2021, 13, 461. https://doi.org/10.3390/pharmaceutics13040461
Babgi BA, Alsayari J, Alenezi HM, Abdellatif MH, Eltayeb NE, Emwas A-HM, Jaremko M, Hussien MA. Alteration of Anticancer and Protein-Binding Properties of Gold(I) Alkynyl by Phenolic Schiff Bases Moieties. Pharmaceutics. 2021; 13(4):461. https://doi.org/10.3390/pharmaceutics13040461
Chicago/Turabian StyleBabgi, Bandar A., Jalal Alsayari, Hana M. Alenezi, Magda H. Abdellatif, Naser E. Eltayeb, Abdul-Hamid M. Emwas, Mariusz Jaremko, and Mostafa A. Hussien. 2021. "Alteration of Anticancer and Protein-Binding Properties of Gold(I) Alkynyl by Phenolic Schiff Bases Moieties" Pharmaceutics 13, no. 4: 461. https://doi.org/10.3390/pharmaceutics13040461
APA StyleBabgi, B. A., Alsayari, J., Alenezi, H. M., Abdellatif, M. H., Eltayeb, N. E., Emwas, A.-H. M., Jaremko, M., & Hussien, M. A. (2021). Alteration of Anticancer and Protein-Binding Properties of Gold(I) Alkynyl by Phenolic Schiff Bases Moieties. Pharmaceutics, 13(4), 461. https://doi.org/10.3390/pharmaceutics13040461