
Immunotherapy for Triple-Negative Breast Cancer


Abstract
:1. Immunotherapy in Triple-Negative Breast Cancer (TNBC)
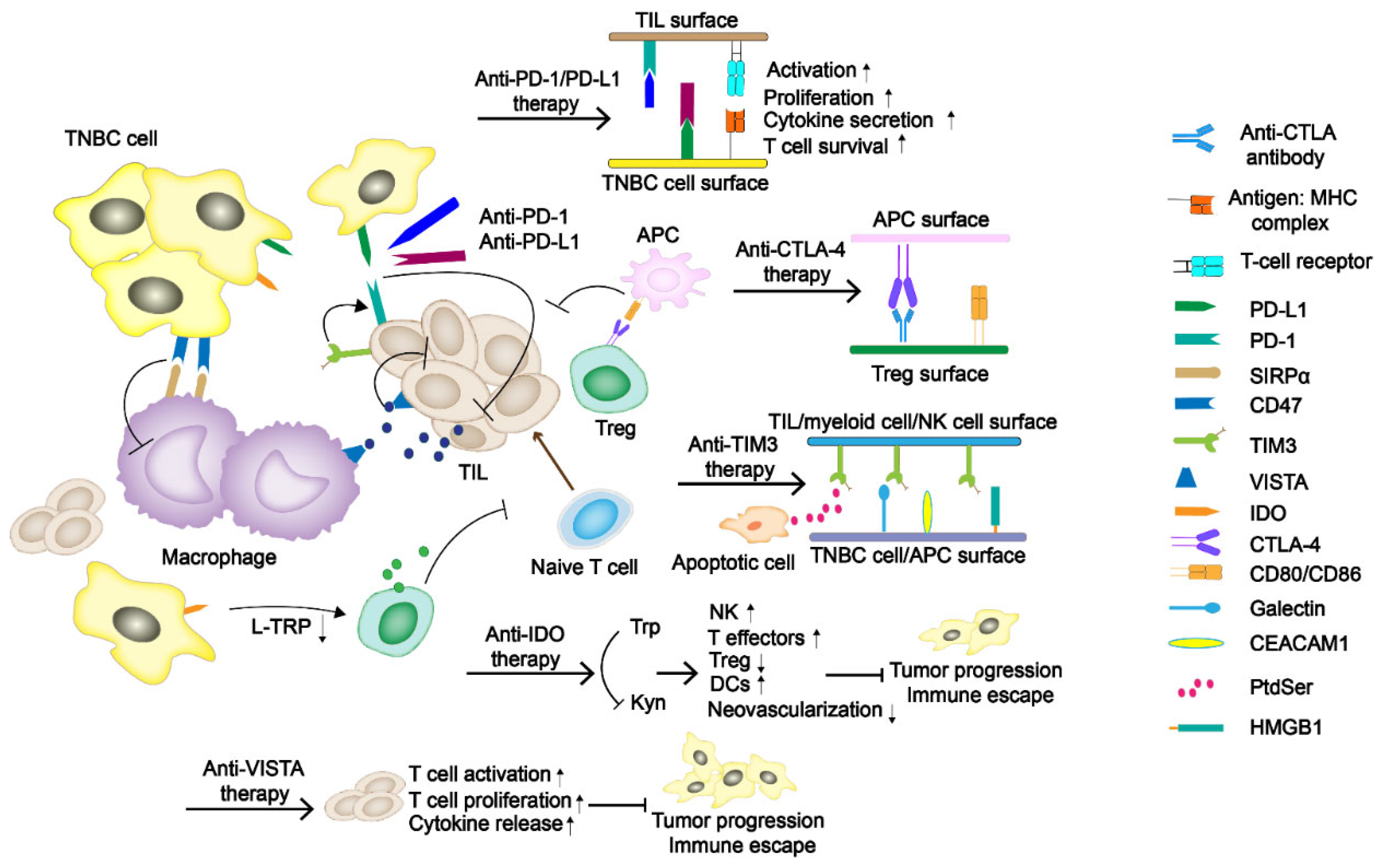
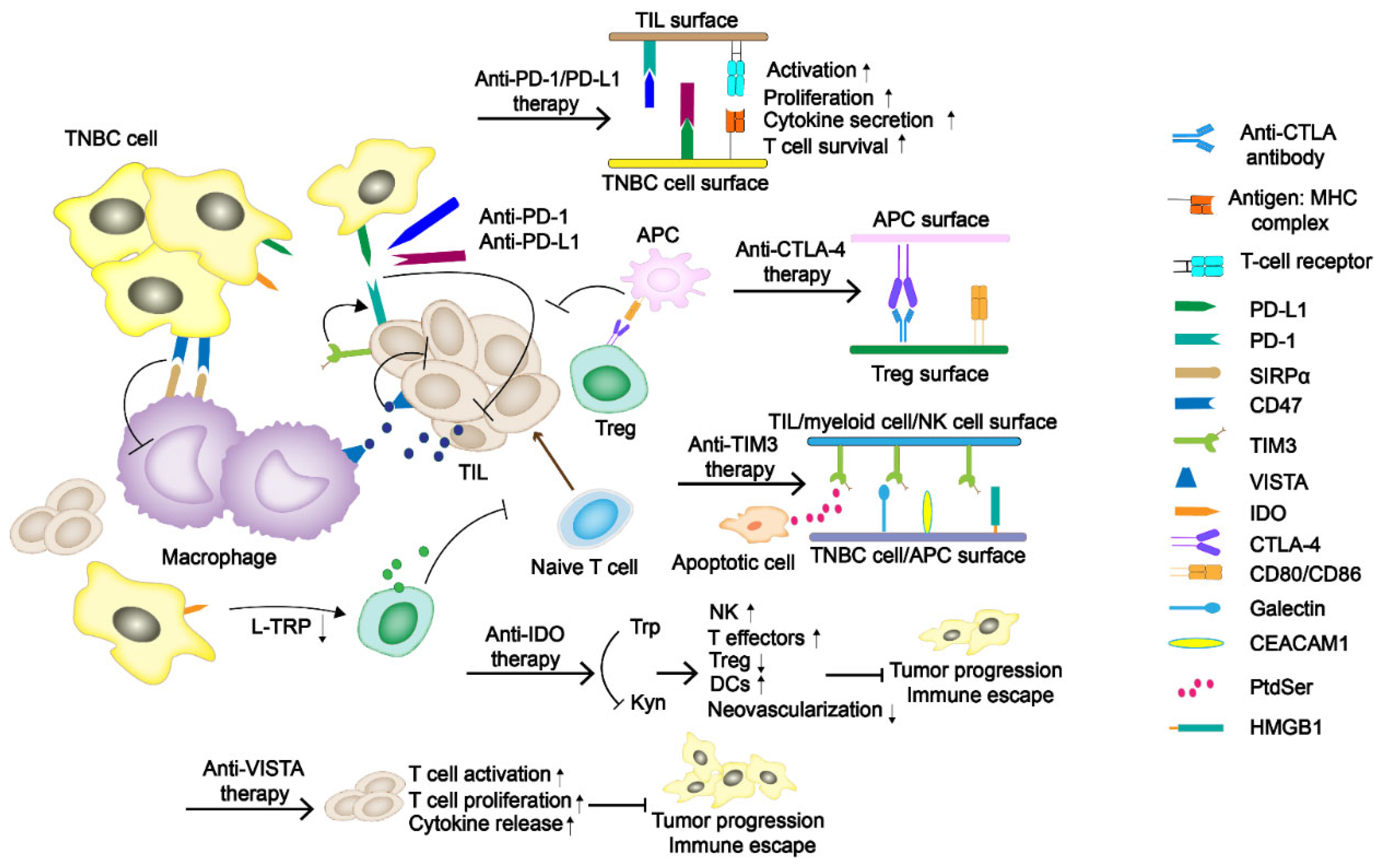
1.1. Immune Checkpoint Blockades PD-1/PD-L1 and CTLA-4
1.2. T-Cell Immunoglobulin Domain and Mucin Domain-3 (TIM-3)
1.3. Indoleamine 2,3-Dioxygenase (IDO)
1.4. V Domain Ig Suppressor of T-Cell Activation (VISTA)
1.5. Adoptive T-Cell Immunotherapy
1.6. Tumor Vaccine Immunotherapy
1.7. Immunotherapy-Involved Combination Therapies
2. Nanocarriers for the Immunotherapeutic Treatment of TNBC
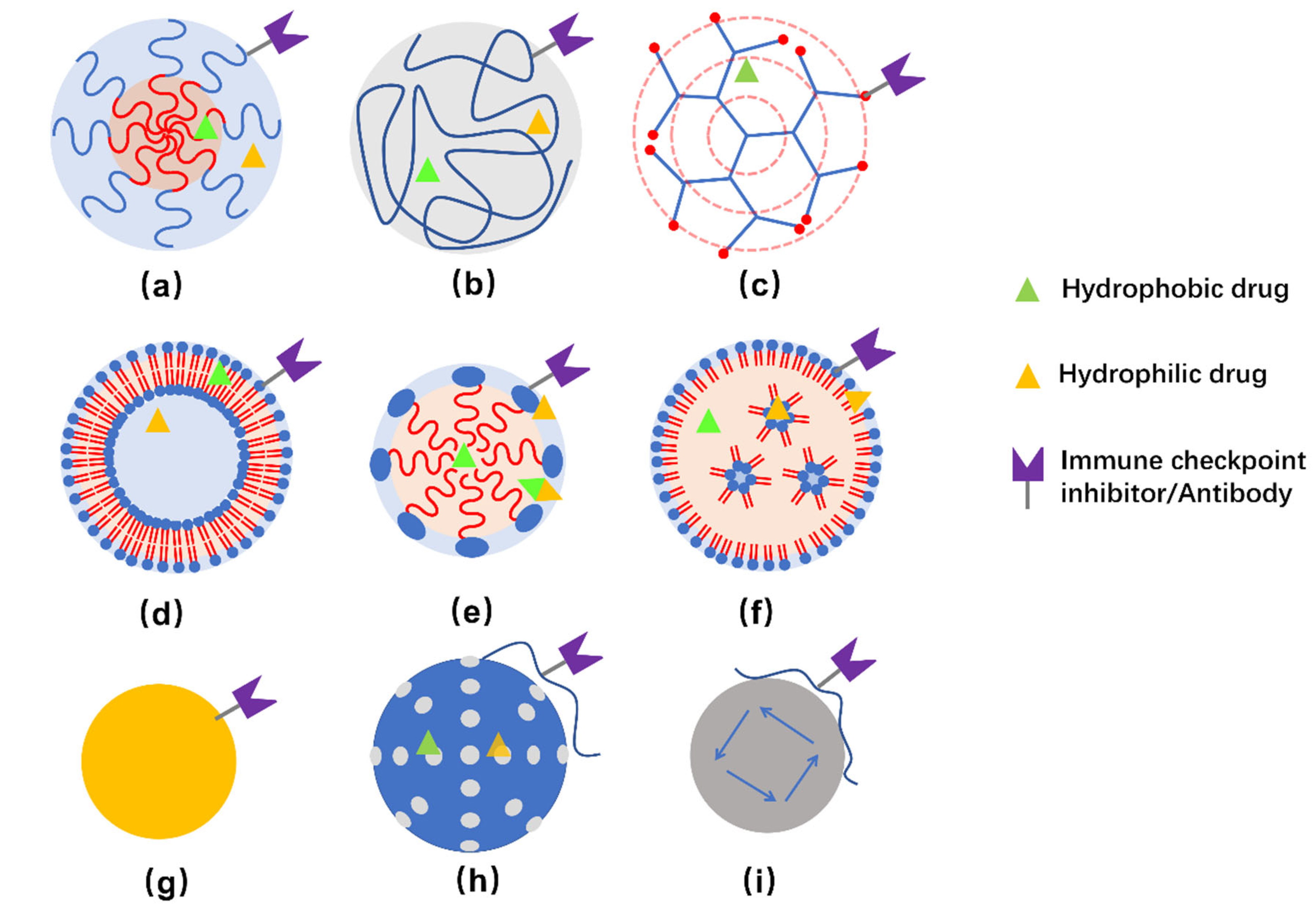
2.1. Nanomaterials for Delivering the Immunotherapeutic Agents of TNBC
2.2. Nanocarriers for the Delivery of Immune Checkpoint Blockade Molecules
2.3. Nanoparticles for the Delivery of Combination Therapy Agents
2.3.1. Immunotherapy Combined with PTT
2.3.2. Immunotherapy Combined with PDT
2.3.3. Immunotherapy Combined with SDT
2.3.4. Others
3. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [Green Version]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [Green Version]
- Makhoul, I.; Atiq, M.; Alwbari, A.; Kieber-Emmons, T. Breast cancer immunotherapy: An update. Breast Cancer (Auckl) 2018, 12, 1–15. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Emens, L.A. Breast cancer immunotherapy: Facts and hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Deepak, K.G.K.; Vempati, R.; Nagaraju, G.P.; Dasari, V.R.; Nagini, S.; Rao, D.N.; Malla, R.R. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 2020, 153, 104683. [Google Scholar] [CrossRef]
- Belli, C.; Trapani, D.; Viale, G.; D’Amico, P.; Duso, B.A.; Della Vigna, P.; Orsi, F.; Curigliano, G. Targeting the microenvironment in solid tumors. Cancer Treat. Rev. 2018, 65, 22–32. [Google Scholar] [CrossRef]
- Shihab, I.; Khalil, B.A.; Elemam, N.M.; Hachim, I.Y.; Hachim, M.Y.; Hamoudi, R.A.; Maghazachi, A.A. Understanding the role of innate immune cells and identifying genes in breast cancer microenvironment. Cancers 2020, 12, 2226. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. FOXP3+ Treg as a therapeutic target for promoting anti-tumor immunity. Expert Opin. Ther. Targets 2018, 22, 353–363. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Targeting Treg cells in cancer immunotherapy. Eur. J. Immunol. 2019, 49, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Wang, S.; Xie, K.; Liu, T. Cancer immunotherapies: From efficacy to resistance mechanisms-Not only checkpoint matters. Front. Immunol. 2021, 12, 690112. [Google Scholar] [CrossRef]
- Hosseini, A.; Gharibi, T.; Marofi, F.; Babaloo, Z.; Baradaran, B. CTLA-4: From mechanism to autoimmune therapy. Int. ImmunoPharmacol. 2020, 80, 106221. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Longo, D.L. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.; Nianqing, X.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. ImmunoPharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Markham, A.; Duggan, S. Cemiplimab: First global approval. Drugs 2018, 78, 1841–1846. [Google Scholar] [CrossRef]
- Kwok, G.; Yau, T.C.; Chiu, J.W.; Tse, E.; Kwong, Y.L. Pembrolizumab (Keytruda). Hum. Vaccin Immunother. 2016, 12, 2777–2789. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Shen, J.; Ivaturi, V.; Gopalakrishnan, M.; Feng, Y.; Schmidt, B.J.; Statkevich, P.; Goodman, V.; Gobburu, J.; Bello, A.; et al. Model-based evaluation of the efficacy and safety of nivolumab once every 4 weeks across multiple tumor types. Ann. Oncol. 2020, 31, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.T.; Lee, J.Y.; Lim, H.; Lee, S.H.; Moon, Y.J.; Pyo, H.J.; Ryu, S.E.; Shin, W.; Heo, Y.S. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci. Rep. 2017, 7, 5532. [Google Scholar] [CrossRef] [Green Version]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef]
- Mediratta, K.; El-Sahli, S.; D’Costa, V.; Wang, L. Current progresses and challenges of immunotherapy in triple-negative breast cancer. Cancers 2020, 12, 3529. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in patients with advanced triple-negative breast cancer: Phase Ib KEYNOTE-012 study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 2 November 2021).
- Joller, N.; Kuchroo, V.K. Tim-3, Lag-3, and TIGIT. Curr Top. Microbiol. Immunol. 2017, 410, 127–156. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a target for cancer immunotherapy and mechanisms of action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Byun, K.D.; Hwang, H.J.; Park, K.J.; Kim, M.C.; Cho, S.H.; Ju, M.H.; Lee, J.H.; Jeong, J.S. T-cell immunoglobulin mucin 3 expression on tumor infiltrating lymphocytes as a positive prognosticator in triple-negative breast cancer. J. Breast Cancer 2018, 21, 406–414. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Iain, A.; Murray, A.D.P.; Gary, H. Perdew. AH receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef]
- Wei, L.; Zhu, S.; Li, M.; Li, F.; Wei, F.; Liu, J.; Ren, X. High indoleamine 2,3-dioxygenase is correlated with microvessel density and worse prognosis in breast cancer. Front. Immunol. 2018, 9, 724. [Google Scholar] [CrossRef]
- Asghar, K.; Loya, A.; Rana, I.A.; Tahseen, M.; Ishaq, M.; Farooq, A.; Bakar, M.A.; Masood, I. Indoleamine 2,3-dioxygenase expression and overall survival in patients diagnosed with breast cancer in Pakistan. Cancer Manag. Res. 2019, 11, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Wu, Y.H.; Song, Y.; Yu, B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 68. [Google Scholar] [CrossRef]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Slater, B.T.; Han, X.; Chen, L.; Xiong, Y. Structural insight into T cell coinhibition by PD-1H (VISTA). Proc. Natl. Acad. Sci. USA 2020, 117, 1648–1657. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, J.; Shi, Z.; Liu, W.; Hu, X.; Qie, C.; Chen, W.; Wang, Y.; Wang, L.; Jiang, J.; et al. The expression pattern and clinical significance of the immune checkpoint regulator VISTA in human breast cancer. Front. Immunol. 2020, 11, 563044. [Google Scholar] [CrossRef]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Ren, X.; Zhou, Y.; Mao, F.; Lin, Y.; Wu, H.; Sun, Q. VISTA expression on immune cells correlates with favorable prognosis in patients with triple-negative breast cancer. Front. Oncol. 2020, 10, 583966. [Google Scholar] [CrossRef]
- Wei, H.; Wang, Z.; Kuang, Y.; Wu, Z.; Zhao, S.; Zhang, Z.; Li, H.; Zheng, M.; Zhang, N.; Long, C.; et al. Intercellular adhesion molecule-1 as target for CAR-T-cell therapy of triple-negative breast cancer. Front. Immunol. 2020, 11, 573823. [Google Scholar] [CrossRef]
- Cummins, K.D.; Gill, S. Anti-CD123 chimeric antigen receptor T-cells (CART): An evolving treatment strategy for hematological malignancies, and a potential ace-in-the-hole against antigen-negative relapse. Leuk. Lymphoma 2018, 59, 1539–1553. [Google Scholar] [CrossRef]
- Wei, J.; Han, X.; Bo, J.; Han, W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Song, D.G.; Ye, Q.; Poussin, M.; Chacon, J.A.; Figini, M.; Powell, D.J., Jr. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J. Hematol. Oncol. 2016, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front. Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef] [Green Version]
- Xia, L.; Zheng, Z.; Liu, J.Y.; Chen, Y.J.; Ding, J.; Hu, G.S.; Hu, Y.H.; Liu, S.; Luo, W.X.; Xia, N.S.; et al. Targeting triple-negative breast cancer with combination therapy of EGFR CAR T cells and CDK7 inhibition. Cancer Immunol. Res. 2021, 9, 707–722. [Google Scholar] [CrossRef]
- Stuber, T.; Monjezi, R.; Wallstabe, L.; Kuhnemundt, J.; Nietzer, S.L.; Dandekar, G.; Wockel, A.; Einsele, H.; Wischhusen, J.; Hudecek, M. Inhibition of TGF-beta-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000676. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Zhang, Z.; Lu, M.; Qin, Y.; Gao, W.; Tao, L.; Su, W.; Zhong, J. Neoantigen: A new breakthrough in tumor immunotherapy. Front. Immunol. 2021, 12, 672356. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Miao, L.; Liu, Q.; Musetti, S.; Li, J.; Huang, L. Combination immunotherapy of MUC1 mRNA nano-vaccine and CTLA-4 blockade effectively inhibits growth of triple negative breast cancer. Mol. Ther. 2018, 26, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Pack, C.D.; Bommireddy, R.; Munoz, L.E.; Patel, J.M.; Bozeman, E.N.; Dey, P.; Radhakrishnan, V.; Vartabedian, V.F.; Venkat, K.; Ramachandiran, S.; et al. Tumor membrane-based vaccine immunotherapy in combination with anti-CTLA-4 antibody confers protection against immune checkpoint resistant murine triple-negative breast cancer. Hum. Vaccin Immunother. 2020, 16, 3184–3193. [Google Scholar] [CrossRef]
- Ho, A.Y.; Barker, C.A.; Arnold, B.B.; Powell, S.N.; Hu, Z.I.; Gucalp, A.; Lebron-Zapata, L.; Wen, H.Y.; Kallman, C.; D’Agnolo, A.; et al. A Phase 2 clinical trial assessing the efficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer 2020, 126, 850–860. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Bernier, C.; Soliman, A.; Gravel, M.; Dankner, M.; Savage, P.; Petrecca, K.; Park, M.; Siegel, P.M.; Shore, G.C.; Roulston, A. DZ-2384 has a superior preclinical profile to taxanes for the treatment of triple-negative breast cancer and is synergistic with anti-CTLA-4 immunotherapy. Anticancer Drugs 2018, 29, 774–785. [Google Scholar] [CrossRef]
- Li, M.; Xing, S.; Zhang, H.; Shang, S.; Li, X.; Ren, B.; Li, G.; Chang, X.; Li, Y.; Li, W. A matrix metalloproteinase inhibitor enhances anti-cytotoxic T lymphocyte antigen-4 antibody immunotherapy in breast cancer by reprogramming the tumor microenvironment. Oncol. Rep. 2016, 35, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, X.; Liu, X.; Pan, W.; Li, N.; Tang, B. Designing and engineering of nanocarriers for bioapplication in cancer immunotherapy. ACS Appl. Bio Mater. 2020, 3, 8321–8337. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [Green Version]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Gu, Z.; Da Silva, C.G.; Van der Maaden, K.; Ossendorp, F.; Cruz, L.J. Liposome-based drug delivery systems in cancer immunotherapy. Pharmaceutics 2020, 12, 1054. [Google Scholar] [CrossRef]
- Johnson, R.; Sabnis, N.; McConathy, W.J.; Lacko, A.G. The potential role of nanotechnology in therapeutic approaches for triple negative breast cancer. Pharmaceutics 2013, 5, 353–370. [Google Scholar] [CrossRef] [Green Version]
- Jain, V.; Kumar, H.; Anod, H.V.; Chand, P.; Gupta, N.V.; Dey, S.; Kesharwani, S.S. A review of nanotechnology-based approaches for breast cancer and triple-negative breast cancer. J. Control. Release 2020, 326, 628–647. [Google Scholar] [CrossRef]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef]
- Bertrand, N.; Grenier, P.; Mahmoudi, M.; Lima, E.M.; Appel, E.A.; Dormont, F.; Lim, J.M.; Karnik, R.; Langer, R.; Farokhzad, O.C. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat. Commun. 2017, 8, 777. [Google Scholar] [CrossRef]
- Hickey, J.W.; Santos, J.L.; Williford, J.M.; Mao, H.Q. Control of polymeric nanoparticle size to improve therapeutic delivery. J. Control. Release 2015, 219, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Zielinska, A.; Carreiro, F.; Oliveira, A.M.; Neves, A.; Pires, B.; Venkatesh, D.N.; Durazzo, A.; Lucarini, M.; Eder, P.; Silva, A.M.; et al. Polymeric nanoparticles: Production, characterization, toxicology and ecotoxicology. Molecules 2020, 25, 3731. [Google Scholar] [CrossRef]
- Jones, M.-C.; Leroux, J.-C. Polymeric micelles–A new generation of colloidal drug carriers. Eur. J. Pharm. Biopharm. 1999, 48, 101–111. [Google Scholar] [CrossRef]
- Kesharwani, P.; Jain, K.; Jain, N.K. Dendrimer as nanocarrier for drug delivery. Prog. Polym. Sci. 2014, 39, 268–307. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Tyler, B.; Gullotti, D.; Mangraviti, A.; Utsuki, T.; Brem, H. Polylactic acid (PLA) controlled delivery carriers for biomedical applications. Adv. Drug Deliv. Rev. 2016, 107, 163–175. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces, 3rd ed.; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal drug delivery systems and anticancer drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W. Liposome-based drug delivery in breast cancer treatment. Breast Cancer Res. 2002, 4, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.C.; Barua, S.; Sharma, G.; Dey, S.K.; Rege, K. Inorganic nanoparticles for cancer imaging and therapy. J. Control. Release 2011, 155, 344–357. [Google Scholar] [CrossRef]
- Surapaneni, S.K.; Bashir, S.; Tikoo, K. Gold nanoparticles-induced cytotoxicity in triple negative breast cancer involves different epigenetic alterations depending upon the surface charge. Sci. Rep. 2018, 8, 12295. [Google Scholar] [CrossRef]
- Wosik, J.; Chen, W.; Qin, K.; Ghobrial, R.M.; Kubiak, J.Z.; Kloc, M. Magnetic field changes macrophage phenotype. Biophys. J. 2018, 114, 2001–2013. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.W.; Tsao, H.Y.; Chiang, C.S.; Chen, S.Y. Advances in magnetic nanoparticle-mediated cancer immune-theranostics. Adv. Healthc. Mater. 2021, 10, e2001451. [Google Scholar] [CrossRef]
- Bayda, S.; Hadla, M.; Palazzolo, S.; Riello, P.; Corona, G.; Toffoli, G.; Rizzolio, F. Inorganic nanoparticles for cancer therapy: A transition from lab to clinic. Curr. Med. Chem. 2018, 25, 4269–4303. [Google Scholar] [CrossRef]
- Chen, C.; Guo, Q.; Fu, H.; Yu, J.; Wang, L.; Sun, Y.; Zhang, J.; Duan, Y. Asynchronous blockade of PD-L1 and CD155 by polymeric nanoparticles inhibits triple-negative breast cancer progression and metastasis. Biomaterials 2021, 275, 120988. [Google Scholar] [CrossRef]
- Manieri, N.A.; Chiang, E.Y.; Grogan, J.L. TIGIT: A key inhibitor of the cancer immunity cycle. Trends Immunol. 2017, 38, 20–28. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Madore, J.; Li, X.Y.; Smyth, M.J. Tumor intrinsic and extrinsic immune functions of CD155. Semin. Cancer Biol. 2020, 65, 189–196. [Google Scholar] [CrossRef]
- Zhang, R.; Zhu, Z.; Lv, H.; Li, F.; Sun, S.; Li, J.; Lee, C.S. Immune checkpoint blockade mediated by a small-molecule nanoinhibitor targeting the PD-1/PD-L1 pathway synergizes with photodynamic therapy to elicit antitumor immunity and antimetastatic effects on breast cancer. Small 2019, 15, e1903881. [Google Scholar] [CrossRef]
- Sau, S.; Alsaab, H.O.; Bhise, K.; Alzhrani, R.; Nabil, G.; Iyer, A.K. Multifunctional nanoparticles for cancer immunotherapy: A groundbreaking approach for reprogramming malfunctioned tumor environment. J. Control. Release 2018, 274, 24–34. [Google Scholar] [CrossRef]
- Hou, X.; Tao, Y.; Pang, Y.; Li, X.; Jiang, G.; Liu, Y. Nanoparticle-based photothermal and photodynamic immunotherapy for tumor treatment. Int. J. Cancer 2018, 143, 3050–3060. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Niu, Z.; Hou, B.; Zhou, L.; Li, Y.; Yu, H. Enhancing triple negative breast cancer immunotherapy by ICG-templated self-assembly of Paclitaxel nanoparticles. Adv. Funct. Mater. 2019, 30, 1906605. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Zhang, Z.; Ji, L.; Zhang, J.; Wang, Q.; Guo, T.; Ni, S.; Cai, R.; Mu, X.; et al. Recent progress on NIR-II photothermal therapy. Front. Chem. 2021, 9, 728066. [Google Scholar] [CrossRef]
- Li, X.; Lovell, J.F.; Yoon, J.; Chen, X. Clinical development and potential of photothermal and photodynamic therapies for cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657–674. [Google Scholar] [CrossRef]
- Yasothamani, V.; Karthikeyan, L.; Shyamsivappan, S.; Haldorai, Y.; Seetha, D.; Vivek, R. Synergistic effect of photothermally targeted NIR-responsive nanomedicine-induced immunogenic cell death for effective triple negative breast cancer therapy. Biomacromolecules 2021, 22, 2472–2490. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, Q.; Guo, Z.; Li, M.; Yang, X.; Wan, G.; Chen, H.; Zhang, Q.; Wang, Y. An intelligent biomimetic nanoplatform for holistic treatment of metastatic triple-negative breast cancer via photothermal ablation and immune remodeling. ACS Nano 2020, 14, 15161–15181. [Google Scholar] [CrossRef]
- Khafaji, M.; Zamani, M.; Golizadeh, M.; Bavi, O. Inorganic nanomaterials for chemo/photothermal therapy: A promising horizon on effective cancer treatment. Biophys. Rev. 2019, 11, 335–352. [Google Scholar] [CrossRef]
- Lv, S.; Miao, Y.; Liu, D.; Song, F. Recent development of photothermal agents (PTAs) based on small organic molecular dyes. ChemBioChem 2020, 21, 2098–2110. [Google Scholar] [CrossRef]
- Huang, L.; Li, Y.; Du, Y.; Zhang, Y.; Wang, X.; Ding, Y.; Yang, X.; Meng, F.; Tu, J.; Luo, L.; et al. Mild photothermal therapy potentiates anti-PD-L1 treatment for immunologically cold tumors via an all-in-one and all-in-control strategy. Nat. Commun. 2019, 10, 4871. [Google Scholar] [CrossRef]
- Huang, G.; Huang, H. Application of hyaluronic acid as carriers in drug delivery. Drug Deliv. 2018, 25, 766–772. [Google Scholar] [CrossRef]
- Balakrishnan, P.B.; Sweeney, E.E.; Ramanujam, A.S.; Fernandes, R. Photothermal therapies to improve immune checkpoint blockade for cancer. Int. J. Hyperth. 2020, 37, 34–49. [Google Scholar] [CrossRef]
- Luby, B.M.; Walsh, C.D.; Zheng, G. Advanced photosensitizer activation strategies for smarter photodynamic therapy beacons. Angew. Chem. Int. Ed. Engl. 2019, 58, 2558–2569. [Google Scholar] [CrossRef]
- Ming, L.; Cheng, K.; Chen, Y.; Yang, R.; Chen, D. Enhancement of tumor lethality of ROS in photodynamic therapy. Cancer Med. 2021, 10, 257–268. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, F.; Deng, H.; Lin, L.; Wang, S.; Kang, F.; Yu, G.; Lau, J.; Tian, R.; Zhang, M.; et al. Smart nanovesicle-mediated immunogenic cell death through tumor microenvironment modulation for effective photodynamic immunotherapy. ACS Nano 2020, 14, 620–631. [Google Scholar] [CrossRef]
- Chung, C.-H.; Lu, K.-Y.; Lee, W.-C.; Hsu, W.-J.; Lee, W.-F.; Dai, J.-Z.; Shueng, P.-W.; Lin, C.-W.; Mi, F.-L. Fucoidan-based, tumor-activated nanoplatform for overcoming hypoxia and enhancing photodynamic therapy and antitumor immunity. Biomaterials 2020, 257, 120227. [Google Scholar] [CrossRef]
- Shamay, Y.; Elkabets, M.; Li, H.; Shah, J.; Brook, S.; Wang, F.; Adler, K.; Baut, E.; Scaltriti, M.; Jena, P.V. P-selectin is a nanotherapeutic delivery target in the tumor microenvironment. Sci. Transl. Med. 2016, 8, 345ra87. [Google Scholar] [CrossRef] [Green Version]
- Lafond, M.; Yoshizawa, S.; Umemura, S.I. Sonodynamic therapy: Advances and challenges in clinical translation. J. Ultrasound. Med. 2019, 38, 567–580. [Google Scholar] [CrossRef]
- Canavese, G.; Ancona, A.; Racca, L.; Canta, M.; Dumontel, B.; Barbaresco, F.; Limongi, T.; Cauda, V. Nanoparticle-assisted ultrasound: A special focus on sonodynamic therapy against cancer. Chem. Eng. J. 2018, 340, 155–172. [Google Scholar] [CrossRef]
- Wan, G.Y.; Liu, Y.; Chen, B.W.; Liu, Y.Y.; Wang, Y.S.; Zhang, N. Recent advances of sonodynamic therapy in cancer treatment. Cancer Biol. Med. 2016, 13, 325–338. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Bai, L.; Wang, H.; Wu, Q.; Wang, H.; Liu, S.; Xu, B.; Shi, X.; Liu, H. Metal-organic-framework-derived carbon nanostructure augmented sonodynamic cancer therapy. Adv. Mater. 2018, 30, e1800180. [Google Scholar] [CrossRef]
- Son, S.; Kim, J.H.; Wang, X.; Zhang, C.; Yoon, S.A.; Shin, J.; Sharma, A.; Lee, M.H.; Cheng, L.; Wu, J.; et al. Multifunctional sonosensitizers in sonodynamic cancer therapy. Chem. Soc. Rev. 2020, 49, 3244–3261. [Google Scholar] [CrossRef]
- Chen, H.; Liu, L.; Ma, A.; Yin, T.; Chen, Z.; Liang, R.; Qiu, Y.; Zheng, M.; Cai, L. Noninvasively immunogenic sonodynamic therapy with manganese protoporphyrin liposomes against triple-negative breast cancer. Biomaterials 2021, 269, 120639. [Google Scholar] [CrossRef]
- Xu, C.; Yu, Y.; Sun, Y.; Kong, L.; Yang, C.; Hu, M.; Yang, T.; Zhang, J.; Hu, Q.; Zhang, Z. Transformable nanoparticle-enabled synergistic elicitation and promotion of immunogenic cell death for triple-negative breast cancer immunotherapy. Adv. Funct. Mater. 2019, 29, 1905213. [Google Scholar] [CrossRef]
- Xu, H.; Hu, M.; Liu, M.; An, S.; Guan, K.; Wang, M.; Li, L.; Zhang, J.; Li, J.; Huang, L. Nano-puerarin regulates tumor microenvironment and facilitates chemo- and immunotherapy in murine triple negative breast cancer model. Biomaterials 2020, 235, 119769. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Agent | Target | ClinicalTrials.Gov Identifier | Combinatorial Agent(s) | Phase | Recruitment Status |
|---|---|---|---|---|---|
| Atezolizumab | PD-L1 | NCT02530489 | Nab-paclitaxe | Phase II | Active, not recruiting |
| Pembrolizumab | PD-1 | NCT02622074 | Nab-paclitaxel + Doxorubicin + Cyclophosphamide, Nab-paclitaxel + Doxorubicin + Cyclophosphamide + Carboplatin, Doxorubicin + Cyclophosphamide + Carboplatin + Paclitaxel | Phase I | Completed |
| Pembrolizumab | PD-1 | NCT02734290 | Paclitaxel, Capecitabine | Phase I Phase II | Active, not recruiting |
| Pembrolizumab | PD-1 | NCT02768701 | Cyclophosphamide | Phase II | Active, not recruiting |
| Pembrolizumab | PD-1 | NCT02977468 | Intraoperative radiation therapy (IORT) | Phase I | Recruiting |
| Pembrolizumab | PD-1 | NCT02981303 | Imprime PGG | Phase II | Completed |
| Pembrolizumab | PD-1 | NCT03012230 | Ruxolitinib Phosphate | Phase I | Recruiting |
| Pembrolizumab | PD-1 | NCT03036488 | Carboplatin + Paclitaxel + Doxorubicin or Epirubicin + Cyclophosphamide + Granulocyte colony-stimulating factor (G-CSF) | Phase III | Active, not recruiting |
| Atezolizumab | PD-L1 | NCT03125902 | Paclitaxel | Phase III | Active, not recruiting |
| Atezolizumab | PD-L1 | NCT03164993 | Pegylated liposomal doxorubicin, Cyclophosphamide | Phase II | Recruiting |
| Durvalumab | PD-L1 | NCT03199040 | Neoantigen DNA vaccine | Phase I | Active, not recruiting |
| Atezolizumab | PD-L1 | NCT03206203 | Carboplatin | Phase II | Active, not recruiting |
| Atezolizumab | PD-L1 | NCT03281954 | Paclitaxel + Carboplatin, Doxorubicin + Cyclophosphamide or Epirubicin + Cyclophosphamide | Phase III | Active, not recruiting |
| Atezolizumab | PD-L1 | NCT03371017 | Gemcitabine + Capecitabine or Carboplatin | Phase III | Recruiting |
| Atezolizumab | PD-L1 | NCT03424005 | Nab-paclitaxel, Nab-paclitaxel + Tocilizumab, Sacituzumab Govitecan, Ipatasertib, Landiratuzumab vedotin (SGN-LIV1A), Selicrelumab + Bevacizumab, Chemo (Gemcitabine + Carboplatin or Eribulin) | Phase I Phase II | Recruiting |
| Nivolumab | PD-1 | NCT03487666 | Capecitabine | Phase II | Active, not recruiting |
| Atezolizumab | PD-L1 | NCT03498716 | Chemo (Paclitaxel, Dose-dense Doxorubicin or dose-dense Epirubicin), Cyclophosphamide | Phase III | Recruiting |
| Pembrolizumab | PD-1 | NCT03639948 | Carboplatin + Docetaxel + Pegfilgrastim | Phase II | Recruiting |
| Durvalumab | PD-L1 | NCT03742102 | Paclitaxel, Paclitaxel + Capivasertib, Paclitaxel + Oleclumab, Trastuzumab deruxtecan, Datopotamab deruxtecan | Phase I Phase II | Recruiting |
| Pembrolizumab | PD-1 | NCT03752723 | Cyclophosphamide + efineptakin alfa (GX-I7) | Phase I Phase II | Recruiting |
| Atezolizumab | PD-L1 | NCT03756298 | Capecitabine | Phase II | Recruiting |
| Durvalumab | PD-L1 | NCT03801369 | Olaparib | Phase II | Recruiting |
| Nivolumab | PD-1 | NCT03818685 | Ipilimumab | Phase II | Recruiting |
| Atezolizumab | PD-L1 | NCT03853707 | Ipatasertib + Carboplatin | Phase I Phase II | Suspended |
| Pembrolizumab | PD-1 | NCT04095689 | Docetaxel + Interleukin-12 gene therapy, Docetaxel + NG-monomethyl-L-arginine (L-NMMA) | Phase II | Recruiting |
| Camrelizumab | PD-1 | NCT04129996 | Nab-paclitaxel + famitinib | Phase II | Recruiting |
| Atezolizumab | PD-L1 | NCT04148911 | Nab-paclitaxel | Phase III | Recruiting |
| Atezolizumab | PD-L1 | NCT04177108 | Ipatasertib | Phase III | Active, not recruiting |
| Pembrolizumab | PD-1 | NCT04191135 | Carboplatin + Gemcitabine, Carboplatin + Gemcitabine + Olaparib | Phase II Phase III | Active, not recruiting |
| Camrelizumab | PD-1 | NCT04331067 | Nivolumab + Paclitaxel + Carboplatin | Phase I Phase II | Recruiting |
| Camrelizumab | PD-1 | NCT04335006 | Nab-paclitaxel + Apatinib, Nab-paclitaxel | Phase III | Recruiting |
| Camrelizumab | PD-1 | NCT04481763 | Radiotherapy | Phase I Phase II | Recruiting |
| Tiragolumab and Atezolizumab | PD-L1 | NCT04584112 | Nab-paclitaxel, Nab-paclitaxel + Carboplatin + Doxorubicin + Cyclophosphamide + G-CSF or Granulocyte-macrophage colony-stimulating factor (GM-CSF), Nab-paclitaxel + Doxorubicin + Cyclophosphamide + G-CSF + GM-CSF | Phase I | Recruiting |
| Camrelizumab | PD-1 | NCT04613674 | Chemotherapy | Phase III | Recruiting |
| Camrelizumab | PD-1 | NCT04676997 | Nab-paclitaxel + Epirubicin + Cyclophosphamide | Phase II | Recruiting |
| Pembrolizumab | PD-1 | NCT04683679 | Olaparib + Radiation, Radiation | Phase II | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Chen, C.; Tao, Y.; Lin, W.; Wang, P. Immunotherapy for Triple-Negative Breast Cancer. Pharmaceutics 2021, 13, 2003. https://doi.org/10.3390/pharmaceutics13122003
Cao Y, Chen C, Tao Y, Lin W, Wang P. Immunotherapy for Triple-Negative Breast Cancer. Pharmaceutics. 2021; 13(12):2003. https://doi.org/10.3390/pharmaceutics13122003
Chicago/Turabian StyleCao, Yifeng, Chuyang Chen, Yi Tao, Weifeng Lin, and Ping Wang. 2021. "Immunotherapy for Triple-Negative Breast Cancer" Pharmaceutics 13, no. 12: 2003. https://doi.org/10.3390/pharmaceutics13122003
APA StyleCao, Y., Chen, C., Tao, Y., Lin, W., & Wang, P. (2021). Immunotherapy for Triple-Negative Breast Cancer. Pharmaceutics, 13(12), 2003. https://doi.org/10.3390/pharmaceutics13122003