
Targeting the Gut Mucosal Immune System Using Nanomaterials


Abstract
1. Introduction
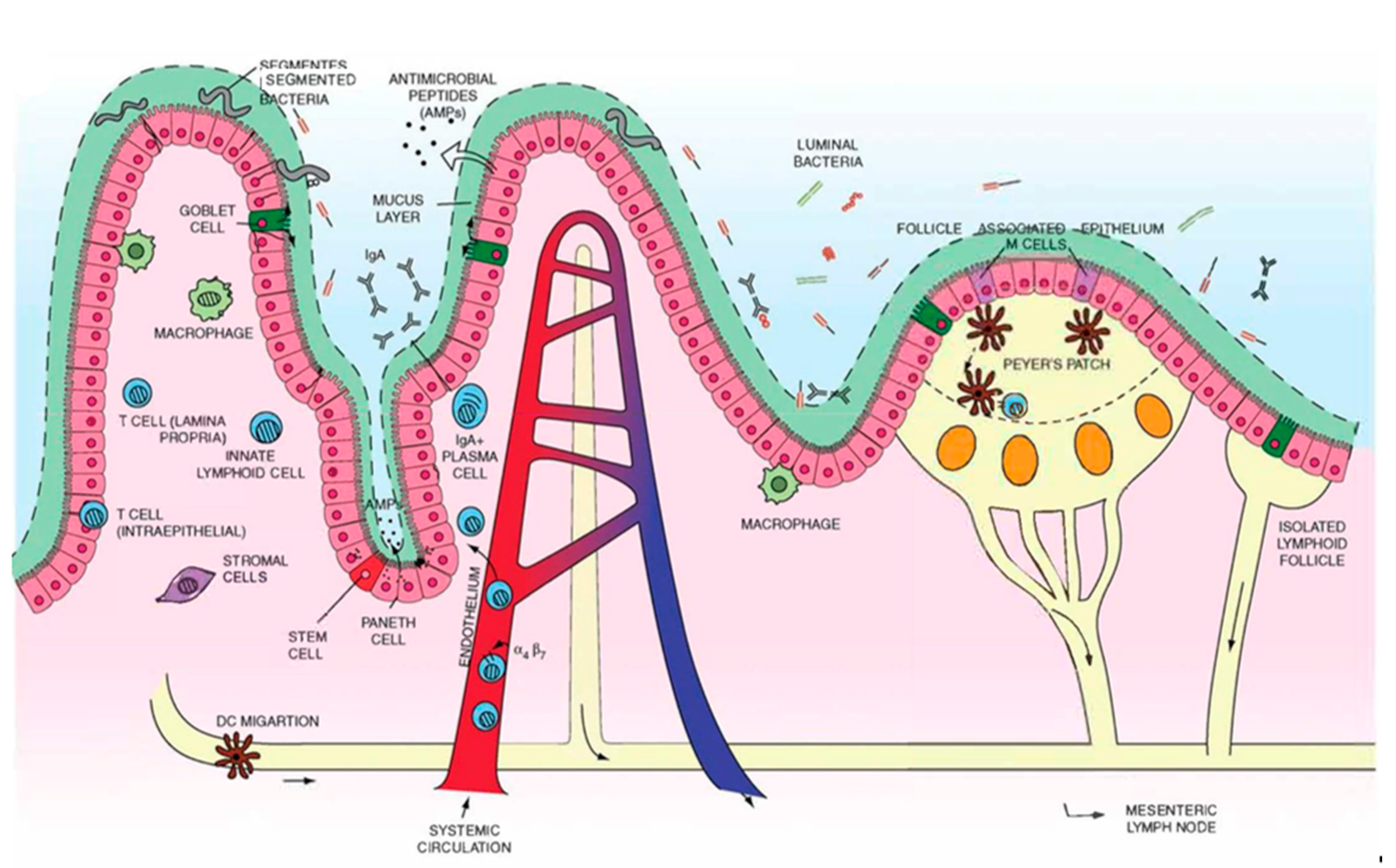
2. Overview of Gut Anatomy
2.1. Mucus and Epithelium
2.2. Lymphatics
2.3. Interstitium
3. Overview of Gut Immunity
3.1. Gut-Associated Lymphoid Tissues (GALT)
3.1.1. Multi-Follicular Lymphoid Tissues
3.1.2. Isolated Lymphoid Follicles
3.2. Lymph Nodes
3.3. Lymphatic Endothelial Cells Transport Antigens and Modulate Immunity
3.4. Oral Tolerance
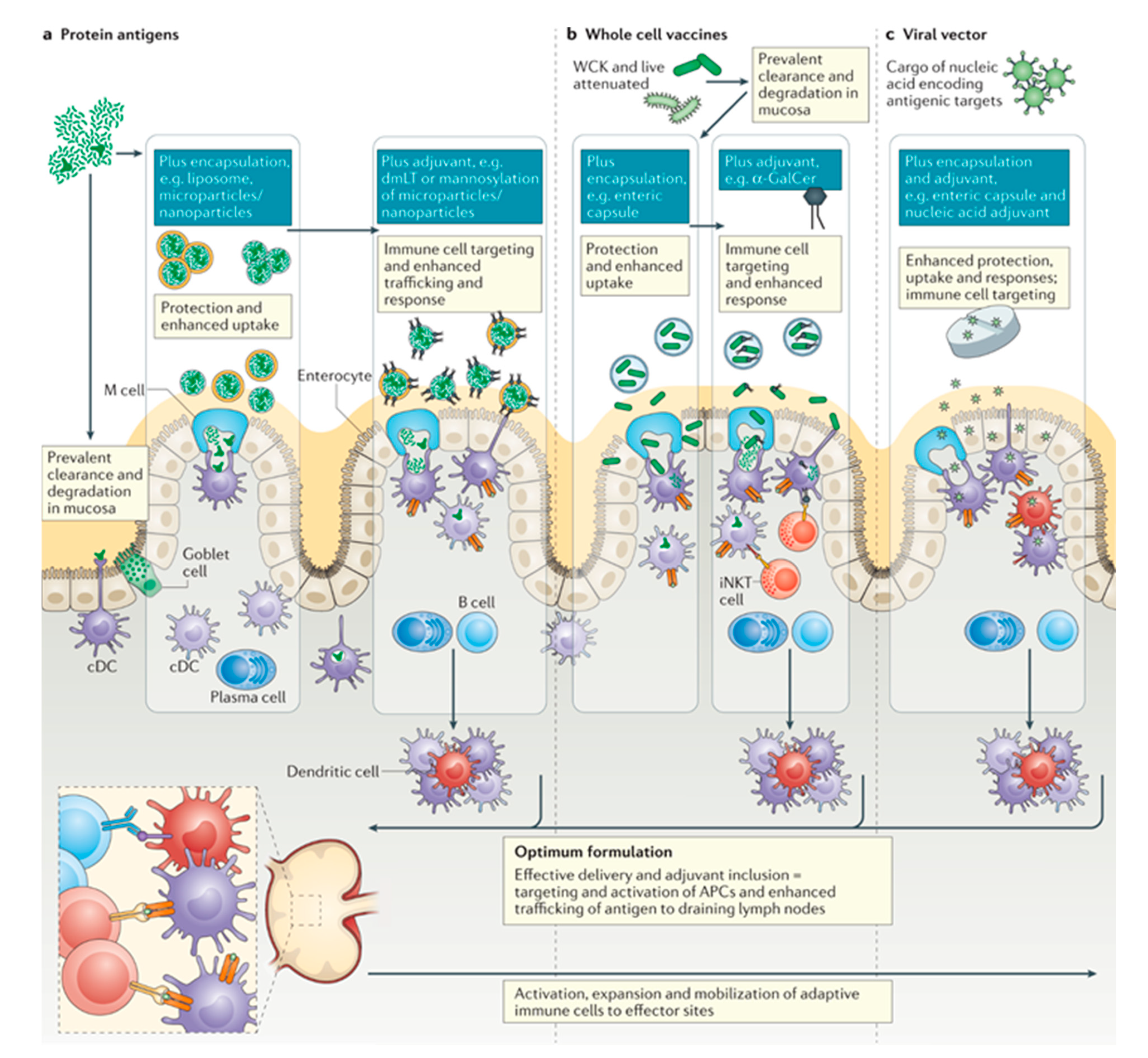
4. Nanomaterials for Targeting the Gut Mucosal Immune System
4.1. GALT Targeting
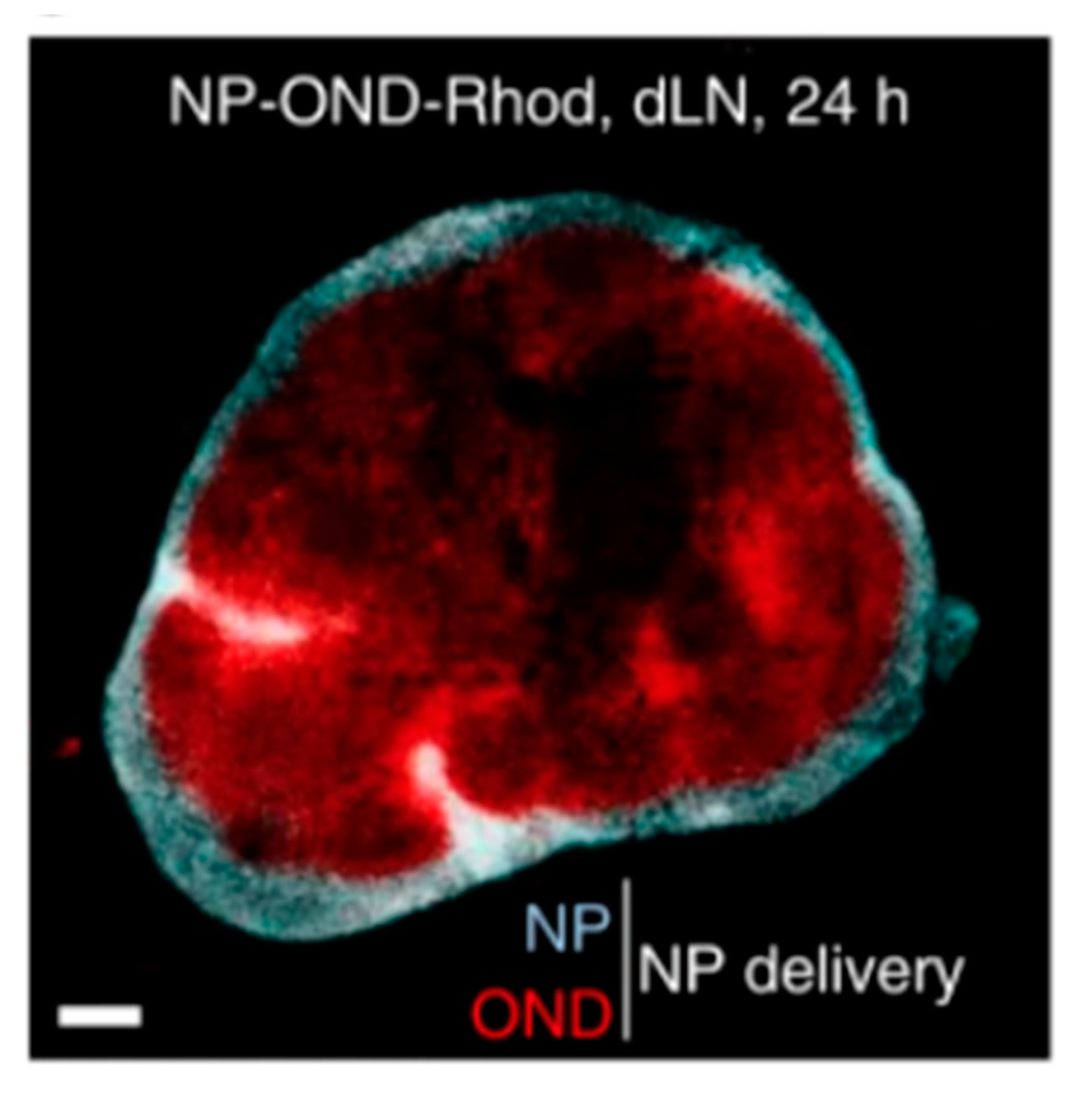
4.2. Lymph Node and Lymphatic Targeting
4.2.1. Lipid-Based Delivery Systems
4.2.2. Non-Lipid-Based Nanomaterials
4.3. Immune Cell Targeting

{kind=link}
{kind=link}
{kind=link}
| Nanomaterial | Dimension | Mechanism of Targeting | Targeted Cell Type/Region | Sources |
|---|---|---|---|---|
| Thiol-organosilica nanoparticles | <700 nm | Transcellular and paracellular transport pathways | M cells and CD11+ cells | [70] |
| Lipid–polymer hybrid nanoparticle | 300–400 nm | Mucus sticking | Peyer’s patches | [71,72] |
| Chitosan nanoparticle | <300 nm | Permeation enhancer | M cells | [73] |
| Targeting peptide nanoparticle | <250 nm | Adherence to specific M cell sugar residues | M cells | [75,76,77,78,79,80,81,82] |
| Lipid nanoparticles | <500 nm | Chylomicron formation | Enterocytes; intestinal lymphatics | [85,86,87,88,89,90,91,92,93,94] |
| Exosomes | 50 nm | Receptor targeting | Targeting receptors on dendritic cells (i.e., CD206) | [95] |
| Poly(propylene sulfide) nanoparticles | <250 nm | Cleaving of linkers | Cortex and paracortex of lymph node | [96] |
4.4. Taking Advantage of Oral Tolerance
4.5. Oral Vaccines
4.6. Outlook
4.7. Literature Search Method
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and regulation of the mucus barrier in the gut. Tissue Barriers 2015, 3, e982426. [Google Scholar] [CrossRef]
- Cone, R.A. Mucus. In Mucosal Immunology; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Kesimer, M.; Sheehan, J.K. Mass spectrometric analysis of mucin core proteins. Methods Mol. Biol. 2012, 842, 67–79. [Google Scholar]
- McCright, J.C.; Maisel, K. Engineering drug delivery systems to overcome mucosal barriers for immunotherapy and vaccination. Tissue Barriers 2020, 8, 1695476. [Google Scholar] [CrossRef]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Zhang, Y.H.; Zhang, W. Regulation of Intestinal Epithelial Cells Properties and Functions by Amino Acids. Biomed. Res. Int. 2018, 2018, 2819154. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, W.L. The lymphatic system in body homeostasis: Physiological conditions. Lymphat. Res. Biol. 2003, 1, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A. The physiology of the lymphatic system. Adv. Drug Deliv. Rev. 2001, 50, 3–20. [Google Scholar] [CrossRef]
- Randolph, G.J.; Ivanov, S.; Zinselmeyer, B.H.; Scallan, J.P. The Lymphatic System: Integral Roles in Immunity. Annu. Rev. Immunol. 2017, 35, 31–52. [Google Scholar] [CrossRef]
- Miller, M.J.; McDole, J.R.; Newberry, R.D. Microanatomy of the intestinal lymphatic system. Ann. N. Y. Acad. Sci. 2010, 1207 (Suppl. 1), E21–E28. [Google Scholar] [CrossRef]
- Cueni, L.N.; Detmar, M. The lymphatic system in health and disease. Lymphat. Res. Biol. 2008, 6, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Oliver, G.; Kipnis, J.; Randolph, G.J.; Harvey, N.L. The Lymphatic Vasculature in the 21(st) Century: Novel Functional Roles in Homeostasis and Disease. Cell 2020, 182, 270–296. [Google Scholar] [CrossRef] [PubMed]
- Benias, P.C.; Wells, R.G.; Sackey-Aboagye, B.; Klavan, H.; Reidy, J.; Buonocore, D.; Miranda, M.; Kornacki, S.; Wayne, M.; Carr-Locke, D.L.; et al. Structure and Distribution of an Unrecognized Interstitium in Human Tissues. Sci. Rep. 2018, 8, 4947. [Google Scholar] [CrossRef] [PubMed]
- Wiig, H.; Swartz, M.A. Interstitial fluid and lymph formation and transport: Physiological regulation and roles in inflammation and cancer. Physiol. Rev. 2012, 92, 1005–1060. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Mucosal Immunity: Induction, Dissemination, and Effector Functions. Scand. J. Immunol. 2009, 70, 505–515. [Google Scholar] [CrossRef]
- Mörbe, U.M.; Jørgensen, P.B.; Fenton, T.M.; von Burg, N.; Riis, L.B.; Spencer, J.; Agace, W.W. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol. 2021, 14, 793–802. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef]
- Brandtzaeg, P.; Kiyono, H.; Pabst, R.; Russell, M.W. Terminology: Nomenclature of mucosa-associated lymphoid tissue. Mucosal Immunol. 2008, 1, 31–37. [Google Scholar] [CrossRef]
- Cader, M.Z.; Kaser, A. Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammation. Gut 2013, 62, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Cornes. Number, size, and distribution of Peyer’s patches in the human small intestine: Part II The effect of age on Peyer’s patches. Gut 1965, 6, 230–233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kobayashi, N.; Takahashi, D.; Takano, S.; Kimura, S.; Hase, K. The Roles of Peyer’s Patches and Microfold Cells in the Gut Immune System: Relevance to Autoimmune Diseases. Front. Immunol. 2019, 10, 2345. [Google Scholar] [CrossRef]
- Jung, C.; Hugot, J.-P.; Barreau, F. Peyer’s Patches: The Immune Sensors of the Intestine. Int. J. Inflamm. 2010, 2010, 823710. [Google Scholar] [CrossRef]
- Van Kruiningen, H.J.; West, A.B.; Freda, B.J.; Holmes, K.A. Distribution of Peyer’s Patches in the Distal Ileum. Inflamm. Bowel Dis. 2002, 8, 180–185. [Google Scholar] [CrossRef]
- Langman, J.M.; Rowland, R. The number and distribution of lymphoid follicles in the human large intestine. J. Anat. 1986, 149, 189–194. [Google Scholar]
- Pabst, O.; Herbrand, H.; Worbs, T.; Friedrichsen, M.; Yan, S.; Hoffmann, M.W.; Körner, H.; Bernhardt, G.; Pabst, R.; Förster, R. Cryptopatches and isolated lymphoid follicles: Dynamic lymphoid tissues dispensable for the generation of intraepithelial lymphocytes. Eur. J. Immunol. 2005, 35, 98–107. [Google Scholar] [CrossRef]
- Rosner, A.J.; Keren, D.F. Demonstration of M cells in the specialized follicle-associated epithelium overlying isolated lymphoid follicles in the gut. J. Leukoc. Biol. 1984, 35, 397–404. [Google Scholar] [CrossRef]
- Jørgensen, P.B.; Fenton, T.M.; Mörbe, U.M.; Riis, L.B.; Jakobsen, H.L.; Nielsen, O.H.; Agace, W.W. Identification, isolation and analysis of human gut-associated lymphoid tissues. Nat. Protoc. 2021, 16, 2051–2067. [Google Scholar] [CrossRef]
- Gasteiger, G.; Ataide, M.; Kastenmüller, W. Lymph node—An organ for T-cell activation and pathogen defense. Immunol. Rev. 2016, 271, 200–220. [Google Scholar] [CrossRef]
- Jalkanen, S.; Salmi, M. Lymphatic endothelial cells of the lymph node. Nat. Rev. Immunol. 2020, 20, 566–578. [Google Scholar] [CrossRef]
- Girard, J.-P.; Moussion, C.; Förster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef]
- Podgrabinska, S.; Braun, P.; Velasco, P.; Kloos, B.; Pepper, M.S.; Skobe, M. Molecular characterization of lymphatic endothelial cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16069–16074. [Google Scholar] [CrossRef]
- Oliver, G.; Sosa-Pineda, B.; Geisendorf, S.; Spana, E.P.; Doe, C.Q.; Gruss, P. Prox 1, a prospero-related homeobox gene expressed during mouse development. Mech. Dev. 1993, 44, 3–16. [Google Scholar] [CrossRef]
- Nibbs, R.J.; Kriehuber, E.; Ponath, P.D.; Parent, D.; Qin, S.; Campbell, J.D.; Henderson, A.; Kerjaschki, D.; Maurer, D.; Graham, G.J.; et al. The beta-chemokine receptor D6 is expressed by lymphatic endothelium and a subset of vascular tumors. Am. J. Pathol. 2001, 158, 867–877. [Google Scholar] [CrossRef]
- Dieterich, L.C.; Ikenberg, K.; Cetintas, T.; Kapaklikaya, K.; Hutmacher, C.; Detmar, M. Tumor-Associated Lymphatic Vessels Upregulate PDL1 to Inhibit T-Cell Activation. Front. Immunol. 2017, 8, 66. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Dubrot, J.; Duraes, F.V.; Potin, L.; Capotosti, F.; Brighouse, D.; Suter, T.; LeibundGut-Landmann, S.; Garbi, N.; Reith, W.; Swartz, M.A.; et al. Lymph node stromal cells acquire peptide–MHCII complexes from dendritic cells and induce antigen-specific CD4+ T cell tolerance. J. Exp. Med. 2014, 211, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Tordesillas, L.; Berin, M.C. Mechanisms of Oral Tolerance. Clin. Rev. Allergy Immunol. 2018, 55, 107–117. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Smith, K. Mesenteric lymph nodes at the center of immune anatomy. J. Exp. Med. 2006, 203, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Pelaseyed, T.; Bergström, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.H.; Schütte, A.; van der Post, S.; Svensson, F.; Rodríguez-Piñeiro, A.M.; Nyström, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Garside, P.; Steel, M.; Liew, F.Y.; Mowat, A.M. CD4+ but not CD8+T cells are required for the induction of oral tolerance. Int. Immunol. 1995, 7, 501–504. [Google Scholar] [CrossRef]
- Trevaskis, N.L.; Kaminskas, L.M.; Porter, C.J.H. From sewer to saviour—Targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discov. 2015, 14, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Esterhazy, D.; Canesso, M.C.C.; Mesin, L.; Muller, P.A.; de Castro, T.B.R.; Lockhart, A.; ElJalby, M.; Faria, A.M.C.; Mucida, D. Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 2019, 569, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.L.; Tang, W.H.; Chen, W.C.; Diako, C.; Ross, C.F.; Li, S.D. Development of a Rapidly Dissolvable Oral Pediatric Formulation for Mefloquine Using Liposomes. Mol. Pharm. 2017, 14, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Lin, Y.; Zhang, X.; Feng, C.; Lu, Y.; Gao, Y.; Dong, C. Cyclic RGD peptide-modified liposomal drug delivery system for targeted oral apatinib administration: Enhanced cellular uptake and improved therapeutic effects. Int. J. Nanomed. 2017, 12, 1941–1958. [Google Scholar]
- Liu, Y.; Luo, X.; Xu, X.; Gao, N.; Liu, X. Preparation, characterization and in vivo pharmacokinetic study of PVP-modified oleanolic acid liposomes. Int. J. Pharm. 2017, 517, 1–7. [Google Scholar]
- Verma, A.K.; Sharma, S.; Gupta, P.; Singodia, D.; Kansal, S.; Sharma, V.; Mishra, P.R. Vitamin B12 Grafted Layer-by-Layer Liposomes Bearing HBsAg Facilitate Oral Immunization: Effect of Modulated Biomechanical Properties. Mol. Pharm. 2016, 13, 2531–2542. [Google Scholar] [CrossRef]
- Chen, W.L.; Yuan, Z.Q.; Liu, Y.; Yang, S.D.; Zhang, C.G.; Li, J.Z.; Zhu, W.J.; Li, F.; Zhou, X.F.; Lin, Y.M.; et al. Liposomes coated with N-trimethyl chitosan to improve the absorption of harmine in vivo and in vitro. Int. J. Nanomed. 2016, 11, 325–336. [Google Scholar]
- Uhl, P.; Helm, F.; Hofhaus, G.; Brings, S.; Kaufman, C.; Leotta, K.; Urban, S.; Haberkorn, U.; Mier, W.; Fricker, G. A liposomal formulation for the oral application of the investigational hepatitis B drug Myrcludex B. Eur. J. Pharm. Biopharm. 2016, 103, 159–166. [Google Scholar] [CrossRef]
- Jensen, S.M.; Christensen, C.J.; Petersen, J.M.; Treusch, A.H.; Brandl, M. Liposomes containing lipids from Sulfolobus islandicus withstand intestinal bile salts: An approach for oral drug delivery? Int. J. Pharm. 2015, 493, 63–69. [Google Scholar] [CrossRef]
- Chen, H.; Wu, J.; Sun, M.; Guo, C.; Yu, A.; Cao, F.; Zhao, L.; Tan, Q.; Zhai, G. N-trimethyl chitosan chloride-coated liposomes for the oral delivery of curcumin. J. Liposome Res. 2012, 22, 100–109. [Google Scholar]
- Hecq, J.; Amighi, K.; Goole, J. Development and evaluation of insulin-loaded cationic solid lipid nanoparticles for oral delivery. J. Drug Deliv. Sci. Technol. 2016, 36, 192–200. [Google Scholar] [CrossRef]
- Feng, H.; Zhu, Y.; Fu, Z.; Li, D. Preparation, characterization, and in vivo study of rhein solid lipid nanoparticles for oral delivery. Chem. Biol. Drug Des. 2017, 90, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Chen, C.; Guo, H.; Xu, J.; Zhang, J.; Zhu, X.; Yang, Y.; Zhou, Z.; Li, L.; Huang, Y. Design and evaluation of solid lipid nanoparticles modified with peptide ligand for oral delivery of protein drugs. Eur. J. Pharm. Biopharm. 2014, 88, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Dudhipala, N.; Janga, K.Y.; Gorre, T. Comparative study of nisoldipine-loaded nanostructured lipid carriers and solid lipid nanoparticles for oral delivery: Preparation, characterization, permeation and pharmacokinetic evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Diwan, R.; Ravi, P.R.; Pathare, N.S.; Aggarwal, V. Pharmacodynamic, pharmacokinetic and physical characterization of cilnidipine loaded solid lipid nanoparticles for oral delivery optimized using the principles of design of experiments. Colloids Surf. B Biointerfaces 2020, 193, 111073. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.L.; Bajaj, P.; Whitehead, K.A. Oral delivery of siRNA lipid nanoparticles: Fate in the GI tract. Sci. Rep. 2018, 8, 2178. [Google Scholar] [CrossRef]
- Baek, J.S.; Cho, C.W. Surface modification of solid lipid nanoparticles for oral delivery of curcumin: Improvement of bioavailability through enhanced cellular uptake, and lymphatic uptake. Eur. J. Pharm. Biopharm. 2017, 117, 132–140. [Google Scholar] [CrossRef]
- Sutradhar, K.B.; Amin, M.L. Nanoemulsions: Increasing possibilities in drug delivery. Eur. J. Nanomed. 2013, 5, 97–110. [Google Scholar] [CrossRef]
- Choi, S.J.; McClements, D.J. Nanoemulsions as delivery systems for lipophilic nutraceuticals: Strategies for improving their formulation, stability, functionality and bioavailability. Food Sci. Biotechnol. 2020, 29, 149–168. [Google Scholar] [CrossRef]
- Barahona, M.J.; Baratta, V.; Ollodart, J.; Mulligan, D.; Geibel, J.P. Design and implementation of novel nutraceuticals and derivatives for treating intestinal disorders. Future Med. Chem. 2019, 11, 847–855. [Google Scholar] [CrossRef]
- Yin, L.; Song, Z.; Qu, Q.; Kim, K.H.; Zheng, N.; Yao, C.; Chaudhury, I.; Tang, H.; Gabrielson, N.P.; Uckun, F.M.; et al. Supramolecular self-assembled nanoparticles mediate oral delivery of therapeutic TNF-α siRNA against systemic inflammation. Angew. Chem. Int. Ed. Engl. 2013, 52, 5757–5761. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, K.B.; Russell-Jones, G.J.; Yandrapu, S.K.; Diwan, P.V.; Jain, S.K. A novel vitamin B12-nanosphere conjugate carrier system for peroral delivery of insulin. J. Control. Release 2007, 117, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Bakhru, S.H.; Furtado, S.; Morello, A.P.; Mathiowitz, E. Oral delivery of proteins by biodegradable nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Sonaje, K.; Chuang, E.Y.; Lin, K.J.; Yen, T.C.; Su, F.Y.; Tseng, M.T.; Sung, H.W. Opening of epithelial tight junctions and enhancement of paracellular permeation by chitosan: Microscopic, ultrastructural, and computed-tomographic observations. Mol. Pharm. 2012, 9, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Sonaje, K.; Lin, Y.H.; Juang, J.H.; Wey, S.P.; Chen, C.T.; Sung, H.W. In vivo evaluation of safety and efficacy of self-assembled nanoparticles for oral insulin delivery. Biomaterials 2009, 30, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Furtado, S.; Abramson, D.; Burrill, R.; Olivier, G.; Gourd, C.; Bubbers, E.; Mathiowitz, E. Oral delivery of insulin loaded poly(fumaric-co-sebacic) anhydride microspheres. Int. J. Pharm. 2008, 347, 149–155. [Google Scholar] [CrossRef]
- Makhlof, A.; Werle, M.; Tozuka, Y.; Takeuchi, H. A mucoadhesive nanoparticulate system for the simultaneous delivery of macromolecules and permeation enhancers to the intestinal mucosa. J. Control. Release 2011, 149, 81–88. [Google Scholar] [CrossRef]
- Damgé, C.; Vranckx, H.; Balschmidt, P.; Couvreur, P. Poly(alkyl cyanoacrylate) nanospheres for oral administration of insulin. J. Pharm. Sci. 1997, 86, 1403–1409. [Google Scholar] [CrossRef]
- Awaad, A.; Nakamura, M.; Ishimura, K. Imaging of size-dependent uptake and identification of novel pathways in mouse Peyer’s patches using fluorescent organosilica particles. Nanomedicine 2012, 8, 627–636. [Google Scholar] [CrossRef]
- Bachhav, S.S.; Dighe, V.D.; Kotak, D.; Devarajan, P.V. Rifampicin Lipid-Polymer hybrid nanoparticles (LIPOMER) for enhanced Peyer’s patch uptake. Int. J. Pharm. 2017, 532, 612–622. [Google Scholar] [CrossRef]
- Bachhav, S.S.; Dighe, V.D.; Devarajan, P.V. Exploring Peyer’s Patch Uptake as a Strategy for Targeted Lung Delivery of Polymeric Rifampicin Nanoparticles. Mol. Pharm. 2018, 15, 4434–4445. [Google Scholar] [CrossRef] [PubMed]
- Kadiyala, I.; Loo, Y.; Roy, K.; Rice, J.; Leong, K.W. Transport of chitosan-DNA nanoparticles in human intestinal M-cell model versus normal intestinal enterocytes. Eur. J. Pharm. Sci. 2010, 39, 103–109. [Google Scholar] [CrossRef]
- Higgins, L.M.; Lambkin, I.; Donnelly, G.; Byrne, D.; Wilson, C.; Dee, J.; Smith, M.; O’Mahony, D.J. In vivo phage display to identify M cell-targeting ligands. Pharm. Res. 2004, 21, 695–705. [Google Scholar] [CrossRef]
- Clark, M.A.; Jepson, M.A.; Hirst, B.H. Exploiting M cells for drug and vaccine delivery. Adv. Drug Deliv. Rev. 2001, 50, 81–106. [Google Scholar] [CrossRef]
- Jepson, M.A.; Clark, M.A.; Hirst, B.H. M cell targeting by lectins: A strategy for mucosal vaccination and drug delivery. Adv. Drug Deliv. Rev. 2004, 56, 511–525. [Google Scholar] [CrossRef]
- Du, L.; Yu, Z.; Pang, F.; Xu, X.; Mao, A.; Yuan, W.; He, K.; Li, B. Targeted Delivery of GP5 Antigen of PRRSV to M Cells Enhances the Antigen-Specific Systemic and Mucosal Immune Responses. Front. Cell Infect. Microbiol. 2018, 8, 7. [Google Scholar] [CrossRef]
- Malik, B.; Goyal, A.K.; Markandeywar, T.S.; Rath, G.; Zakir, F.; Vyas, S.P. Microfold-cell targeted surface engineered polymeric nanoparticles for oral immunization. J. Drug Target 2012, 20, 76–84. [Google Scholar] [CrossRef]
- Lee, D.Y.; Nurunnabi, M.; Kang, S.H.; Nafiujjaman, M.; Huh, K.M.; Lee, Y.K.; Kim, Y.C. Oral Gavage Delivery of PR8 Antigen with beta-Glucan-Conjugated GRGDS Carrier to Enhance M-Cell Targeting Ability and Induce Immunity. Biomacromolecules 2017, 18, 1172–1179. [Google Scholar] [CrossRef]
- Shima, H.; Watanabe, T.; Fukuda, S.; Fukuoka, S.; Ohara, O.; Ohno, H. A novel mucosal vaccine targeting Peyer’s patch M cells induces protective antigen-specific IgA responses. Int. Immunol. 2014, 26, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Singh, B.; Li, H.S.; Kim, Y.K.; Kang, S.K.; Nah, J.W.; Choi, Y.J.; Cho, C.S. Targeted oral delivery of BmpB vaccine using porous PLGA microparticles coated with M cell homing peptide-coupled chitosan. Biomaterials 2014, 35, 2365–2373. [Google Scholar] [CrossRef]
- Islam, M.A.; Firdous, J.; Badruddoza, A.M.; Reesor, E.; Azad, M.; Hasan, A.; Lim, M.; Cao, W.J.; Guillemette, S.; Cho, C.S. M cell targeting engineered biomaterials for effective vaccination. Biomaterials 2019, 192, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Schudel, A.; Francis, D.M.; Thomas, S.N. Material design for lymph node drug delivery. Nat. Rev. Mater. 2019, 4, 415–428. [Google Scholar] [CrossRef]
- Lee, G.; Han, S.; Lu, Z.; Hong, J.; Phillips, A.R.J.; Windsor, J.A.; Porter, C.J.H.; Trevaskis, N.L. Intestinal delivery in a long-chain fatty acid formulation enables lymphatic transport and systemic exposure of orlistat. Int. J. Pharm. 2021, 596, 120247. [Google Scholar] [CrossRef]
- Giammanco, A.; Cefalù, A.B.; Noto, D.; Averna, M.R. The pathophysiology of intestinal lipoprotein production. Front. Physiol. 2015, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, N.L.; Caliph, S.M.; Nguyen, G.; Tso, P.; Charman, W.N.; Porter, C.J.H. A Mouse Model to Evaluate the Impact of Species, Sex, and Lipid Load on Lymphatic Drug Transport. Pharm. Res. 2013, 30, 3254–3270. [Google Scholar] [CrossRef][Green Version]
- Trevaskis, N.L.; McEvoy, C.L.; McIntosh, M.P.; Edwards, G.A.; Shanker, R.M.; Charman, W.N.; Porter, C.J.H. The Role of the Intestinal Lymphatics in the Absorption of Two Highly Lipophilic Cholesterol Ester Transfer Protein Inhibitors (CP524,515 and CP532,623). Pharm. Res. 2010, 27, 878–893. [Google Scholar] [CrossRef]
- Caliph, S.M.; Faassen, F.W.; Porter, C.J.H. The influence of intestinal lymphatic transport on the systemic exposure and brain deposition of a novel highly lipophilic compound with structural similarity to cholesterol. J. Pharm. Pharmacol. 2014, 66, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Kochappan, R.; Cao, E.; Han, S.; Hu, L.; Quach, T.; Senyschyn, D.; Ferreira, V.I.; Lee, G.; Leong, N.; Sharma, G.; et al. Targeted delivery of mycophenolic acid to the mesenteric lymph node using a triglyceride mimetic prodrug approach enhances gut-specific immunomodulation in mice. J. Control. Release 2021, 332, 636–651. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yang, J.; Pan, Y.; Guo, Z.; Gao, Y.; Huang, L.; Zhou, D.; Ge, Y.; Guo, F.; Zhu, W.; et al. Chylomicrons-Simulating Sustained Drug Release in Mesenteric Lymphatics for the Treatment of Crohn’s-Like Colitis. J. Crohn’s Colitis 2020, 15, 631–646. [Google Scholar] [CrossRef]
- Muntoni, E.; Marini, E.; Ahmadi, N.; Milla, P.; Ghè, C.; Bargoni, A.; Capucchio, M.T.; Biasibetti, E.; Battaglia, L. Lipid nanoparticles as vehicles for oral delivery of insulin and insulin analogs: Preliminary ex vivo and in vivo studies. Acta Diabetol. 2019, 56, 1283–1292. [Google Scholar] [CrossRef]
- Obinu, A.; Burrai, G.P.; Cavalli, R.; Galleri, G.; Migheli, R.; Antuofermo, E.; Rassu, G.; Gavini, E.; Giunchedi, P. Transmucosal Solid Lipid Nanoparticles to Improve Genistein Absorption via Intestinal Lymphatic Transport. Pharmaceutics 2021, 13, 267. [Google Scholar] [CrossRef]
- Du, Y.; Xia, Y.; Zou, Y.; Hu, Y.; Fu, J.; Wu, J.; Gao, X.D.; Ma, G. Exploiting the Lymph-Node-Amplifying Effect for Potent Systemic and Gastrointestinal Immune Responses via Polymer/Lipid Nanoparticles. ACS Nano 2019, 13, 13809–13817. [Google Scholar] [CrossRef]
- Yu, G.; Jung, H.; Kang, Y.Y.; Mok, H. Comparative evaluation of cell- and serum-derived exosomes to deliver immune stimulators to lymph nodes. Biomaterials 2018, 162, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.S.; Song, J.; Kang, Y.Y.; Mok, H. Mannose-Modified Serum Exosomes for the Elevated Uptake to Murine Dendritic Cells and Lymphatic Accumulation. Macromol. Biosci. 2019, 19, 1900042. [Google Scholar] [CrossRef] [PubMed]
- Schudel, A.; Chapman, A.P.; Yau, M.-K.; Higginson, C.J.; Francis, D.M.; Manspeaker, M.P.; Avecilla, A.R.C.; Rohner, N.A.; Finn, M.G.; Thomas, S.N. Programmable multistage drug delivery to lymph nodes. Nat. Nanotechnol. 2020, 15, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.C.; Curiel, D.T.; Pereboeva, L. Cell vehicle targeting strategies. Gene Ther. 2008, 15, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.; Li, M.O. Tissue-Resident Lymphocytes Across Innate and Adaptive Lineages. Front. Immunol. 2018, 9, 2104. [Google Scholar] [CrossRef]
- Wang, B.; Zhuang, X.; Deng, Z.-B.; Jiang, H.; Mu, J.; Wang, Q.; Xiang, X.; Guo, H.; Zhang, L.; Dryden, G.; et al. Targeted drug delivery to intestinal macrophages by bioactive nanovesicles released from grapefruit. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 522–534. [Google Scholar] [CrossRef]
- Dammes, N.; Goldsmith, M.; Ramishetti, S.; Dearling, J.L.J.; Veiga, N.; Packard, A.B.; Peer, D. Conformation-sensitive targeting of lipid nanoparticles for RNA therapeutics. Nat. Nanotechnol. 2021, 16, 1030–1038. [Google Scholar] [CrossRef]
- Neumann, U.H.; Ho, J.S.S.; Chen, S.; Tam, Y.Y.C.; Cullis, P.R.; Kieffer, T.J. Lipid nanoparticle delivery of glucagon receptor siRNA improves glucose homeostasis in mouse models of diabetes. Mol. Metab. 2017, 6, 1161–1172. [Google Scholar] [CrossRef]
- Xu, Y.; Van Hul, M.; Suriano, F.; Preat, V.; Cani, P.D.; Beloqui, A. Novel strategy for oral peptide delivery in incretin-based diabetes treatment. Gut 2020, 69, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, J.; Wang, J.; Zhang, W.; Xu, B.; Xu, X.; Zong, L. Targeted delivery of antigen to intestinal dendritic cells induces oral tolerance and prevents autoimmune diabetes in NOD mice. Diabetologia 2018, 61, 1384–1396. [Google Scholar] [CrossRef]
- Lee, W.K.; Park, J.Y.; Jung, S.; Yang, C.W.; Kim, W.U.; Kim, H.Y.; Park, J.H.; Park, J.S. Preparation and characterization of biodegradable nanoparticles entrapping immunodominant peptide conjugated with PEG for oral tolerance induction. J. Control Release 2005, 105, 77–88. [Google Scholar] [CrossRef]
- Kim, W.U.; Lee, W.K.; Ryoo, J.W.; Kim, S.H.; Kim, J.; Youn, J.; Min, S.Y.; Bae, E.Y.; Hwang, S.Y.; Park, S.H.; et al. Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen: A novel treatment strategy for induction of oral tolerance. Arthritis Rheum. 2002, 46, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Moon, J.H.; Jeong, S.U.; Jung, H.H.; Park, C.S.; Hwang, B.Y.; Lee, C.K. Induction of antigen-specific immune tolerance using biodegradable nanoparticles containing antigen and dexamethasone. Int. J. Nanomed. 2019, 14, 5229–5242. [Google Scholar] [CrossRef] [PubMed]
- De, S.R.J.; Irache, J.M.; Camacho, A.I.; Gastaminza, G.; Sanz, M.L.; Ferrer, M.; Gamazo, C. Immunogenicity of peanut proteins containing poly(anhydride) nanoparticles. Clin. Vaccine Immunol. 2014, 21, 1106–1112. [Google Scholar]
- Brotons-Canto, A.; Gamazo, C.; Martin-Arbella, N.; Abdulkarim, M.; Matias, J.; Gumbleton, M.; Irache, J.M. Evaluation of nanoparticles as oral vehicles for immunotherapy against experimental peanut allergy. Int. J. Biol. Macromol. 2018, 110, 328–335. [Google Scholar] [CrossRef]
- Gamazo, C.; Garcia-Azpiroz, M.; Souza Reboucas, J.; Gastaminza, G.; Ferrer, M.; Irache, J.M. Oral immunotherapy using polymeric nanoparticles loaded with peanut proteins in a murine model of fatal anaphylaxis. Immunotherapy 2017, 9, 1205–1217. [Google Scholar] [CrossRef]
- Srivastava, K.D.; Siefert, A.; Fahmy, T.M.; Caplan, M.J.; Li, X.M.; Sampson, H.A. Investigation of peanut oral immunotherapy with CpG/peanut nanoparticles in a murine model of peanut allergy. J. Allergy Clin. Immunol. 2016, 138, 536–543.e4. [Google Scholar] [CrossRef]
- Church, J.A.; Parker, E.P.; Kirkpatrick, B.D.; Grassly, N.C.; Prendergast, A.J. Interventions to improve oral vaccine performance: A systematic review and meta-analysis. Lancet Infect. Dis. 2019, 19, 203–214. [Google Scholar] [CrossRef]
- Tate, J.E.; Burton, A.H.; Boschi-Pinto, C.; Parashar, U.D.; for the World Health Organization–Coordinated Global Rotavirus Surveillance Network; Agocs, M.; Serhan, F.; de Oliveira, L.; Mwenda, J.M.; Mihigo, R. Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000–2013. Clin. Infect. Dis. 2016, 62, S96–S105. [Google Scholar] [PubMed]
- Parker, E.P.; Ramani, S.; Lopman, B.A.; Church, J.A.; Iturriza-Gomara, M.; Prendergast, A.J.; Grassly, N.C. Causes of impaired oral vaccine efficacy in developing countries. Future Microbiol. 2018, 13, 97–118. [Google Scholar] [CrossRef]
- Lavelle, E.C.; Ward, R.W. Mucosal vaccines—Fortifying the frontiers. Nat. Rev. Immunol. 2021, 1–15, online only. [Google Scholar]
- Davitt, C.J.H.; Lavelle, E.C. Delivery strategies to enhance oral vaccination against enteric infections. Adv. Drug Deliv. Rev. 2015, 91, 52–69. [Google Scholar] [CrossRef]
- Panda, S.; Colonna, M. Innate lymphoid cells in mucosal immunity. Front Immunol. 2019, 10, 861. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Hepworth, M.R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 2019, 19, 599–613. [Google Scholar] [CrossRef]
- Jazayeri, S.D.; Lim, H.X.; Shameli, K.; Yeap, S.K.; Poh, C.L. Nano and Microparticles as Potential Oral Vaccine Carriers and Adjuvants Against Infectious Diseases. Front. Pharmacol. 2021, 12, 682286. [Google Scholar] [CrossRef] [PubMed]
- Frizzell, H.; Woodrow, K.A. Biomaterial Approaches for Understanding and Overcoming Immunological Barriers to Effective Oral Vaccinations. Adv. Funct. Mater. 2020, 30, 1907170. [Google Scholar] [CrossRef]



| Advantages | Disadvantages | GI Application | Sources | |
|---|---|---|---|---|
 Liposomes |
|
| Liposomes can be formulated to release poorly soluble agents in GI regions of interest. Liposomes are useful to encapsulate poorly soluble drugs. | [44,45,46,47,48,49,50,51] |
 Solid Lipid Nanoparticles |
|
| Attachment of homing molecules (i.e., bile salts) have been used to enhance uptake in GI tissues. Solid lipid nanoparticles are useful to encapsulate poorly soluble drugs. | [52,53,54,55,56,57,58] |
 Nanoemulsion |
|
| Nanoemulsions can greatly improve solubility of both hydrophilic and hydrophobic agents. | [59,60] |
 Polymeric Nanoparticle Polymeric Nanoparticle |
|
| Attachment of homing molecules, including bile salts and vitamin B12, have been used to enhance uptake in GI tissues. | [61,62,63,64,65,66,67,68,69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCright, J.; Ramirez, A.; Amosu, M.; Sinha, A.; Bogseth, A.; Maisel, K. Targeting the Gut Mucosal Immune System Using Nanomaterials. Pharmaceutics 2021, 13, 1755. https://doi.org/10.3390/pharmaceutics13111755
McCright J, Ramirez A, Amosu M, Sinha A, Bogseth A, Maisel K. Targeting the Gut Mucosal Immune System Using Nanomaterials. Pharmaceutics. 2021; 13(11):1755. https://doi.org/10.3390/pharmaceutics13111755
Chicago/Turabian StyleMcCright, Jacob, Ann Ramirez, Mayowa Amosu, Arnav Sinha, Amanda Bogseth, and Katharina Maisel. 2021. "Targeting the Gut Mucosal Immune System Using Nanomaterials" Pharmaceutics 13, no. 11: 1755. https://doi.org/10.3390/pharmaceutics13111755
APA StyleMcCright, J., Ramirez, A., Amosu, M., Sinha, A., Bogseth, A., & Maisel, K. (2021). Targeting the Gut Mucosal Immune System Using Nanomaterials. Pharmaceutics, 13(11), 1755. https://doi.org/10.3390/pharmaceutics13111755