
Amorphous Solid Dispersions (ASDs): The Influence of Material Properties, Manufacturing Processes and Analytical Technologies in Drug Product Development

 ,
,
Abstract
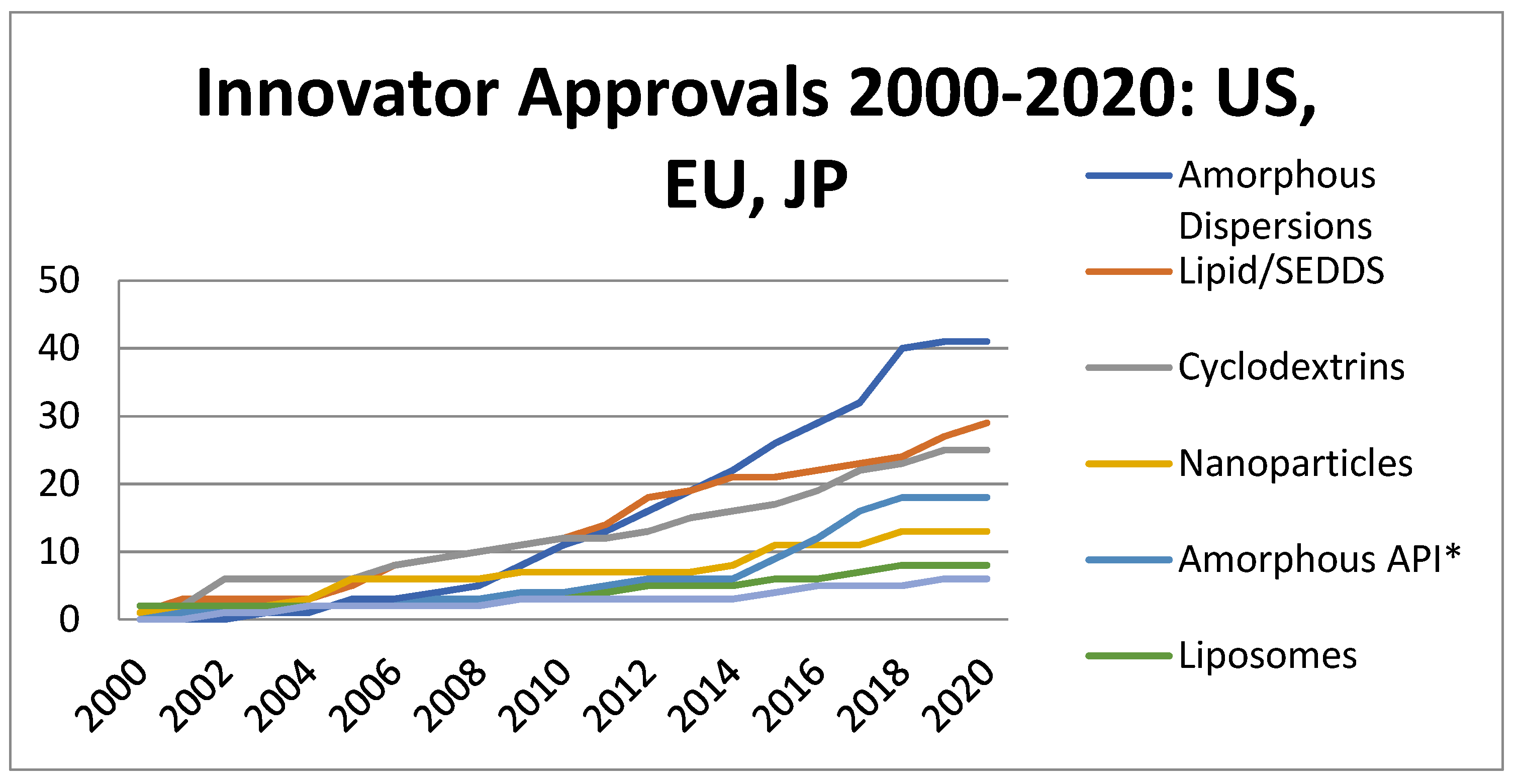
1. Introduction
2. Theoretical Considerations
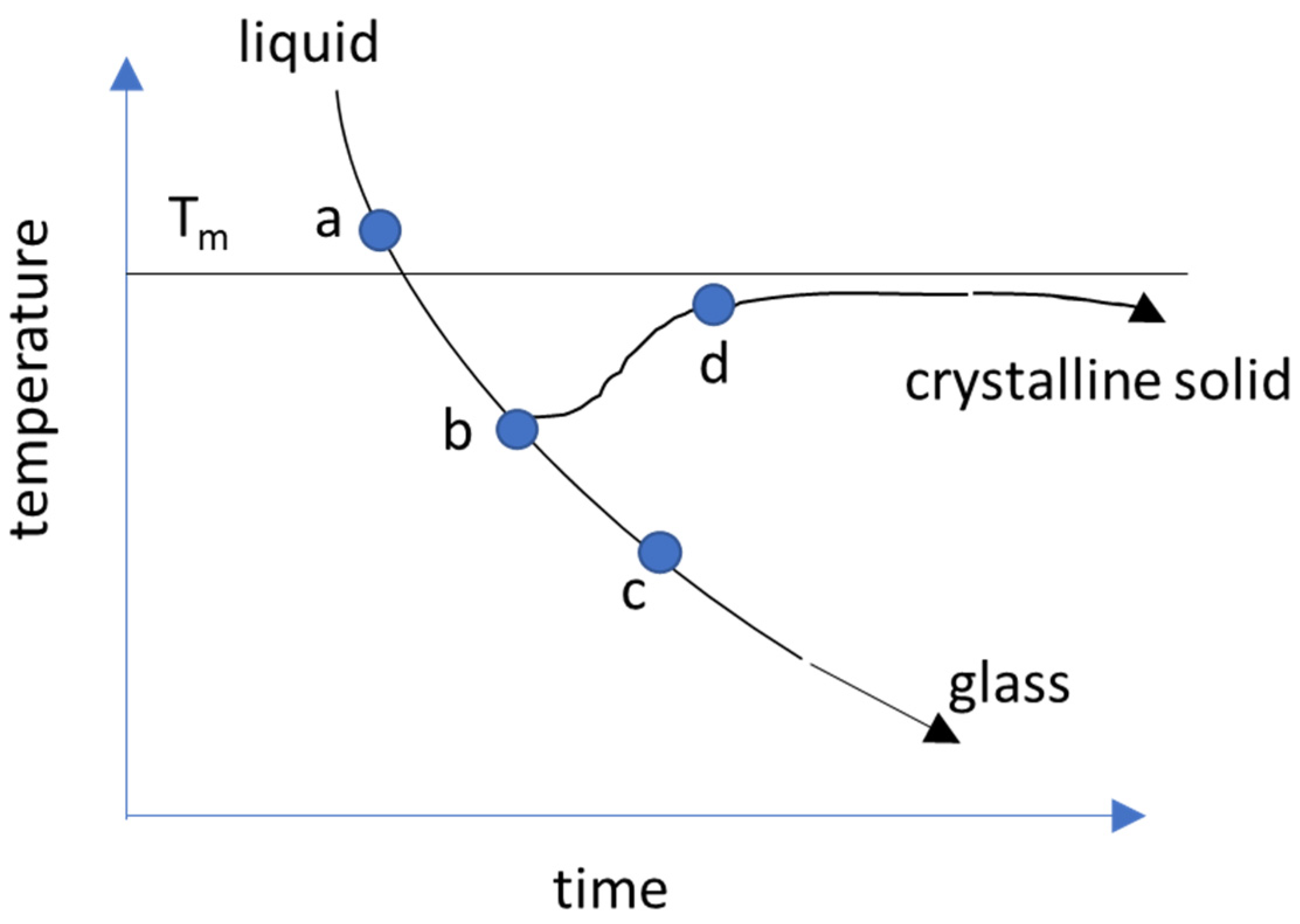
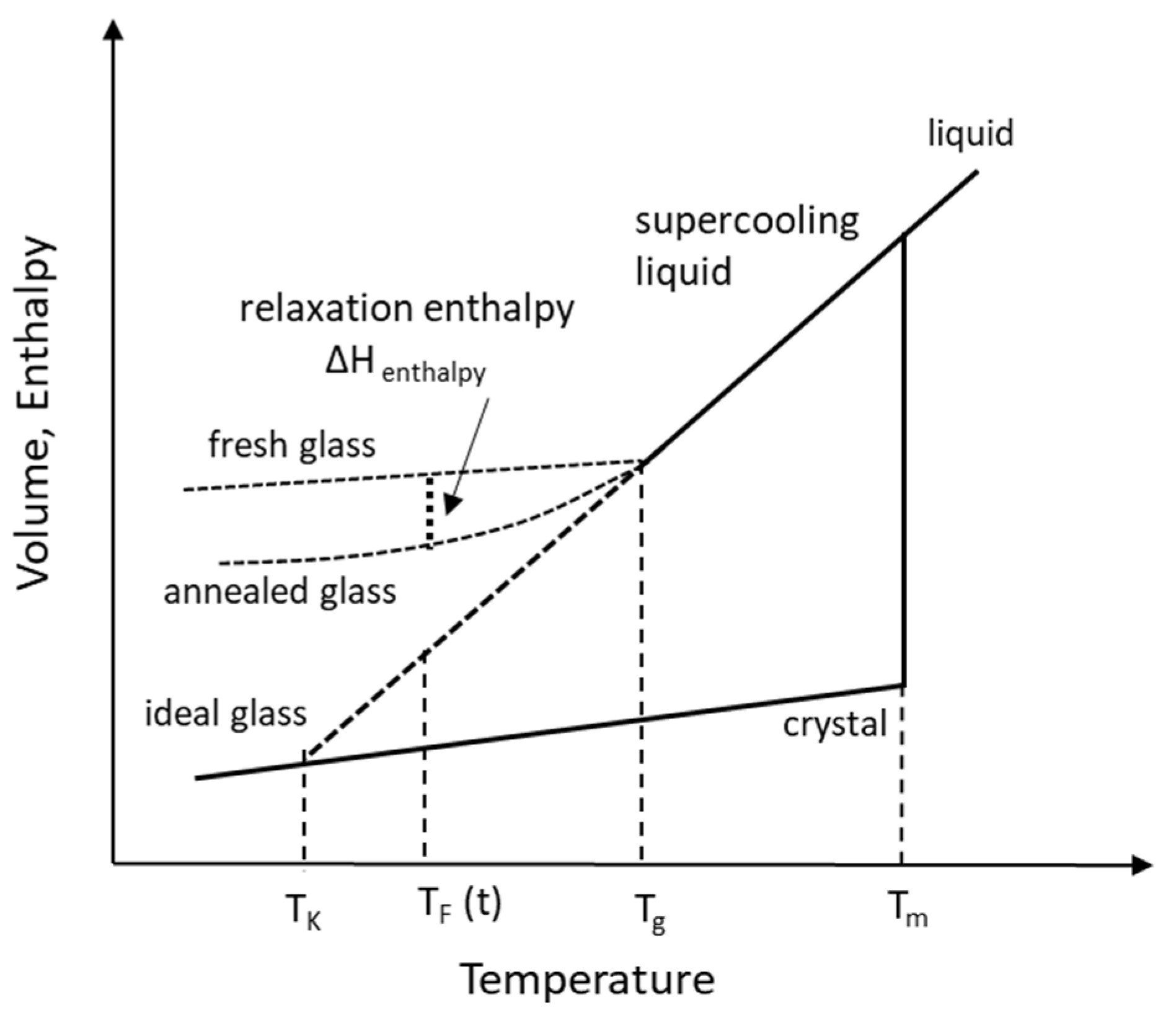
2.1. Temperatures in the Supercooling of Amorphous Solid
2.1.1. Melting Temperature, Tm
2.1.2. Glass Transition Temperature, Tg
2.1.3. Kauzmann Temperature, TK
2.1.4. Fictive Temperature, TF
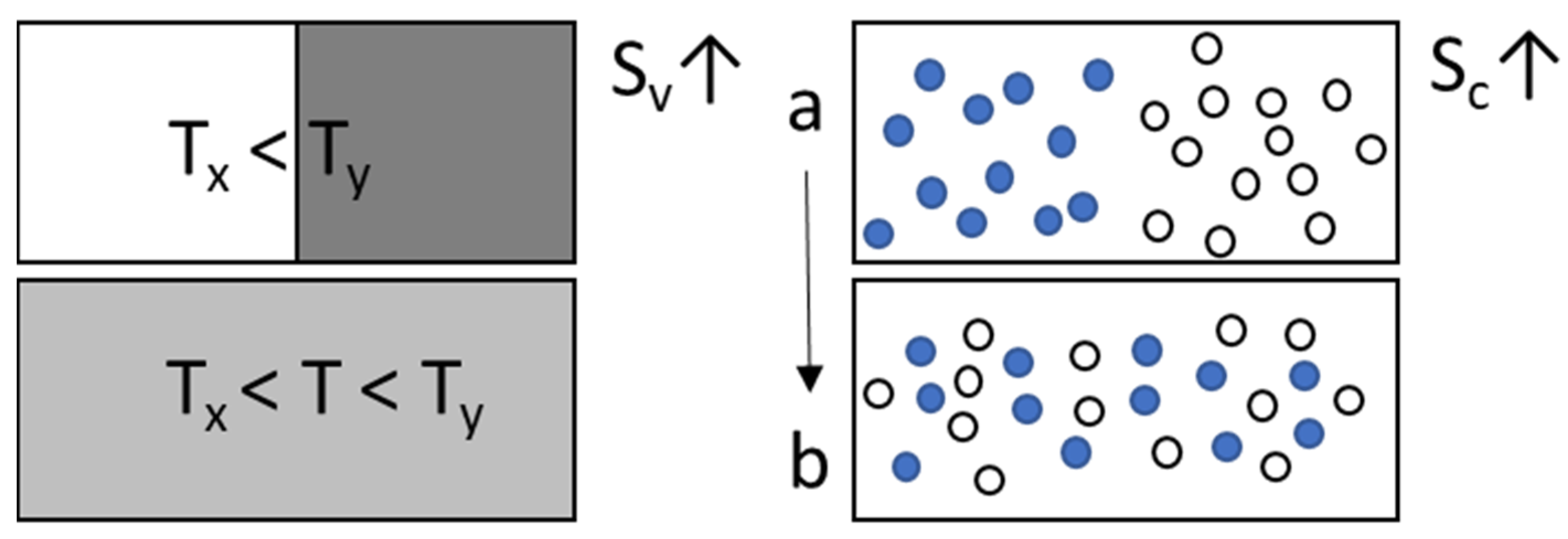
2.2. Molecular Mobility, Structural Relaxation and Relaxation Time
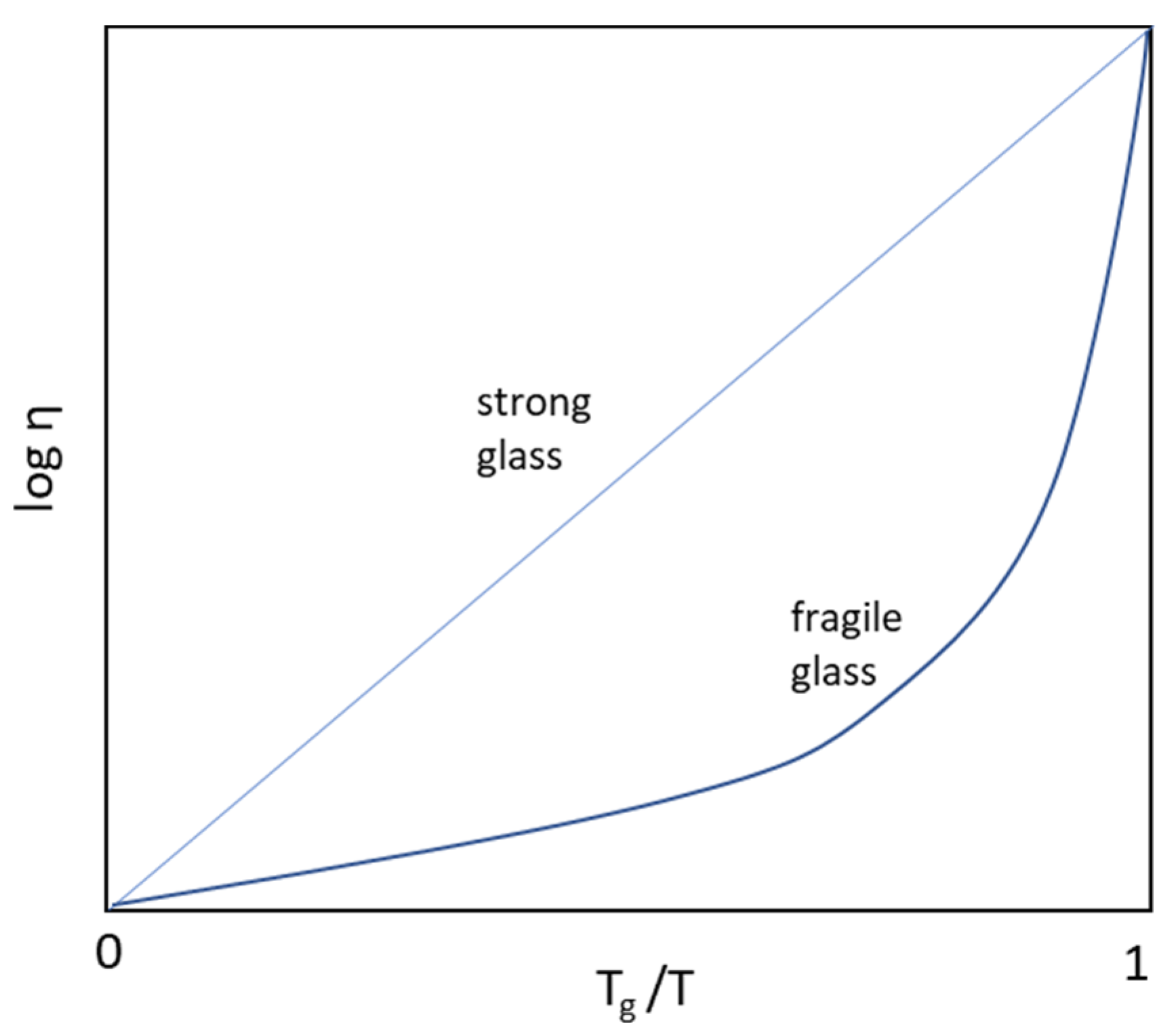
2.3. Fragility
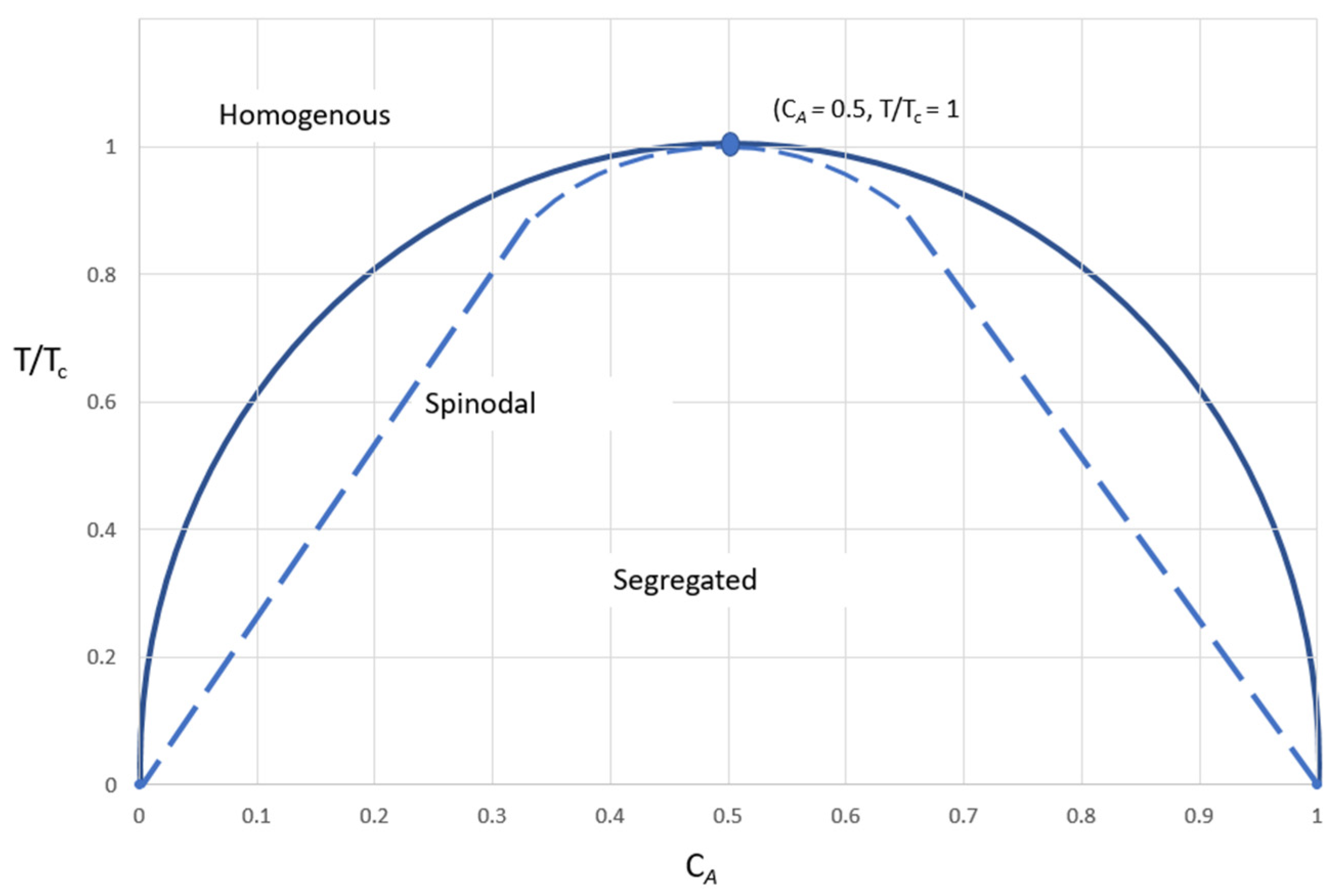
2.4. Theories of Glass Formation
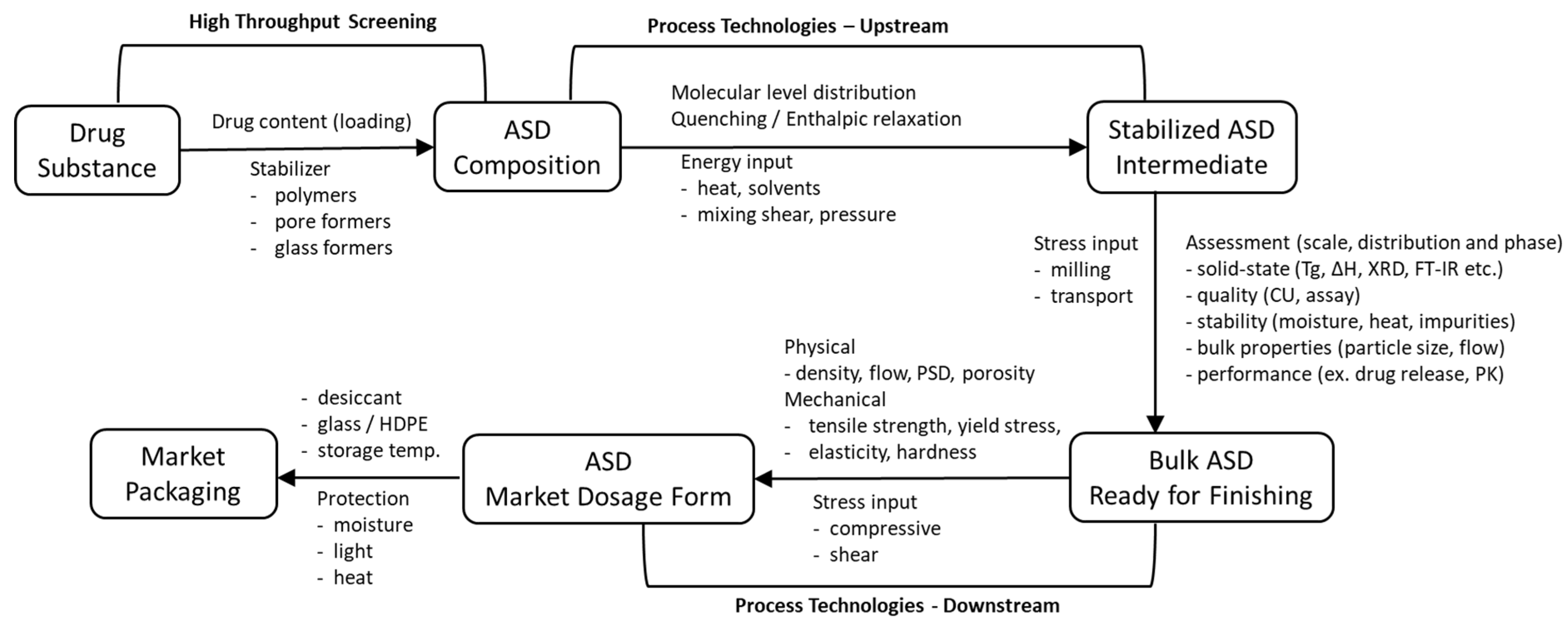
3. Product Development Strategy
3.1. Formulation Development
3.1.1. Drug Substance Considerations
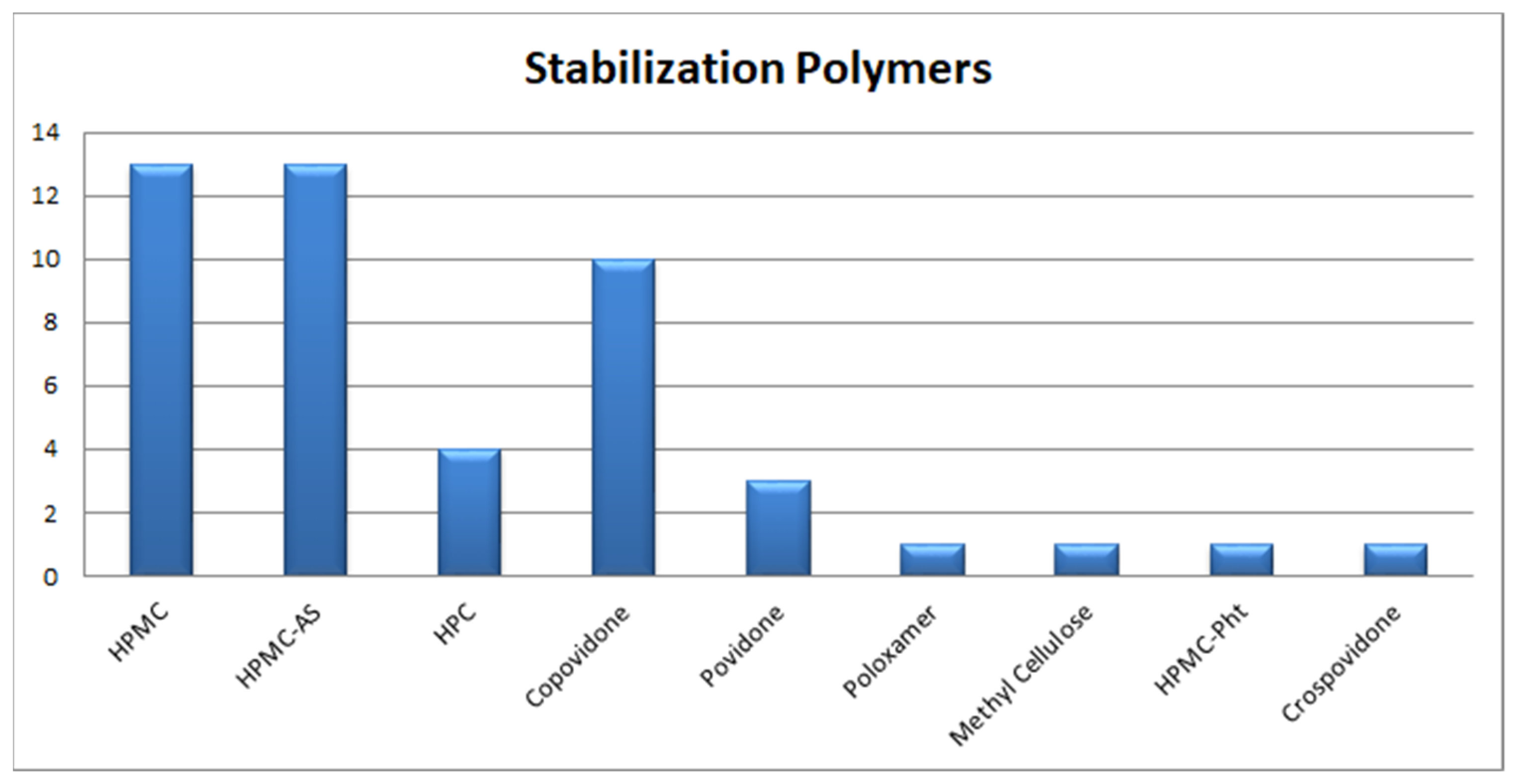
3.1.2. Polymeric Stabilization of ASD
Selection of Polymeric Stabilizers in ASD
- Cellulose derivatives (ethers/esters) are preferred due to their wide safety and functionalities obtained with alkyl-substituted hydroxyl groups e.g., methoxy, hydroxypropyl, phthaloyl, acetyl and succinyl groups that provide different aqueous dissolution profiles (e.g., hypromellose, HPMC-AS);
- Polyvinylpyrrolidones and vinyl acetate balanced co-polymers are also used in ASDs due to their relative ease in processing. Their functionalities are limited to balancing the hydrophilic and hydrophobic monomers and degree of polymerization (e.g., PVP, copovidone);
- Methacrylic acid and methacrylate esters are another class of polymers that offer functionalities tailored for dissolution at different pH of the aqueous medium e.g., with containing carboxyl or amino groups (e.g., Eudragit L or E).
Plasticization
Drug–Polymer Interactions in ASD
3.1.3. Early Phase Testing
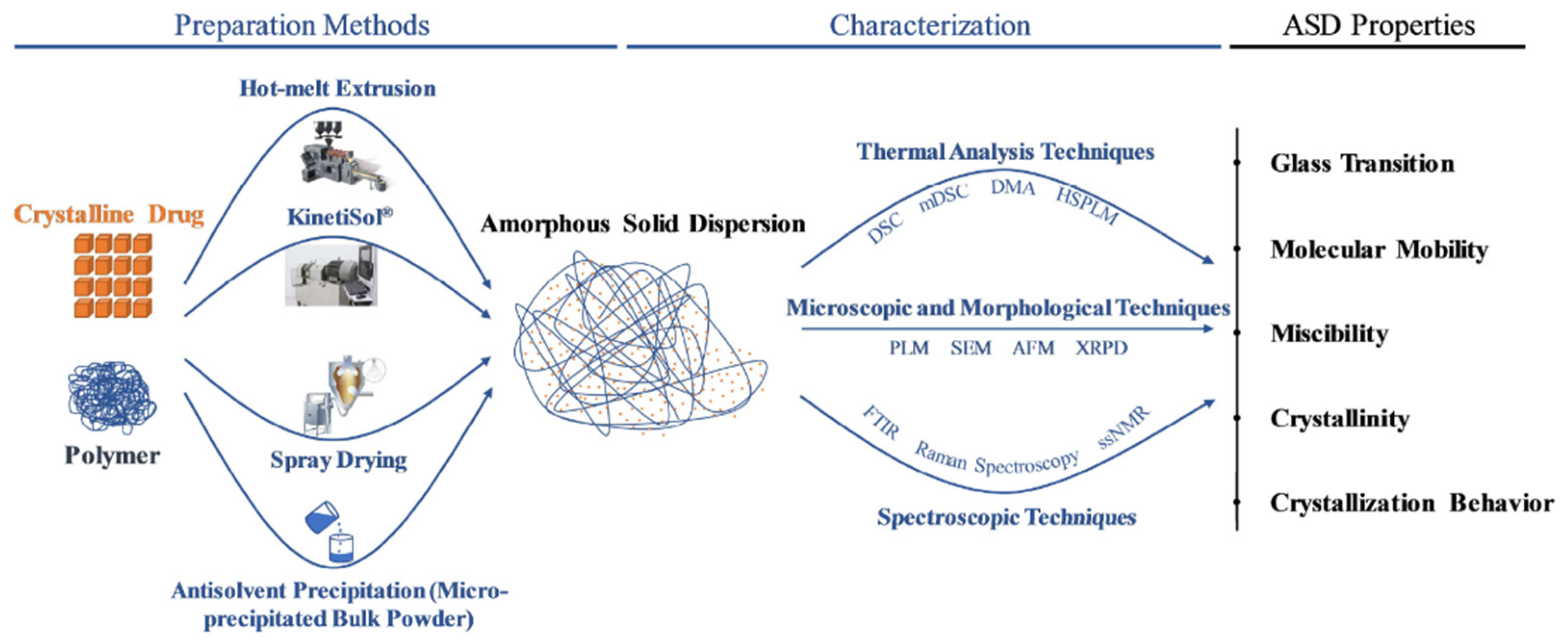
3.2. Analytical Testing and Characterization of ASD
3.2.1. Solid-State Characterization
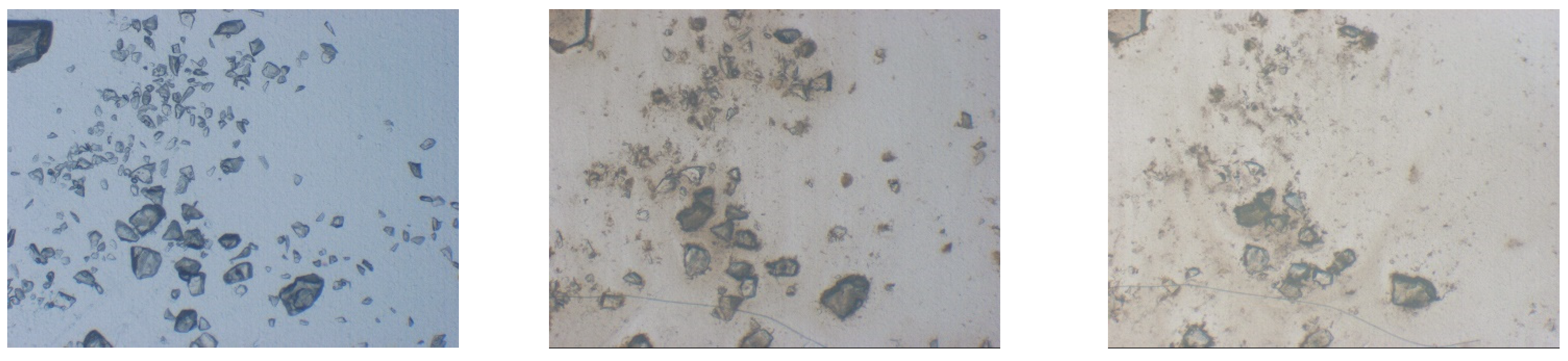
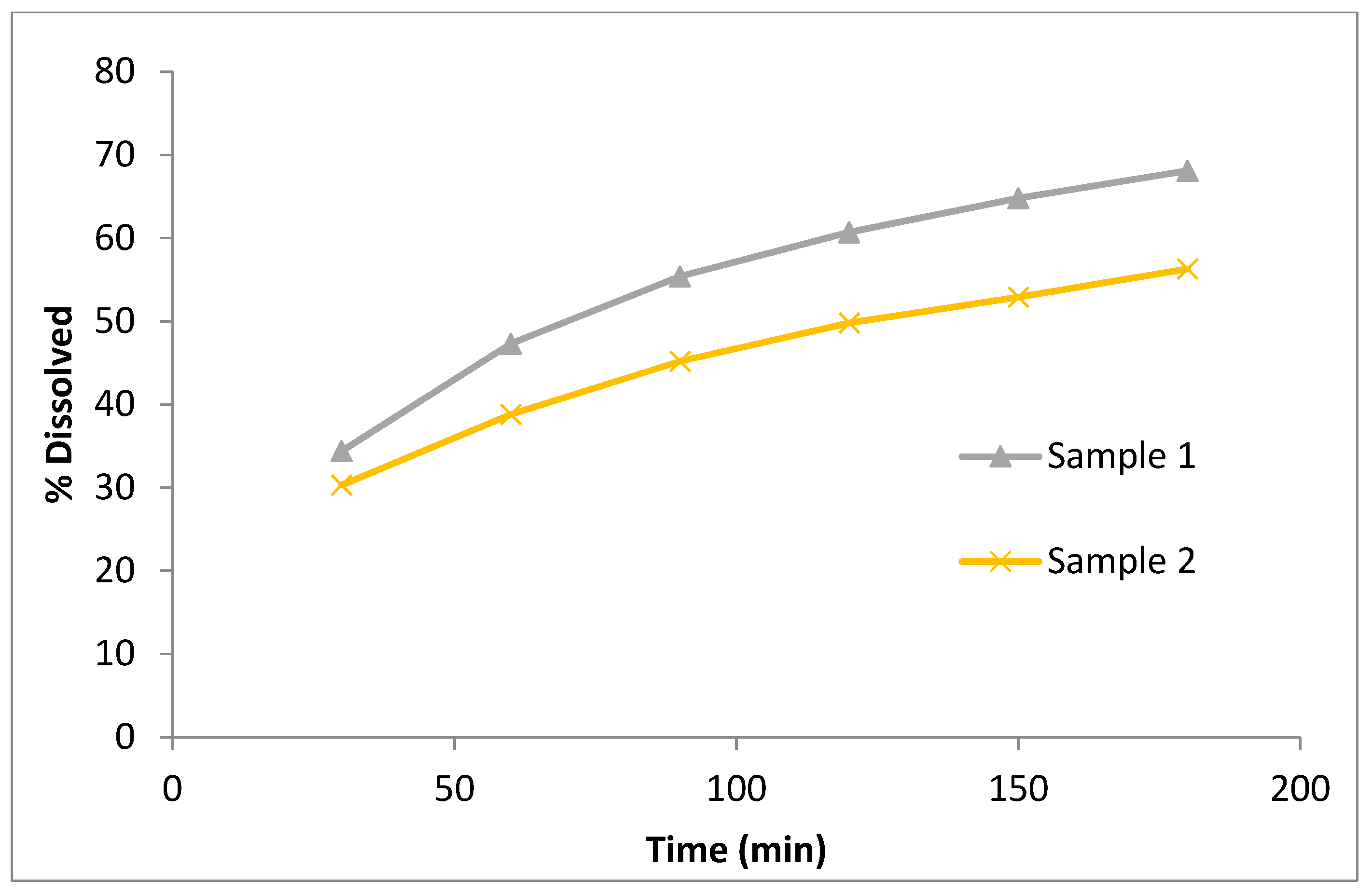
3.2.2. Dissolution Testing and Drug Release from ASD
4. Manufacturing Technologies and Scale-Up
- Maintaining room humidity conditions at a relatively low level <40% RH;
- Packaging that prevents or significantly reduces moisture penetration such as sealed Alu bags for bulk storage, glass/HDPE bottles with desiccant and Alu-Alu blister packaging;
- Storage and transport under “cold/refrigerated” conditions to maintain ASD stability (cold storage reduces mobility via lower entropy) especially for handling in tropical countries (Africa and South East Asia) classified under climactic zones III and IV.
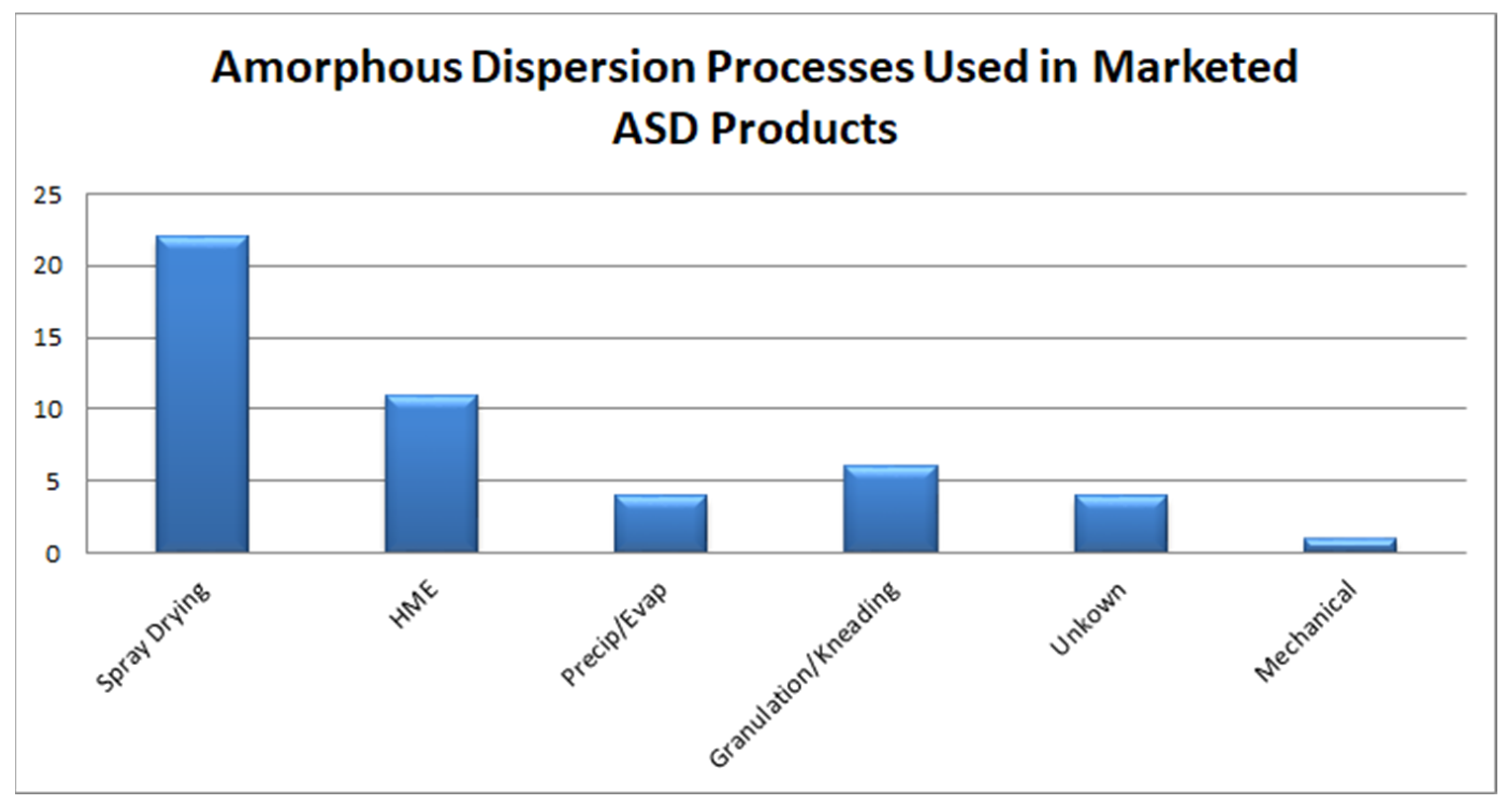
4.1. Overview of ASD Manufacturing Technologies
4.2. Technologies Based on Melting
4.3. Technologies Based on Use of Solvents
4.4. Current Technologies for Commercial Manufacturing of ASDs
4.4.1. Spray Drying (SD) and Fluid-Bed Layering (FBL)
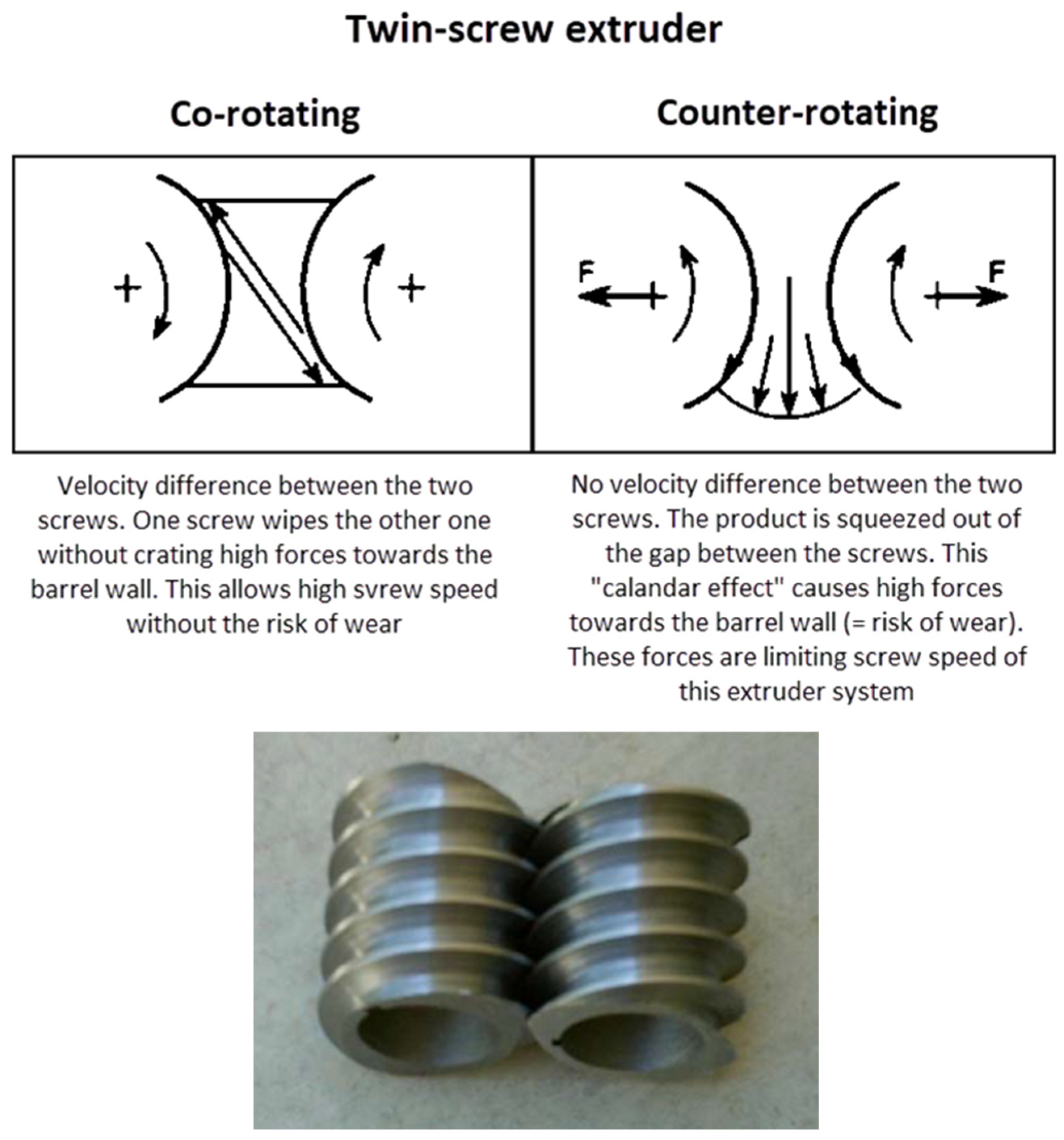
4.4.2. Hot-Melt Extrusion (HME)
4.4.3. Impact of Physico-Mechanical Properties of ASD on Manufacturing Processes
4.5. Considerations in Scale-Up and Technology Transfer of SD
- Identify key response variables for each transformation based on atomization, drying and gas-solid separation;
- Use correlations with non-dimensional parameters;
- Develop fundamental dimensional variables to ensure similarity across scale.
- The Tout is measured directly and is kept constant during the scale-up;
- The next part in scaling-up is to obtain the same particle size of powder through the scales. The particle size of dried powder is dependent on the droplet particle size. The droplet size obtained from an atomization process is estimated from empirical correlations that are often product and nozzle dependent and may not describe adequately the complex fluid dynamic processes [167]. Extrapolation should therefore be done carefully since the particle size may be valid only within the local experimental space. Nevertheless, the mean droplet diameter obtained empirically is still a good approximation and can be a good guide for scale-up purposes;
- The Pfeed is the major driver in determining the particle size of the final powder. A higher relative velocity between the air and the liquid increases the shear forces and provides a higher atomizing energy, reducing the mean droplet size. The droplet (and particle) size is smaller at laboratory scale due to the relatively small drying chamber and shorter drying times. A larger droplet size requires a larger drying chamber with longer drying times to obtain larger solid particles. Naturally, this requires a reduction of the Pfeed and typically also needs an adjustment of the operating temperatures to maintain the same residual moisture content in the final powder;
- The next step is to calculate the evaporation rate required at the pilot scale to achieve the same vapor concentration in the outlet air as in the laboratory scale, thus providing the same level of residual solvent in the spray dried powder at both scales. This is achieved by using a simple mass balance with the following parameters: Ffeed, total solids content in the feed and the residual water content in powder. From the evaporation rate at laboratory scale, Ffeed and Tin are calculated for pilot scale. This is particularly important when producing amorphous materials since the residual solvent level in the ASD significantly affects its glass transition temperature (Tg).
5. Novel ASD Polymer Considerations and Challenge
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AFM | Atomic force microscopy |
| AG | Adam-Gibbs equation |
| AGV | Adam-Gibbs-Vogel equation |
| API | Active pharmaceutical ingredient |
| ASD | Amorphous solid dispersion |
| BCS | Biopharmaceutical classification system |
| BID | Bis in die (two a day) |
| BLA | Biological license application |
| bRo5 | Beyond the rule of 5 molecules properties |
| Cfeed | Feed concentration |
| CRR | Cooperative rearranging regions |
| cGMP | Current good manufacturing practice regulations |
| CMAs | Critical material attributes |
| CPPs | Critical process parameters |
| cps | Capsules |
| CQAs | Critical quality attributes |
| DMA | Dynamic Mechanical Analyzer |
| DOSS | Dioctyl sulfosuccinate |
| DR | Delayed release |
| DSC | Differential Scanning Calorimetry |
| DTI | Dimensionless tableting indices |
| DVS | Dynamic vapor sorption |
| EDS | Energy-dispersive X-ray spectroscopy |
| ER | Extended release |
| FaSSIF | Fasted State Simulated Intestinal Fluid |
| FBL | Fluid-bed layering |
| FeSSIF | Fed State Simulated Intestinal Fluid |
| FDA | Food and Drug Administration |
| Fdrying | Feed flow rate |
| Ffeed | Feed flow |
| GRAS | Generally recognized as safe |
| GT | Gordon-Taylor parameter |
| HME | Hot-melt extrusion |
| HP-β-CD | Hydroxypropyl-beta-cyclodextrin |
| ICH | International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human use |
| IND | Investigational New Drug application |
| IVIVC | In-vitro in-vivo correlation |
| LLPS | Liquid-liquid phase separation |
| NDA | New drug application |
| NQR | Nuclear quadrupole resonance |
| mDSC | Modulated differential scanning calorimetry |
| PAT | Process analytical technology |
| Pfeed | Atomization pressure air |
| PROTAC | Proteolysis-targeting chimeras |
| pXRD | Powder X-ray diffraction |
| QbD | Quality by design |
| QC | Quality control |
| QD | Quaque die (once a day) |
| SAED | Selected angle electron diffraction |
| SEDDS | Self-emulsifying drug delivery system |
| SEM | Scanning electron microscopy |
| SD | Solid dispersion |
| SGF | Simulated gastric fluid |
| SONICC | Second-order nonlinear optical imaging of chiral crystals |
| susp | Suspension |
| SEM | Scanning Electron Microscopy |
| SMEDDS | Self microemulsifying drug delivery system |
| tbl | tablets |
| TEM | Transmission Electron Microscopy |
| Tin | Inlet temperature |
| Tfeed | Feed temperature |
| Tg | Glass transition temperature |
| TGA | Thermogravimetric analysis |
| TNO | Is a Netherlands organization specializing oral absorption and bioavailability |
| Tout | Outlet temperature |
| USP II w/w | United States Pharmacopeia dissolution apparatus II Weight/weight |
| VTF | Vogel–Tammann–Fulcher equation |
| XRPD | X-Ray Powder Diffraction |
References
- FDA. Guidance for Industry, Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System; FDA. U.S. Department of Health and Human Services, Center for Drug Evaluation and Research (CDER): Beltsville, MD, USA, 2000; p. 13.
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- United States Pharmacopoeis, USP-NF Online, 2021, General Notices and Requirements, 5.30 Description and Solubility. Available online: https://www.uspnf.com/sites/default/files/usp_pdf/EN/USPNF/generalNoticesandRequirementsFinal.pdf (accessed on 29 January 2021).
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Carter, G.T. Drug-like property concepts in pharmaceutical design. Curr. Pharm. Des. 2009, 15, 2184–2194. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Fish, P.V.; Mano, T. Bridging solubility between drug discovery and development. Drug Discov. Today 2012, 17, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Effects on drug permeation through biological membranes. J. Pharm. Pharmacol. 2011, 63, 1119–1135. [Google Scholar] [CrossRef]
- Liu, R. Water-Insoluble Drug Formulation, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Rodriguez-Aller, M.; Guillarme, D.; Veuthey, J.-V.; Gurny, R. Strategies for formulating and delivering poorly water-soluble drugs. J. Drug Deliv. Sci. Technol. 2015, 30, 342–351. [Google Scholar] [CrossRef]
- Noyes, A.; Whitney, W. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Keck, C.M.; Müller, R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur. J. Pharm. Biopharm. 2006, 62, 3–16. [Google Scholar] [CrossRef]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- Hauss, D.J. Oral Lipid-Based Formulations; Informa Healthcare: New York, NY, USA, 2007. [Google Scholar]
- Armstrong, N.A.; James, K.C. Drug release from lipid-based dosage forms. Int. J. Pharm. 1980, 6, 195–204. [Google Scholar] [CrossRef]
- Narang, A.S.; Delmarre, D.; Gao, D. Stable drug encapsulation in micelles and microemulsions. Int. J. Pharm. 2007, 345, 9–25. [Google Scholar] [CrossRef]
- Shah, N.; Iyer, R.M.; Mair, H.J.; Choi, D.; Tian, H.; Diodone, R.; Fahnrich, K.; Pabst-Ravot, A.; Tang, K.; Scheubel, E. Improved human bioavailability of vemurafenib, a practically insoluble drug, using an amorphous polymer-stabilized solid dispersion prepared by a solvent-controlled coprecipitation process. J. Pharm. Sci. 2013, 102, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Malick, A.W. Amorphous solild dispersions. In CRS Advances in Delivery Science and Technology Series; Springer: New York, NY, USA, 2014. [Google Scholar]
- Serajuddin, A.T. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Stella, V.; Borchardt, R.; Hageman, M.; Oliyai, R.; Maag, H.; Tilley, J. Prodrugs: Challenges and Rewards; Springer: New York, NY, USA, 2007. [Google Scholar]
- Chan, O.H.; Schmid, H.L.; Stilgenbauer, L.A.; Howson, W.; Horwell, D.C.; Stewart, B.H. Evaluation of a targeted prodrug strategy of enhance oral absorption of poorly water-soluble compounds. Pharm. Res. 1998, 15, 1012–1018. [Google Scholar] [CrossRef]
- Erxleben, A. Cocrystal applications in drug delivery. Pharmaceutics 2020, 12, 834. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, R.J.; Sandhu, H.K.; Iyer, R.M.; Shah, N.H.; Malick, A.W.; Zia, H. Characterization of physico-mechanical properties of indomethacin and polymers to assess their suitability for hot-melt extrusion processs as a means to manufacture solid dispersion/solution. J. Pharm. Sci. 2005, 94, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Snyder, J.; Vippagunta, R.; Alvine, M.; Vakil, R.; Tong, W.Q.T.; Vippagunta, S. Comparison of HPMC based polymers performance as carriers for manufacture of solid dispersions using the melt extruder. Int. J. Pharm. 2011, 419, 12–19. [Google Scholar] [CrossRef]
- Law, D.; Schmitt, E.A.; Marsh, K.C.; Everitt, E.A.; Wang, W.; Fort, J.J.; Krill, S.L.; Qiu, Y. Ritonavir-PEG 8000 amorphous solid dispersions: In vitro and in vivo evaluations. J. Pharm. Sci. 2004, 93, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Debenedetti, P.G.; Stillinger, F.H. Supercooled liquids and the glass transition. Nature 2001, 410, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Hilden, L.R.; Morris, K.R. Physics of amorphous solids. J. Pharm. Sci. 2004, 93, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Stachurski, Z.H. On structure and properties of amorphous materials. Materials 2011, 4, 1564–1598. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A Review of amorphization, crystallization, stabilization, solid-state characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Duong, T.V.; Van den Mooter, G. The role of the carrier in the formulation of pharmaceutical solid dispersions. Part II: Amorphous carriers. Expert Opin. Drug Deliv. 2016, 13, 1681–1694. [Google Scholar] [CrossRef]
- Righetti, M.C. Crystallization of polymers investigated by temperature-modulated DSC. Materials 2017, 10, 442. [Google Scholar] [CrossRef]
- Tran, P.; Pyo, Y.C.; Kim, D.H.; Lee, S.E.; Kim, J.K.; Park, J.S. Overview of the manufacturing methods of solid dispersion technology for improving the solubility of poorly water-soluble drugs and application to anticancer drugs. Pharmaceutics 2019, 11, 132. [Google Scholar] [CrossRef]
- Pandi, P.; Bulusu, R.; Kommineni, N.; Khan, W.; Singh, M. Amorphous solid dispersions: An update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int. J. Pharm. 2020, 586, 119560. [Google Scholar] [CrossRef]
- Schittny, A.; Huwyler, J.; Puchkov, M. Mechanisms of increased bioavailability through amorphous solid dispersions: A review. Drug Deliv. 2020, 27, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Noggle, J.H. Physical Chemistry, 3rd ed.; Little, Brown & Company: Toronto, ON, Canada, 1985; p. 118. [Google Scholar]
- Hoffman, J.D. Thermodynamic driving force in nucleation and growth processes. J. Chem. Phys. 1958, 29, 1192–1193. [Google Scholar] [CrossRef]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Adam, G.; Gibbs, J.H. On the temperature dependence of cooperative relaxation properties in glass-forming liquids. J. Chem. Phys. 1965, 43, 139–146. [Google Scholar] [CrossRef]
- Angell, C.A. Relaxation in liquids, polymers and plastic crystals—Strong/fragile patterns and problems. J. Non-Cryst. Solids 1991, 131, 13–31. [Google Scholar] [CrossRef]
- Kauzmann, W. The nature of the glassy state and the behavior of liquids at low temperatures. Chem. Rev. 1948, 43, 219–256. [Google Scholar] [CrossRef]
- Rastogi, S.; Vega, J.F.; van Ruth, N.J.L.; Terry, A.E. Non-linear changes in the specific volume of the amorphous phase of poly(4-methyl-1-pentene); Kauzmann curves, inverse melting, fragility. Polymer 2006, 47, 5555–5565. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, G.G.; Law, D.; Grant, D.J.; Schmitt, E.A. Physical stability of amorphous pharmaceuticals: Importance of configurational thermodynamic quantities and molecular mobility. J. Pharm. Sci. 2002, 91, 1863–1872. [Google Scholar] [CrossRef]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Rades, T. The role of configurational entropy in amorphous systems. Pharmaceutics 2010, 2, 224–244. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef]
- Hancock, B.C.; Dalton, C.R.; Pikal, M.J.; Shamblin, S.L. A pragmatic test of a simple calorimetric method for determining the fragility of some amorphous pharmaceutical materials. Pharm. Res. 1998, 15, 762–767. [Google Scholar] [CrossRef]
- Baird, J.A.; Taylor, L.S. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv. Drug Deliv. Rev. 2012, 64, 396–421. [Google Scholar] [CrossRef]
- Zheng, Q.; Mauro, J.C. Viscosity of glass-forming systems. J. Am. Ceram. Soc. 2017, 100, 6–25. [Google Scholar] [CrossRef]
- Bohmer, R.; Ngai, K.L.; Angell, C.A.; Plazek, D.J. Nonexponential relaxations in strong and fragile glass formers. J. Chem. Phys. 1993, 99, 4201–4209. [Google Scholar] [CrossRef]
- Hatley, R.H. Glass fragility and the stability of pharmaceutical preparations—Excipient selection. Pharm. Dev. Technol. 1997, 2, 257–264. [Google Scholar] [CrossRef]
- Angell, C.A. Formation of glasses from liquids and biopolymers. Science 1995, 267, 1924–1935. [Google Scholar] [CrossRef]
- Gibbs, J.H.; DiMarzio, E.A. Nature of the glass transition and the glassy state. J. Chem. Phys. 1958, 28, 373–384. [Google Scholar] [CrossRef]
- Gaskell, D.R.; Laughlin, D.E. Introduction to the Thermodynamics of Materials; CRC Press; Taylor & Francis Group: Boca Raton, FL, USA, 2017; pp. 93–119. [Google Scholar]
- Hodge, I.M. Effects of annealing and prior history on enthalpy relaxation in glassy polymers—Adam-Gibbs formulation of nonlinearity. Macromolecules 1987, 20, 2897–2908. [Google Scholar] [CrossRef]
- DiMarzio, E.A. Equilibrium theory of glasses. In Annals of the New York Academy of Sciences; New York Academy of Sciences: New York, NY, USA, 1981; Volume 371. [Google Scholar]
- Ma, X.; Williams, R.O., III. Characterization of amorphous solid dispersions: An update. J. Drug Deliv. Sci. Technol. 2019, 50, 113–124. [Google Scholar] [CrossRef]
- Hsieh, Y.L.; Ilevbare, G.A.; Van Eerdenbrugh, B.; Box, K.J.; Sanchez-Felix, M.V.; Taylor, L.S. pH-Induced precipitation behavior of weakly basic compounds: Determination of extent and duration of supersaturation using potentiometric titration and correlation to solid state properties. Pharm. Res. 2012, 29, 2738–2753. [Google Scholar] [CrossRef]
- Raina, S.A.; Van Eerdenbrugh, B.; Alonzo, D.E.; Mo, H.; Zhang, G.G.; Gao, Y.; Taylor, L.S. Trends in the precipitation and crystallization behavior of supersaturated aqueous solutions of poorly water-soluble drugs assessed using synchrotron radiation. J. Pharm. Sci. 2015, 104, 1981–1992. [Google Scholar] [CrossRef]
- Raina, S.A.; Zhang, G.G.; Alonzo, D.E.; Wu, J.; Zhu, D.; Catron, N.D.; Gao, Y.; Taylor, L.S. Enhancements and limits in drug membrane transport using supersaturated solutions of poorly water soluble drugs. J. Pharm. Sci. 2014, 103, 2736–2748. [Google Scholar] [CrossRef]
- Glass Transition Temperature of Cellulose Derivatives by TMA Methods G-001; Technical Information by Shin-Etsu Chemical Co., Ltd.: Tokyo, Japan, 2010; Available online: https://www.shinetsu.co.jp/cellulose/en/pharmaceutical/technical.html (accessed on 10 September 2010).
- Glass Transition Temperature of Hypromellose G-018; Technical Information by Shin-Etsu Chemical Co., Ltd.: Tokyo, Japan, 2016; Available online: https://www.shinetsu.co.jp/cellulose/en/pharmaceutical/technical.html (accessed on 29 January 2021).
- Kolter, K.; Karl, M.; Gryczke, A. Hot-Melt Extrusion with BASF Pharma Polymers, 2nd ed.; Pharma Ingredient & Services: Ludwighafen, Germany, 2012. [Google Scholar]
- Thakral, S.; Thakral, N.K.; Majumdar, D.K. Eudragit: A technology evaluation. Expert Opin. Drug Deliv. 2013, 10, 131–149. [Google Scholar] [CrossRef]
- Paudel, A.; Meeus, J.; Van den Mooter, G. Structural characterization of amorphous solid dispersions. In Amorphous Solid Dispersions, Theory and Practice; Shah, N., Sandhu, H., Choi, D.S., Chokshi, H., Malick, A.W., Eds.; Springer: New York, NY, USA, 2014. [Google Scholar]
- Rowe, R.C.; Sheskey, P.J.; Quinn, M.E. Handbook of Pharmaceutical Excipients; Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Davey, R.J.; Schroeder, S.L.; ter Horst, J.H. Nucleation of organic crystals—A molecular perspective. Angew. Chem. Int. Ed. Engl. 2013, 52, 2166–2179. [Google Scholar] [CrossRef]
- Hursthouse, M.B.; Huth, L.S.; Threlfall, T.L. Why do organic compounds crystallise well or badly or ever so slowly? Why is crystallisation nevertheless such a good purification technique? Org. Process. Res. Dev. 2009, 13, 1231–1240. [Google Scholar]
- Tsarfati, Y.; Rosenne, S.; Weissman, H.; Shimon, L.J.; Gur, D.; Palmer, B.A.; Rybtchinski, B. Crystallization of organic molecules: Nonclassical mechanism revealed by direct imaging. ACS Cent. Sci. 2018, 4, 1031–1036. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Raina, S.; Hsieh, Y.L.; Augustijns, P.; Taylor, L.S. Classification of the crystallization behavior of amorphous active pharmaceutical ingredients in aqueous environments. Pharm. Res. 2014, 31, 969–982. [Google Scholar] [CrossRef]
- Alonzo, D.E.; Raina, S.; Zhou, D.; Gao, Y.; Zhang, G.G.; Taylor, L.S. Characterizing the Impact of Hydroxypropylmethyl Cellulose on the Growth and Nucleation Kinetics of Felodipine from Supersaturated Solutions. Cryst. Growth Des. 2012, 12, 1538–1547. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Maintaining supersaturation in aqueous drug solutions: Impact of different polymers on induction times. Cryst. Growth Des. 2013, 13, 740–751. [Google Scholar] [CrossRef]
- Pinto, J.M.O.; Leão, A.F.; Riekes, M.K.; França, M.T.; Stulzer, H.K. HPMCAS as an effective precipitation inhibitor in amorphous solid dispersions of the poorly soluble drug candesartan cilexetil. Carbohydr. Polym. 2018, 184, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Nair, A.; Kuentz, M.; Dressman, J.; Saal, C. Calculation of drug-polymer mixing enthalpy as a new screening method of precipitation inhibitors for supersaturating pharmaceutical formulations. Eur. J. Pharm. Sci. 2019, 132, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.E.; James, M.B.; Forster, A.H.; Rades, T. Melt extrusion and spray drying of carbamazepine and dipyridamole with polyvinylpyrrolidone/vinyl acetate copolymers. Drug Dev. Ind. Pharm. 2008, 34, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Wadhawan, V. Kinetics of Phase Transitions; CRC Press; Taylor & Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Brouwers, J.; Brewster, M.E.; Augustijns, P. Supersaturating drug delivery systems: The answer to solubility-limited oral bioavailability? J. Pharm. Sci. 2009, 98, 2549–2572. [Google Scholar] [CrossRef] [PubMed]
- Borbás, E.; Kádár, S.; Tsinman, K.; Tsinman, O.; Csicsák, D.; Takács-Novák, K.; Völgyi, G.; Sinkó, B.; Pataki, H. Prediction of bioequivalence and food effect using flux-and solubility-based methods. Mol. Pharm. 2019, 16, 4121–4130. [Google Scholar] [CrossRef] [PubMed]
- Hate, S.S.; Reutzel-Edens, S.M.; Taylor, L.S. Absorptive dissolution testing of supersaturating systems: Impact of absorptive sink conditions on solution phase behavior and mass transport. Mol. Pharm. 2017, 14, 4052–4063. [Google Scholar] [CrossRef]
- Stewart, A.M.; Grass, M.E.; Brodeur, T.J.; Goodwin, A.K.; Morgen, M.M.; Friesen, D.T.; Vodak, D.T. Impact of drug-rich colloids of itraconazole and HPMCAS on membrane flux in vitro and oral bioavailability in rats. Mol. Pharm. 2017, 14, 2437–2449. [Google Scholar] [CrossRef]
- Stewart, A.M.; Grass, M.E.; Mudie, D.M.; Morgen, M.M.; Friesen, D.T.; Vodak, D.T. Development of a biorelevant, material-sparing membrane flux test for rapid screening of bioavailability-enhancing drug product formulations. Mol. Pharm. 2017, 14, 2032–2046. [Google Scholar] [CrossRef]
- Ojo, A.T.; Lee, P.I. Applications of dynamic mechanical analysis in the engineering of amorphous solid dispersions. Pharm. Front. 2020, 2, e55–e63. [Google Scholar] [CrossRef][Green Version]
- Kestur, U.S.; Wanapun, D.; Toth, S.J.; Wegiel, L.A.; Simpson, G.J.; Taylor, L.S. Nonlinear optical imaging for sensitive detection of crystals in bulk amorphous powders. J. Pharm. Sci. 2012, 101, 4201–4213. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, H.; Wang, S.; Liu, C.; Qian, F. A single hydrogen to fluorine substitution reverses the trend of surface composition enrichment of sorafenib amorphous solid dispersion upon moisture exposure. Pharm. Res. 2019, 36, 105. [Google Scholar] [CrossRef] [PubMed]
- Ricarte, R.G.; Lodge, T.P.; Hillmyer, M.A. Detection of pharmaceutical drug crystallites in solid dispersions by transmission electron microscopy. Mol. Pharm. 2015, 12, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Ashwathy, P.; Anto, A.T.; Sudheesh, M.S. A mechanistic review on the dissolution phase behavior and supersaturation stabilization of amorphous solid dispersions. Drug Dev. Ind. Pharm. 2021, 47, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.D.; Wen, H.; Taylor, L.S. Non-sink dissolution conditions for predicting product quality and in vivo performance of supersaturating drug delivery systems. J. Pharm. Sci. 2016, 105, 2477–2488. [Google Scholar] [CrossRef]
- Lu, E.; Li, S.; Wang, Z. Biorelevant test for supersaturable formulation. Asian J. Pharm. Sci. 2017, 12, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Berben, P.; Ashworth, L.; Beato, S.; Bevernage, J.; Bruel, J.L.; Butler, J.; Dressman, J.; Schäfer, K.; Hutchins, P.; Klumpp, L.; et al. Biorelevant dissolution testing of a weak base: Interlaboratory reproducibility and investigation of parameters controlling in vitro precipitation. Eur. J. Pharm. Biopharm. 2019, 140, 141–148. [Google Scholar] [CrossRef]
- Arun Raj, R.; Das, A.C.; Sreerekha, S.; Harindran, J. Formulation and evaluation of verapamil solid dispersion tablets for solubility enhancement. Res. Rev. A J. Pharm. Sci. 2016, 7, 39–54. [Google Scholar]
- Parikh, T.; Sandhu, H.K.; Talele, T.T.; Serajuddin, A.T. Characterization of solid dispersion of itraconazole prepared by solubilization in concentrated aqueous solutions of weak organic acids and drying. Pharm. Res. 2016, 33, 1456–1471. [Google Scholar] [CrossRef]
- Joe, J.H.; Lee, W.M.; Park, Y.J.; Joe, K.H.; Oh, D.H.; Seo, Y.G.; Woo, J.S.; Yong, C.S.; Choi, H.-G. Effect of the solid-dispersion method on the solubility and crystalline property of tacrolimus. Int. J. Pharm. 2010, 395, 161–166. [Google Scholar] [CrossRef]
- Alam, A.A.; Masum, M.A.A.; Islam, R.B.; Sharmin, F.; Islam, S.M.A. Formulation of solid dispersion and surface solid dispersion of nifedipine: A comparative study. Afr. J. Pharm. Pharmacol. 2013, 7, 1707–1718. [Google Scholar] [CrossRef][Green Version]
- Wani, R.J.; Sharma, P.; Zhong, H.A.; Chauhan, H. Preparation and characterization of griseofulvin solid dispersions. Assay Drug Dev. Technol. 2020, 18, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yang, R.; Tang, X.; Zheng, L. Part I: Characterization of solid dispersions of nimodipine prepared by hot-melt extrusion. Drug Dev. Ind. Pharm. 2007, 33, 791–802. [Google Scholar] [PubMed]
- Trasi, N.S.; Bhujbal, S.; Zhou, Q.T.; Taylor, L.S. Amorphous solid dispersion formation via solvent granulation—A case study with ritonavir and lopinavir. Int. J. Pharm. X 2019, 1, 100035. [Google Scholar] [CrossRef]
- Ghosh, M.; Wahed, M.I.I.; Ali, M.A.; Kumar Barman, R. Formulation and characterization of fenofibrate loaded solid dispersion with enhanced dissolution profile. Pharmacol. Pharm. 2019, 10, 343–355. [Google Scholar] [CrossRef]
- Guntaka, P.R.; Lankalapalli, S.R.I.N.I.V.A.S. Solubility and dissolution enhancement of Ivacaftor tablets by using solid dispersion technique of hot-melt extrusion-a design of experimental approach. Asian J. Pharm. Clin. Res. 2019, 12, 356–363. [Google Scholar] [CrossRef]
- Mudie, D.M.; Stewart, A.M.; Biswas, N.; Brodeur, T.J.; Shepard, K.B.; Smith, A.; Morgen, M.M.; Baumann, J.M.; Vodak, D.T. Novel high-drug-loaded amorphous dispersion tablets of posaconazole; in vivo and in vitro assessment. Mol. Pharm. 2020, 17, 4463–4472. [Google Scholar] [CrossRef]
- Jang, S.W.; Kang, M.J. Improved oral absorption and chemical stability of everolimus via preparation of solid dispersion using solvent wetting technique. Int. J. Pharm. 2014, 473, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Zhang, M.; Hou, Q.; Tang, P.; Suo, Z.; Zhu, Y.; Li, H. Solid dispersions of telaprevir with improved solubility prepared by co-milling: Formulation, physicochemical characterization, and cytotoxicity evaluation. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 105, 110012. [Google Scholar] [CrossRef]
- Sinha, S.; Ali, M.; Baboota, S.; Ahuja, A.; Kumar, A.; Ali, J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech 2010, 11, 518–527. [Google Scholar] [CrossRef]
- Alshehri, S.; Imam, S.S.; Hussain, A.; Altamimi, M.A.; Alruwaili, N.K.; Alotaibi, F.; Alanazi, A.; Shakeel, F. Potential of solid dispersions to enhance solubility, bioavailability, and therapeutic efficacy of poorly water-soluble drugs: Newer formulation techniques, current marketed scenario and patents. Drug Deliv. 2020, 27, 1625–1643. [Google Scholar] [CrossRef]
- Zhang, J.; Han, R.; Chen, W.; Zhang, W.; Li, Y.; Ji, Y.; Chen, L.; Pan, H.; Yang, X.; Pan, W.; et al. Analysis of the literature and patents on solid dispersions from 1980 to 2015. Molecules 2018, 23, 1697. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, R.; Ohta, T.; Shiraki, K.; Higashi, K.; Moribe, K. Effects of wet-granulation process parameters on the dissolution and physical stability of a solid dispersion. Int. J. Pharm. 2017, 524, 304–311. [Google Scholar] [CrossRef]
- Ayenew, Z.; Paudel, A.; Van den Mooter, G. Can compression induce demixing in amorphous solid dispersions? A case study of naproxen-PVP K25. Eur. J. Pharm. Biopharm. 2012, 81, 207–213. [Google Scholar] [CrossRef]
- Hancock, B.C.; Zografi, G. The relationship between the glass transition temperature and the water content of amorphous pharmaceutical solids. Pharm. Res. 1994, 11, 471–477. [Google Scholar] [CrossRef]
- Hirasawa, N.; Ishise, S.; Miyata, H.; Danjo, K. Application of nilvadipine solid dispersion to tablet formulation and manufacturing using crospovidone and methylcellulose as dispersion carriers. Chem. Pharm. Bull. 2004, 52, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug. Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Vilhelmsen, T.; Eliasen, H.; Schaefer, T. Effect of a melt agglomeration process on agglomerates containing solid dispersions. Int. J. Pharm. 2005, 303, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Pokharkar, V.B.; Mandpe, L.P.; Padamwar, M.N.; Ambike, A.A.; Mahadik, K.R.; Paradkar, A. Development, characterization and stabilization of amorphous form of a low Tg drug. Powder Technol. 2006, 167, 20–25. [Google Scholar] [CrossRef]
- Sethia, S.; Squillante, E. Physicochemical characterization of solid dispersions of carbamazepine formulated by supercritical carbon dioxide and conventional solvent evaporation method. J. Pharm. Sci. 2002, 91, 1948–1957. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Obi, N. Studies on absorption of eutectic mixture. I. A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem. Pharm. Bull. 1961, 9, 866–872. [Google Scholar]
- Chiou, W.L.; Niazi, S. Phase diagram and dissolution-rate studies on sulfathiazole-urea solid dispersions. J. Pharm. Sci. 1971, 60, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Carstensen, J.T. Pharmaceutical Principles of Solid Dosage Form; Technomic: Lancaster, PA, USA, 1993. [Google Scholar]
- Simonelli, A.P.; Mehta, S.C.; Higuchi, W.I. Dissolution rates of high energy polyvinylpyrrolidone (PVP)-sulfathiazole coprecipitates. J. Pharm. Sci. 1969, 58, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Mayersohn, M.; Gibaldi, M. New method of solid-state dispersion for increasing dissolution rates. J. Pharm. Sci. 1966, 55, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- Bates, T.R. Dissolution characteristics of reserpine-polyvinylpyrrolidone co-precipitates. J. Pharm. Pharmacol. 1969, 21, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Vasanthavada, M.; Tong, W.Q.T.; Joshi, Y.; Kislalioglu, M.S. Phase behavior of amorphous molecular dispersions II: Role of hydrogen bonding in solid solubility and phase separation kinetics. Pharm. Res. 2005, 22, 440–448. [Google Scholar] [CrossRef]
- Rumondor, A.C.; Marsac, P.J.; Stanford, L.A.; Taylor, L.S. Phase behavior of poly(vinylpyrrolidone) containing amorphous solid dispersions in the presence of moisture. Mol. Pharm. 2009, 6, 1492–1505. [Google Scholar] [CrossRef]
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-melt extrusion: From theory to application in pharmaceutical formulation. AAPS PharmSciTech 2016, 17, 20–42. [Google Scholar] [CrossRef]
- Tan, D.K.; Davis, D.A.; Miller, D.A.; Williams, R.O.; Nokhodchi, A. Innovations in thermal processing: Hot-melt extrusion and Kinetisol® dispersing. AAPS PharmSciTech 2020, 21, 312. [Google Scholar] [CrossRef]
- Tachibana, T.; Nakamura, A. A methode for preparing an aqueous colloidal dispersion of organic materials by using water-soluble polymers: Dispersion ofΒ-carotene by polyvinylpyrrolidone. Kolloid-Z. Z. Polym. 1965, 203, 130–133. [Google Scholar] [CrossRef]
- Betageri, G.V.; Makarla, K.R. Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques. Int. J. Pharm. 1995, 126, 155–160. [Google Scholar] [CrossRef]
- Yu, D.G.; Li, J.J.; Williams, G.R.; Zhao, M. Electrospun amorphous solid dispersions of poorly water-soluble drugs: A review. J. Control Release 2018, 292, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yan, E.; Huang, Z.; Wang, C.; Xin, Y.; Zhao, Q.; Tong, Y. Preparation and study of PPV/PVA nanofibers via electrospinning PPV precursor alcohol solution. Eur. Polym. J. 2007, 43, 802–807. [Google Scholar] [CrossRef]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Roy, S.; Kalimuthu, Y.; Khanam, J.; Nanda, A. Solid dispersion: An approach to enhance the bioavailability of poorly water soluble drugs. Int. J. Pharmacol. Pharm. 2012, 1, 37–46. [Google Scholar]
- Singh, A.; Van den Mooter, G. Spray drying formulation of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef]
- Butreddy, A.; Bandari, S.; Repka, M.A. Quality-by-design in hot melt extrusion based amorphous solid dispersions: An industrial perspective on product development. Eur. J. Pharm. Sci. 2021, 158, 105655. [Google Scholar] [CrossRef]
- Ziaee, A.; Albadarin, A.B.; Padrela, L.; Femmer, T.; O’Reilly, E.; Walker, G. Spray drying of pharmaceuticals and biopharmaceuticals: Critical parameters and experimental process optimization approaches. Eur. J. Pharm. Sci. 2019, 127, 300–318. [Google Scholar] [CrossRef]
- He, Y.; Ho, C. Amorphous solid dispersions: Utilization and challenges in drug discovery and development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef]
- Impurities: Guideline for Residual Solvents Q3C (R8) Current Step 4 Version; G. ICH Harmonised Guideline, International Council for Harmonisation, 22 April 2021. Available online: https://database.ich.org/sites/default/files/ICH_Q3C-R8_Guideline_Step4_2021_0422_1.pdf (accessed on 29 January 2021).
- Paudel, A.; Worku, Z.A.; Meeus, J.; Guns, S.; Van den Mooter, G. Manufacturing of solid dispersions of poorly water soluble drugs by spray drying: Formulation and process considerations. Int. J. Pharm. 2013, 453, 253–284. [Google Scholar] [CrossRef]
- Thybo, P.; Hovgaard, L.; Lindeløv, J.S.; Brask, A.; Andersen, S.K. Scaling up the spray drying process from pilot to production scale using an atomized droplet size criterion. Pharm. Res. 2008, 25, 1610–1620. [Google Scholar] [CrossRef]
- Karanth, H.; Shenoy, V.S.; Murthy, R.R. Industrially feasible alternative approaches in the manufacture of solid dispersions: A technical report. AAPS PharmSciTech 2006, 7, 87. [Google Scholar] [CrossRef]
- Thiry, J.; Krier, F.; Evrard, B. A review of pharmaceutical extrusion: Critical process parameters and scaling-up. Int. J. Pharm. 2015, 479, 227–240. [Google Scholar] [CrossRef]
- Alshahrani, S.M.; Morott, J.T.; Alshetaili, A.S.; Tiwari, R.V.; Majumdar, S.; Repka, M.A. Influence of degassing on hot-melt extrusion process. Eur. J. Pharm. Sci. 2015, 80, 43–52. [Google Scholar] [CrossRef]
- Gupta, A.; Ma, K. Hot-melt extrusion: An FDA perspective on product and process understanding. In Hot-Melt Extrusion: Pharmaceutical Applications; Douroumis, D., Ed.; John Wiley & Sons: New York, NY, USA, 2012; pp. 323–331. [Google Scholar]
- Famili, M.H.N. A fast and economical method for producing of self-wipe twin-screw extruder modules. Open Mech. Eng. J. 2008, 2, 93–96. [Google Scholar] [CrossRef]
- Démuth, B.; Nagy, Z.K.; Balogh, A.; Vigh, T.; Marosi, G.; Verreck, G.; Van Assche, I.; Brewster, M.E. Downstream processing of polymer-based amorphous solid dispersions to generate tablet formulations. Int. J. Pharm. 2015, 486, 268–286. [Google Scholar] [CrossRef]
- Vachon, M.G.; Chulia, D. The use of energy indices in estimating powder compaction functionality of mixtures in pharmaceutical tableting. Int. J. Pharm. 1999, 177, 183–200. [Google Scholar] [CrossRef]
- Amidon, G. Physical and mechanical property characterization of powders. In Physical Characterization of Pharmaceutical Solids; Brittan, H., Ed.; CRC Press: New York, NY, USA, 1995; pp. 281–317. [Google Scholar]
- Amidon, G.E.; Secreast, P.J.; Mudie, D. Particle powder and compact characterization. In Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice; Qiu, Y., Chen, Y., Zhang, G.G.Z., Lirong, L., Porter, W.R., Eds.; Academic Press: London, UK, 2009; pp. 163–183. [Google Scholar]
- Carraher, C.E., Jr. Introduction to Polymer Chemistry, 2nd ed.; CRC Press: London, UK; New York, NY, USA, 2010. [Google Scholar]
- Crowley, M.M.; Zhang, F.; Repka, M.A.; Thumma, S.; Upadhye, S.B.; Kumar Battu, S.; McGinity, J.W.; Martin, C. Pharmaceutical applications of hot-melt extrusion: Part I. Drug Dev. Ind. Pharm. 2007, 33, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, S.M. Increased glass transition temperature in motionally constrained semicrystalline polymers. Polym. Adv. Technol. 1998, 9, 169–201. [Google Scholar] [CrossRef]
- Davis, M.; Walker, G. Recent strategies in spray drying for the enhanced bioavailability of poorly water-soluble drugs. J. Control Release 2018, 269, 110–127. [Google Scholar] [CrossRef]
- Davis, M.T.; Potter, C.B.; Mohammadpour, M.; Albadarin, A.B.; Walker, G.M. Design of spray dried ternary solid dispersions comprising itraconazole, soluplus and HPMCP: Effect of constituent compositions. Int. J. Pharm. 2017, 519, 365–372. [Google Scholar] [CrossRef]
- Albadarin, A.B.; Potter, C.B.; Davis, M.T.; Iqbal, J.; Korde, S.; Pagire, S.; Paradkar, A.; Walker, G. Development of stability-enhanced ternary solid dispersions via combinations of HPMCP and Soluplus(®) processed by hot melt extrusion. Int. J. Pharm. 2017, 532, 603–611. [Google Scholar] [CrossRef]
- Iyer, R.; Hegde, S.; Zhang, Y.E.; Dinunzio, J.; Singhal, D.; Malick, A.; Amidon, G. The impact of hot melt extrusion and spray drying on mechanical properties and tableting indices of materials used in pharmaceutical development. J. Pharm. Sci. 2013, 102, 3604–3613. [Google Scholar] [CrossRef][Green Version]
- Davis, M.T.; Potter, C.B.; Walker, G.M. Downstream processing of a ternary amorphous solid dispersion: The impacts of spray drying and hot melt extrusion on powder flow, compression and dissolution. Int. J. Pharm. 2018, 544, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Freeman, R. Measuring the flow properties of consolidated, conditioned and aerated powders—A comparative study using a powder rheometer and a rotational shear cell. Powder Technol. 2007, 174, 25–33. [Google Scholar] [CrossRef]
- Olsson, H.; Nyström, C. Assessing tablet bond types from structural features that affect tablet tensile strength. Pharm. Res. 2001, 18, 203–210. [Google Scholar] [CrossRef] [PubMed]
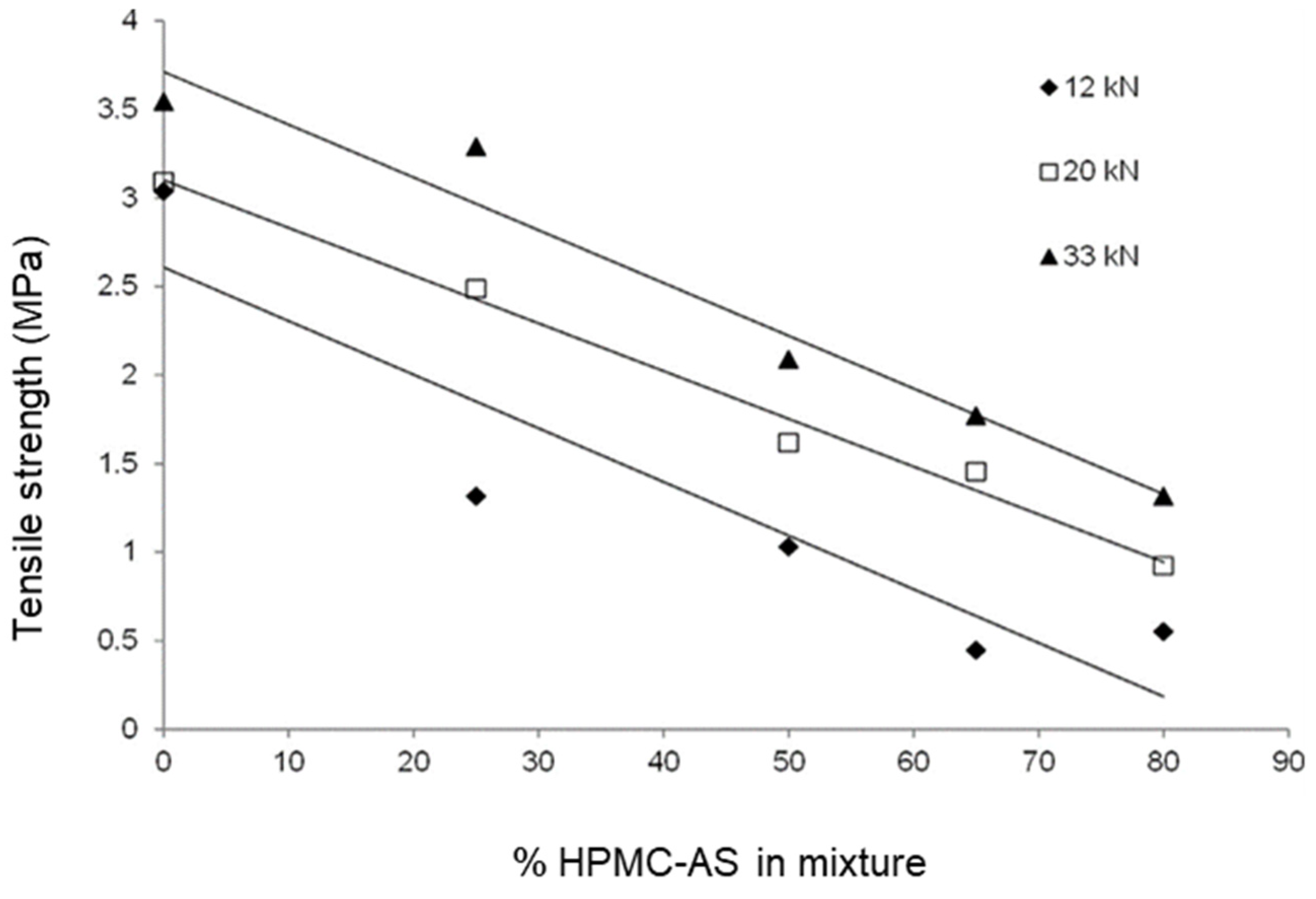
- Hou, H.H.; Rajesh, A.; Pandya, K.M.; Lubach, J.W.; Muliadi, A.; Yost, E.; Jia, W.; Nagapudi, K. Impact of Method of Preparation of Amorphous Solid Dispersions on Mechanical Properties: Comparison of Coprecipitation and Spray Drying. J. Pharm. Sci. 2019, 108, 870–879. [Google Scholar] [CrossRef]
- Boersen, N.; Lee, T.W.Y.; Shen, X.; Hui, H.W. A preliminary assessment of the impact of hot-melt extrusion on the physico-mechanical properties of a tablet. Drug Dev. Ind. Pharm. 2014, 40, 1386–1394. [Google Scholar] [CrossRef]
- Lourdin, D.; Colonna, P.; Brownsey, G.J.; Noel, T.R.; Ring, S.G. Structural relaxation and physical ageing of starchy materials. Carbohydr. Res. 2002, 337, 827–833. [Google Scholar] [CrossRef]
- Iyer, R.M.; Hegde, S.; DiNunzio, J.; Singhal, D.; Malick, W. The impact of roller compaction and tablet compression on physicomechanical properties of pharmaceutical excipients. Pharm. Dev. Technol. 2014, 19, 583–592. [Google Scholar] [CrossRef]
- Flügel, K.; Hennig, R.; Thommes, M. Impact of structural relaxation on mechanical properties of amorphous polymers. Eur. J. Pharm. Biopharm. 2020, 154, 214–221. [Google Scholar] [CrossRef]
- Patel, S.; Kou, X.; Hou, H.H.; Huang, Y.B.; Strong, J.C.; Zhang, G.G.; Sun, C.C. Mechanical properties and tableting behavior of amorphous solid dispersions. J. Pharm. Sci. 2017, 106, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Vicente, J.; Gaspar, V.F. Scale-up methodology for pharmaceutical spray drying. Chem. Today 2010, 28, 18–22. [Google Scholar]
- Petrides, D.P.; Koulouris, A.; Lagonikos, P.T. The role of process simulation in pharmaceutical process development and product commercialization. Pharm. Eng. 2002, 22, 56–65. [Google Scholar]
- Poozesh, S.; Bilgili, E. Scale-up of pharmaceutical spray drying using scale-up rules: A review. Int. J. Pharm. 2019, 562, 271–292. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, A. Atomisation and Spray Technology, 3rd ed.; CRC Press: Boca Raton, FL, USA, 1987. [Google Scholar]
- FDA. Inactive Ingredient Search for Approved Drug Products; US Food and Drug Administration: Silver Spring, MD, USA, 2017.
- FDA GRAS Notices Website. Available online: https://www.cfsanappsexternal.fda.gov/scripts/fdcc/?set=GRASNotices. (accessed on 1 August 2021).
- Kozarewicz, P.; Loftsson, T. Novel excipients—Regulatory challenges and perspectives—The EU insight. Int. J. Pharm. 2018, 546, 176–179. [Google Scholar] [CrossRef]
- FDA. Novel Excipient Review Program. Proposal; Request for Information and Comments; US Food and Drug Administration: Silver Spring, MD, USA, 2019.
- Pottel, J.; Armstrong, D.; Zou, L.; Fekete, A.; Huang, X.P.; Torosyan, H. The activities of drug inactive ingredients on biological targets. Science 2020, 369, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Gadgil, P.; Krishnamurthy, V.R.; Landis, M.; Mallick, P.; Patel, D. The Evolving Druggability and Developability Space: Chemically Modified New Modalities and Emerging Small Molecules. Aaps J. 2020, 22, 21. [Google Scholar] [CrossRef]
- Bhujbal, S.V.; Pathak, V.; Zemlyanov, D.Y.; Taylor, L.S.; Zhou, Q.T. Physical stability and dissolution of lumefantrine amorphous solid dispersions produced by spray anti-solvent precipitation. J. Pharm. Sci. 2020, 110, 2423–2431. [Google Scholar] [CrossRef]
- Xi, Z.; Zhang, W.; Fei, Y.; Cui, M.; Xie, L.; Chen, L.; Xu, L. Evaluation of the solid dispersion system engineered from mesoporous silica and polymers for the poorly water soluble drug indomethacin: In vitro and in vivo. Pharmaceutics 2020, 12, 144. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Technique | Mode of Enhancement | Reference |
|---|---|---|---|
| Physical | Micronization or nanoparticles using sizing-down (top down) or building-up (bottom up) technologies | Increase in diffusional surface area | [13,14] |
| Inclusion complexes of drugs using cyclodextrins | Increase in solubility via H-bonding | [9,15] | |
| Self-emulsifying emulsion and microemulsion systems (SEDDS, SMEDDS) using oils, lipids and/or surfactants | Micellar solubilization, reduced efflux and vesicular entrapment | [16,17,18] | |
| Conversion of crystalline drug into a stabilized amorphous form (solid dispersion, solid solution) | Increase in kinetic solubility and supersaturation | [19,20] | |
| Chemical | Salt forms of poorly soluble acidic or basic drugs | pH-partition (via ionization or dissociation) | [21] |
| Conversion to inactive but soluble pro-drugs with functional groups such as esters for metabolic conversion to parent drug | Reduced lipophilicity and optimized ADME properties | [22,23] | |
| Conversion to co-crystals with a conformer | Higher solubility | [24] |
| Drug (Marketed Product) | Technology (1) | Polymer (2) | Dissolution Approach (3) FDA Recommended Method (  ) vs. Bio-Relevant Approach (∞) ) vs. Bio-Relevant Approach (∞) | Supporting Analytical Techniques | Conclusion |
|---|---|---|---|---|---|
| Reference product of Verapamil: ISOPTIN-SRE, ER tablets Developed formulation (tablets) [93] | HME (ISOPTIN-SRE) Kneading, solvent and co-precipitation method | HPMC/HPC 12 SDs prepared: 1:1, 1:2 and 1:3 API—polymer ratios with the following polymers: PVPK30, β-cyclodextrin, PEG 6000, HPMCK100M | Two-phase dissolution: phase 1: 900 mL SGF without enzyme 60 min; phase 2: withdraw and transfer to 900 mL SIF without enzyme, 7 h 50 rpm, USP II with wire helix ∞Phosphate buffer pH 1.2, 900 mL, 50 rpm, USP I | DSC, PXRD, SEM, FTIR, supersaturation solubility testing, stability studies | Increased dissolution rates of tablets containing SD with API: PEG6000 ratio of 1:3 in comparison to other formulations and marketed tablets due to decreased particle size, increased wettability and dispersibility of verapamil; Drug–carrier interaction observed; Higher polymer concentration gives faster drug release. |
| Reference product of Itraconazole: Sporanox cps, and ONMEL tbl Developed formulation (SD, tablets) [94] | Spray lavering (Sporanox) HME (ONMEL) Solubilization in concentrated aqueous solutions of weak organic acids and drying | HPMC SDs with 2–20% drug load prepared with Glutaric acid | 0.1 N HCl, 900 mL 75 rpm, USP II (tbl) SGF without enzyme, 900 mL, 100 rpm, USP II (cps) ∞0.1 N HCL, 250 mL 75 rpm, USP II (ASD) | DSC, PXRD, ATR-FTIR, pH-solubility studies | Solubility greatly enhanced compared to amorphous form of drug, possible weak drug–acid interactions observed; precipitated as mostly nanoparticles that enable rapid re-dissolution, which might influence absorption. |
| Reference product of Tacrolimus PROGRAF Developed formulation (SD) [95] | Spray drying/fluid bed (PROGRAF) Spray drying via solvent-evaporation method, solvent-wetting method, or surface-attached method – three different processing methodologies | HPMC 3SDs prepared: 10:80:1 API:HP-β-CD:DOSS ratio | 0.005% HPC in Water with 0.50% SLS adjusted to pH 4.5, 900 mL, 100 rpm, USP II (tbl) HPC solution (1 in 20,000), adjusted to pH 4.5 by phosphoric acid, 900 mL50 rpm, USP II (cps) ∞0.005% HPC in Water with 0.50% SLS adjusted to pH 4.5 by phosphoric acid, 500 mL 50 rpm, USP II with sinker (SD) | SEM, DSC, PXRD | The solubility and dissolution were significantly improved by SD preparation method compared to drug powder. |
| Reference product of Nifedipine Afeditab Developed Tablets [96] | Melt/absorb on carrier Co-precipitation | Poloxamer or PVP 12 SDs prepared:1:1, 1:5 and 1:10 API:polymer ratio with all listed polymers: poloxamer, HPMC, PEG 4000 and PEG 6000 [36]; with each API-Polymer ratios of 1:1, 1:5 and 1:10 tested | 0.5% SLS in SGF without enzyme pH 1.2, 900 mL 100 rpm, USP II (tbl ER) SGF without enzyme, 900 mL50 rpm, USP II (cps) ∞SGF without enzyme, 900 mL 50 rpm, USP XXI (SD, tbl) | DSC, FT-IR | SD tablets prepared with PEG 6000 and poloxamer showed better release profile than marketed products. |
| Reference product of Griseofulvin (Gris-PEG) Developed formulation (SD) [97] | HME (Gris-PEG) Solvent evaporation technique | PEG 6000 24 SDs prepared: 3:1, 1:1, 1:2 and 1:9 API:polymer ratios with all polymers: PVP, HPMC, and Eudragit L 100, Eudragit E 100, Eudragit S 100, PEG 8000 | 4.0% SLS in water, 1000 mL 75 rpm, USP II (tbl) 0.54% SLS in water, 1000 mL25 an 50 rpm, USP II (susp) ∞Dissolution studies not performed. | PXRD, mDSC, ATR-IR, Raman spectroscopy | Increased polymer concentration leads to lower drug released because drug binds tighter to the concentrated polymers, however SD is more stable. |
| Reference product of Nimodipine Nimotop Developed formulation (SD) [98] | Spray drying (Nimotop) HME | PEG 9 SDs prepared: 1:2, 3:7 and 1:9 API:polymer ratio with polymers: HPMC, PVP-VA, Eudragit EPO | 0.5% SDS in water, 900 mL 50 rpm, USP II (cps) ∞0.05% SLS in acetate buffer pH 4.5, 900 mL 75 rpm, ZRS-8G (paddle) | DSC, XRPD, FT-IR, SEM | Eudragit EPO and PVP-VA showed better miscibility than HPMC. Drug–polymer hydrogen bonding was observed. |
| Reference formulation of Lopinavir & Ritonavir KALETRA tablets and capsules Developed formulation (SD) [99] | HME (KALETRA) Solvent granulation process | PVP-VA SDs with various API1:API2:PVP-VA ratios | Tier 1:0.06 M polyoxyethyelene 10 lauryl ether with 10 mM sodium phosphate monobasic (pH 6.8)Tier 2: same as tier 1 with no more than 1750 USP units/L of pancreatin, 900 mL (cps) 50 rpm, USP II Test 1: 0.06 M decaethyelene glycolmonododecyl ether in waterTest 2: 37.7 g/L of polyoxyethyelene 10-lauryl ether in water (tbl)75 rpm, USP II ∞10 mM phosphate buffer pH 6.8, 250 mL and 0.1 N HCl, 250 mL 150 rpm, jacketed beaker | XRPD, FT-IR | Molecular mixing of both components into a single amorphous phase negatively impacts ritonavir dissolution performance in comparison with marketed formulation. Amorphous suppression phenomenon observed in pH-shift dissolution method. It is proposed that dissolution of ritonavir from the surface of the particles in acidic media leaves behind a lopinavir-rich surface which acts as a barrier for the remaining ritonavir to dissolve. |
| Reference product of Fenofibrate Fenoglide Developed formulation (SD) [100] | HME (Fenoglide) Solvent evaporation method | PEG/Poloxamer 188 7 SDs prepared: 1:1, 1:2 and 1:3 API:polymer ratio with polymers: Carplex 80 and PEG 4000 and 1:5:6 API:polymer ratio with Carplex 80 and PEG 6000 respectively | 25 mM/50 mM/0.75% SLS in water, 1000/1000/900 mL (40 and 120 mg/48 and 145 mg/54 and 160 mg tbl) 50/50/75 rpm, USP II Phosphate buffer w/2% Tween 80 and 0.1% pancreatin pH 6.8, 900 mL75 rpm, USP II (cps) ∞Demineralized water, 900 mL 50 rpm, USP II | DSC, PXRD, FT-IR, SEM | The most significant improvement of drug dissolution and amorphization was obtained with SD prepared with drug:Carplex:PEG ratio 1:5:6 |
| Reference product of Ivacaftor KALYDECO Developed formulation (SD) [101] | Spray drying (KALYDECO) HME | HPMCAS 9 SDs prepared: 1:1 API: polymer (Soluplus, HPMC, Copovidone), each pair with three surfactants (SLS, poloxamer, polysorbate 70) | 50 mM sodium phosphate buffer with 0.7% SLS pH 6.8, 900 mL (tbl) 65 rpm, USP II with a sinker ∞50 mM sodium phosphate buffer pH 6.8, 900 mL 65 rpm, USP II | XRPD, DSC, FT-IR | Improved solubilization by improved wetting of drug substance by hydrophilic carriers which represent rich microenvironment formed at the surface of the drug substance and this leads to improved dissolution rate. No defined drug–polymer interaction was observed. |
| Reference product of Posaconazole Noxafil Developed formulation (SD, tablets) [102] | HME (Noxafil) Spray drying | HPMCAS 1 SD prepared: 3:1 API:polymer ratio with polymer Eudragit L100 | Acid Stage: 0.01 N HCl, 750 mL; Buffer Stage: 50 mM phosphate buffer, pH 6.8 with 0.37% Polysorbate 80 (after 120 min, to the acid stage, add 250 mL of 0.2 M Phosphate Buffer, 1.46% Polysorbate 80) (tbl DR) 75 rpm, USP II 0.3% SLS, 900 mL25 rpm, USP II (susp) ∞0.01 M HCl with 34 mM NaCl solution and phosphate buffer with SIF powder pH 6.5 CTD apparatus | mDSC, PXRD, SEM, in-vivo study | The in-vitro dissolution data underpredicted in-vivo performance, potentially due to higher driving force for precipitation in-vitro versus in-vivo. Including a concentration-sustaining polymer extragranularly to SD but inside tablet was as effective as including it inside the ASD itself. |
| Reference product of Everolimus CERTICAN and ZORTRESS Developed formulation (SD, tablets) [103] | Melt or spray drying (CERTICAN, ZORTRESS) Solvent-wetting and co-precipitation methods | HPMC SDs with various API:polymer ratios with HPMC were prepared | Water with 0.4% sodium dodecylsulfate, 500 mL (tbl) 50 rpm, USP II ∞0.4% SLS solution in water and distilled water, dissolution media pH 1.2, pH 4.0 and pH 6.8, 900 mL 50 rpm, USP II | XRPD, SEM, particle size analysis, stability and in-vivo studies | The optimized SD consisted of drug:HPMC weight ratio of 1:15. Tablets with SD created with solvent-wetting technique showed identical release rate to that of commercially available product. |
| Reference formulation of Telaprevir INCIVEK Developed formulation (SD) [104] | Spray drying (INCIVEK) Co-milling with polymers | HPMCAS 3 SDs prepared: 1:1 API:polymer ratio with polymers: PVP-K30, PEG 6000, HPMC | 1% SLS in water, 900 mL (tbl) 50 rpm, USP II ∞Distilled water, 0.1 M HCl pH 1.2, phosphate buffer pH 6.8, 900 mL 100 rpm, ZRC-8D (paddle) | XRPD, DSC, SEM, FT-IR, cytotoxicity evaluation, stability studies | Hydrogen bonding drug–polymer interaction observed. Drug–polymer SD did not affect efficacy of the drug and showed no toxic side effects to normal liver cells. No comparison to reference product shown. |
| Reference formulation of Vemurafenib ZELBORAF Developed formulation (SD, capsules) [19] | Co-precipitation method (ZELBORAF) Co-precipitation method | HPMCAS 3 SDs prepared: 2:3 API:polymer ratio with HPMCP, HPMCAS, and Eudragit L 100-55 | 1% Hexadecyltrimethylammonium bromide in 0.05 M phosphate buffer pH 6.8, 900 mL (tbl) 75 rpm, USP II ∞0.05% hexadecyltrimethylammonium bromide in phosphate buffer pH 6.8 10 mL/min, USP IV ∞FaSSIF, 900 mL 75 rpm, USP II [101] | XRPD, DSC, SEM, stability and in-vivo studies | Among used polymers, HPMCAS was found to be the best to prepare stable SD, based on superior physical stability and faster dissolution. No dissolution comparison to reference product shown. |
| Reference product of Ritonavir NORVIR HIV Developed formulation (SD) [105] | HME (NORVIR HIV) Solvent evaporation and melt method | PVP-VA 4 SDs prepared: 1:4 API:polymer ratio polymers Gelucire, sorbitol (with both listed method): | 60 mM polyoxyethyelene 10 lauryl ether, 900 mL (tbl) 75 rpm, USP II 0.1 M HCl with 25 mM polyoxyethyelene 10-lauryl ether (cps)50 rpm, USP II ∞0.1 M HCl, 900 mL FaSSIF pH 6.5, 500 mL ∞FeSSIF pH 5.0, 1000 mL 50 rpm, USP II with sinkers | DSC, XRPD, TEM, FT-IR, in-vivo study | Hydrogen bonding was observed in SD resulting in increased drug solubility as compared to pure drug. Maximum dissolution was obtained with FeSSIF media, which confirmed food-related absorption of drugs. No comparison to reference product available. |
| Methods of ASD Preparation | Principle of ASD Conversion | Limitations |
|---|---|---|
| Melting | ||
| Fusion/melting | The physical mixture of a drug and the carrier (polymer) is heated (above their eutectic point) then melted, then the melt is cooled and solidified. The final solid mass is crushed or milled or sieved. |
|
| Hot-melt extrusion | ||
| High shear blending generating melting (KinetiSol®) | ||
| Solvent Based | ||
| Solvent evaporation by rotary evaporator | Dissolving or suspending the drug and the carrier (polymer) in a solvent or a solvent mixture and then evaporating the solvent using different techniques. |
|
| Lyophilization/Freeze drying | ||
| Electrostatic spinning (similar to forming cotton candy) | ||
| Supercritical Fluid Extraction (SCF) | ||
| Spray drying (SD) | ||
| Fluidized bed-coating/layering (FBL) | ||
| Feed Properties | |
|---|---|
| Tfeed | Defined and controlled at lab scale. |
| Cfeed | Defined at lab scale with minor adjustments as needed. |
| Ffeed | Calculated from the evaporation rate needed in order to achieve the same vapor concentration at lab scale and pilot scale. |
| Drying properties. | |
| Tout | Measured directly and is kept constant during scale-up. |
| Tin | Calculated from the evaporation rate needed in order to achieve the same vapor concentration on lab scale and pilot scale. However, it may not be always possible due to equipment constraints. |
| Fdrying | Use the nominal flow of the equipment (limited range) to maintain correct loading and velocity in the air disperser, powder transporting ducts, cyclone and filters. |
| Atomization. | |
| Pfeed | Adjusted in order to obtain the same droplet size distribution through the different scales. |
| Attributes (Needs) | Differentiation Impact | Comments | Differentiation Targets |
|---|---|---|---|
| Higher drug loading | Lower pill or capsule burden that is known to reduce adherence and increase dropout rate. | Typical drug loading for the commercially available polymer is between 10 and 30% of a ASD product. Taking a hypothetical 1 g drug dose with a 20% w/w drug load, this would equate to 5 g of drug product requiring multiple large capsules or pills. | Drug loading of challenging molecules >40% wt/wt. |
| Longer shelf-life, physical and chemical stability | Reduce development and scale-up risks, enabling acceleration of a product to market. | Multiple companies have experienced stability issues at phase II or III clinical trials with ASD that resulted in delays in development and expensive reformulation. | Increased physical and chemical stability under accelerated condition (40 °C and 40 °C/75% RH). |
| High oral bioavailability | The product requires the solubility advantage offered by an ASD. | Oral bioavailability can be tested in a preclinical species which is known not to have a toxicity issue or extensive high first-pass metabolism which will make it challenging to differentiate between formulations. | Bioavailability in preclinical species is greater than a standard ASD polymer and has acceptable variability. |
| Enabling continuous manufacturing or reducing manufacturing complexity | Reduced risk during scale-up and/or reduce complexity of manufacturing. | The industry is moving towards green technologies and reducing or eliminating organic solvents. Some compounds have poor solubility in the organic solvents that are acceptable for cGMP ASD manufacturing or have stability issues in these solvents. | Compatible with continuous manufacturing process or compatible with established manufacturing technologies (e.g., SD or HME, while allowing use of low toxicity solvents in process). Without losing solubility advantage and ensuring no toxicity. |
| Compound | Compound ASD Challenges | Reference |
|---|---|---|
| lumefantrine |
| [174] |
| vemurafenib |
| [19] |
| indomethacin |
| [175] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, R.; Petrovska Jovanovska, V.; Berginc, K.; Jaklič, M.; Fabiani, F.; Harlacher, C.; Huzjak, T.; Sanchez-Felix, M.V. Amorphous Solid Dispersions (ASDs): The Influence of Material Properties, Manufacturing Processes and Analytical Technologies in Drug Product Development. Pharmaceutics 2021, 13, 1682. https://doi.org/10.3390/pharmaceutics13101682
Iyer R, Petrovska Jovanovska V, Berginc K, Jaklič M, Fabiani F, Harlacher C, Huzjak T, Sanchez-Felix MV. Amorphous Solid Dispersions (ASDs): The Influence of Material Properties, Manufacturing Processes and Analytical Technologies in Drug Product Development. Pharmaceutics. 2021; 13(10):1682. https://doi.org/10.3390/pharmaceutics13101682
Chicago/Turabian StyleIyer, Raman, Vesna Petrovska Jovanovska, Katja Berginc, Miha Jaklič, Flavio Fabiani, Cornelius Harlacher, Tilen Huzjak, and Manuel Vicente Sanchez-Felix. 2021. "Amorphous Solid Dispersions (ASDs): The Influence of Material Properties, Manufacturing Processes and Analytical Technologies in Drug Product Development" Pharmaceutics 13, no. 10: 1682. https://doi.org/10.3390/pharmaceutics13101682
APA StyleIyer, R., Petrovska Jovanovska, V., Berginc, K., Jaklič, M., Fabiani, F., Harlacher, C., Huzjak, T., & Sanchez-Felix, M. V. (2021). Amorphous Solid Dispersions (ASDs): The Influence of Material Properties, Manufacturing Processes and Analytical Technologies in Drug Product Development. Pharmaceutics, 13(10), 1682. https://doi.org/10.3390/pharmaceutics13101682