
Bi-Functional Peptides as a New Therapeutic Tool for Hepatocellular Carcinoma

 , ,
, ,  ,
, 
Abstract
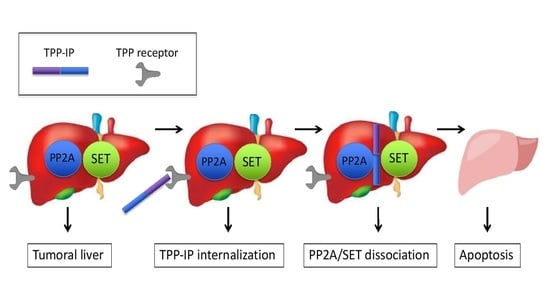
:
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Peptide Synthesis and Sequences
2.3. Isolation and Culture of Primary Human Tumoral Hepatocytes
2.4. Quantification of Cellular Internalization
2.5. Immunohistochemistry
2.6. Detection of Apoptosis by Annexin-V Staining
2.7. Immunoprecipitation and Western Blotting
2.8. Statistical Analysis
3. Results
3.1. Clinical Characteristics of the Patients and Tumor Aggressiveness Classification
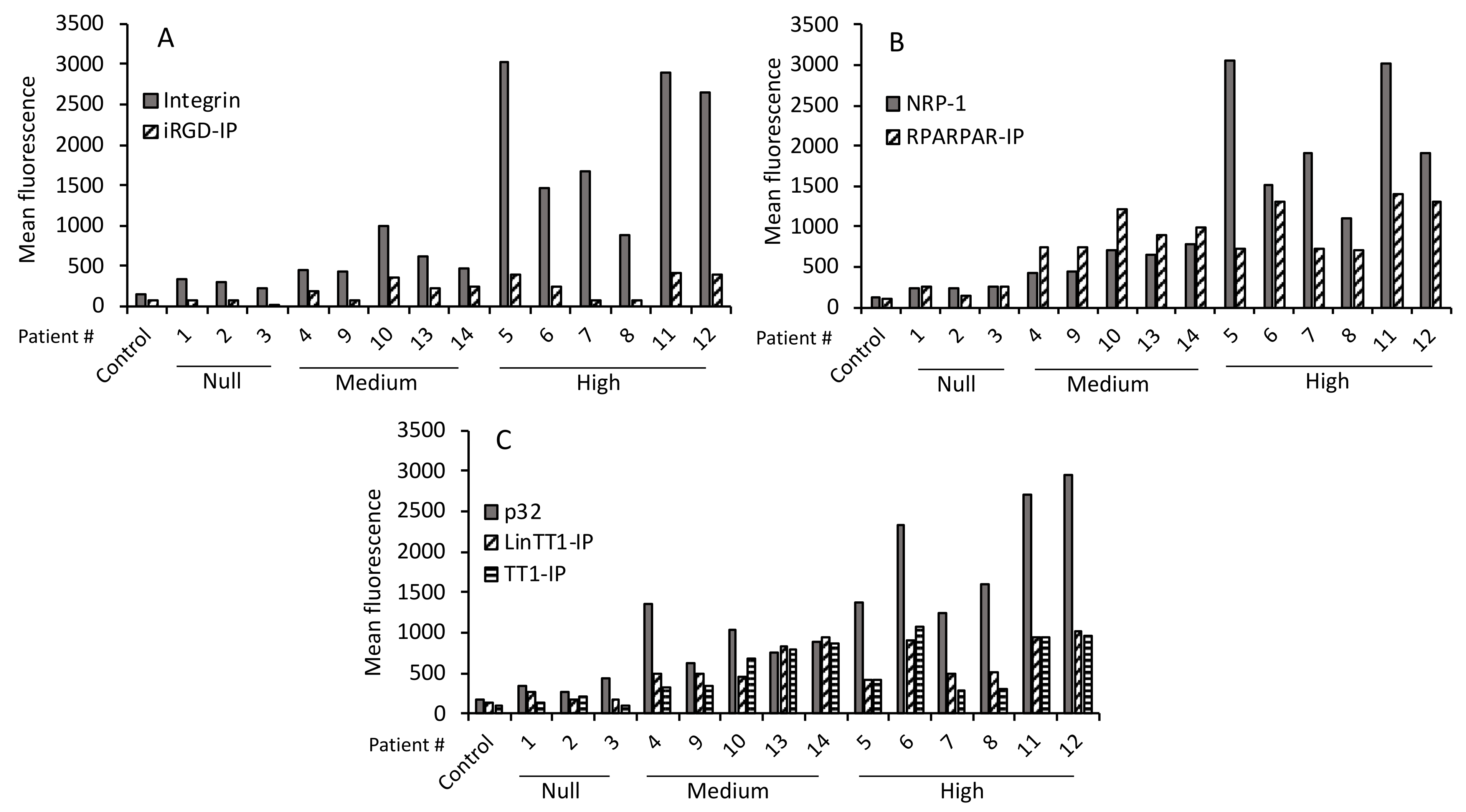
3.2. Immunohistochemical Characteristics of the Patients
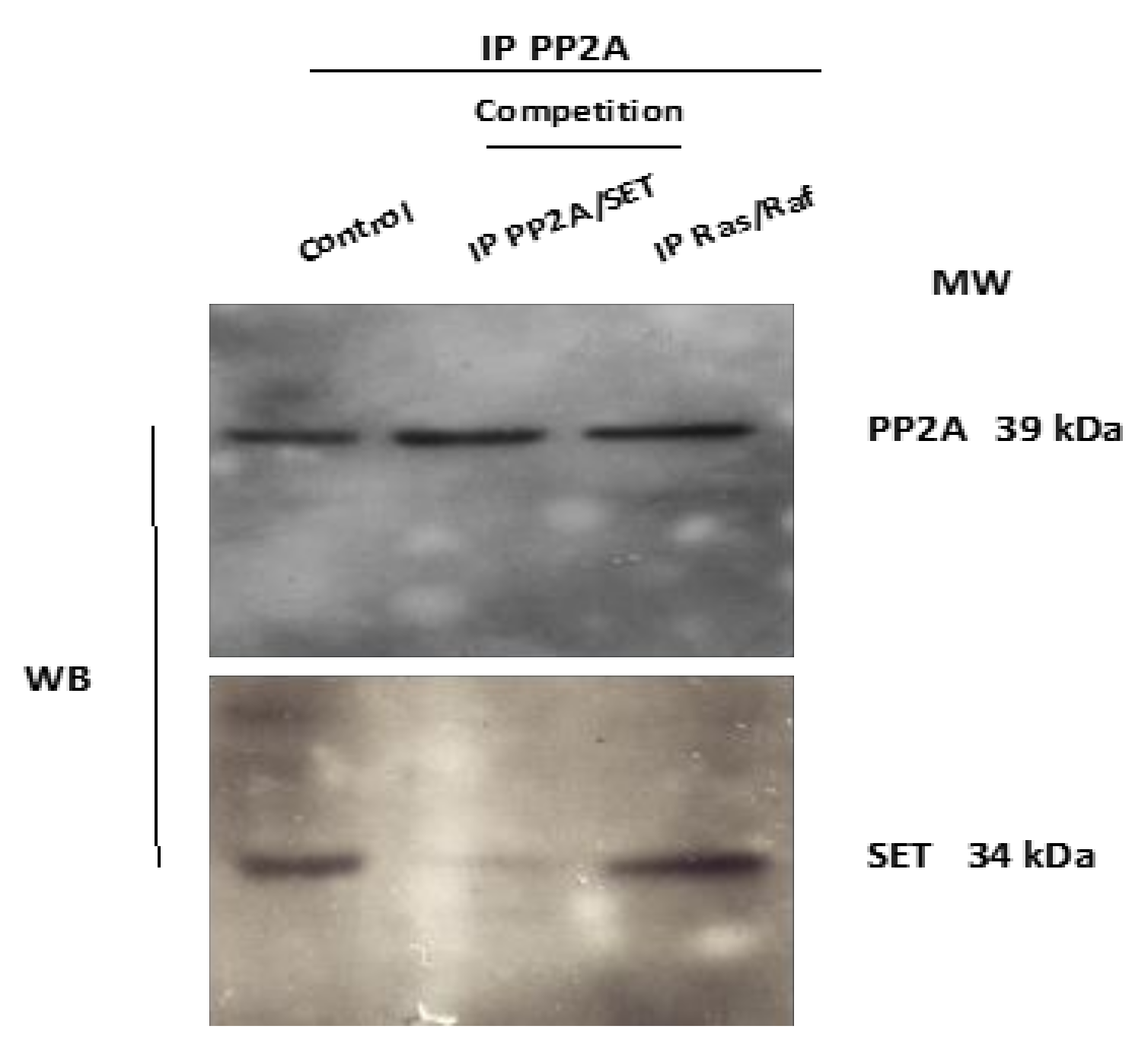
3.3. In Vitro Competition against PP2A/SET Interaction
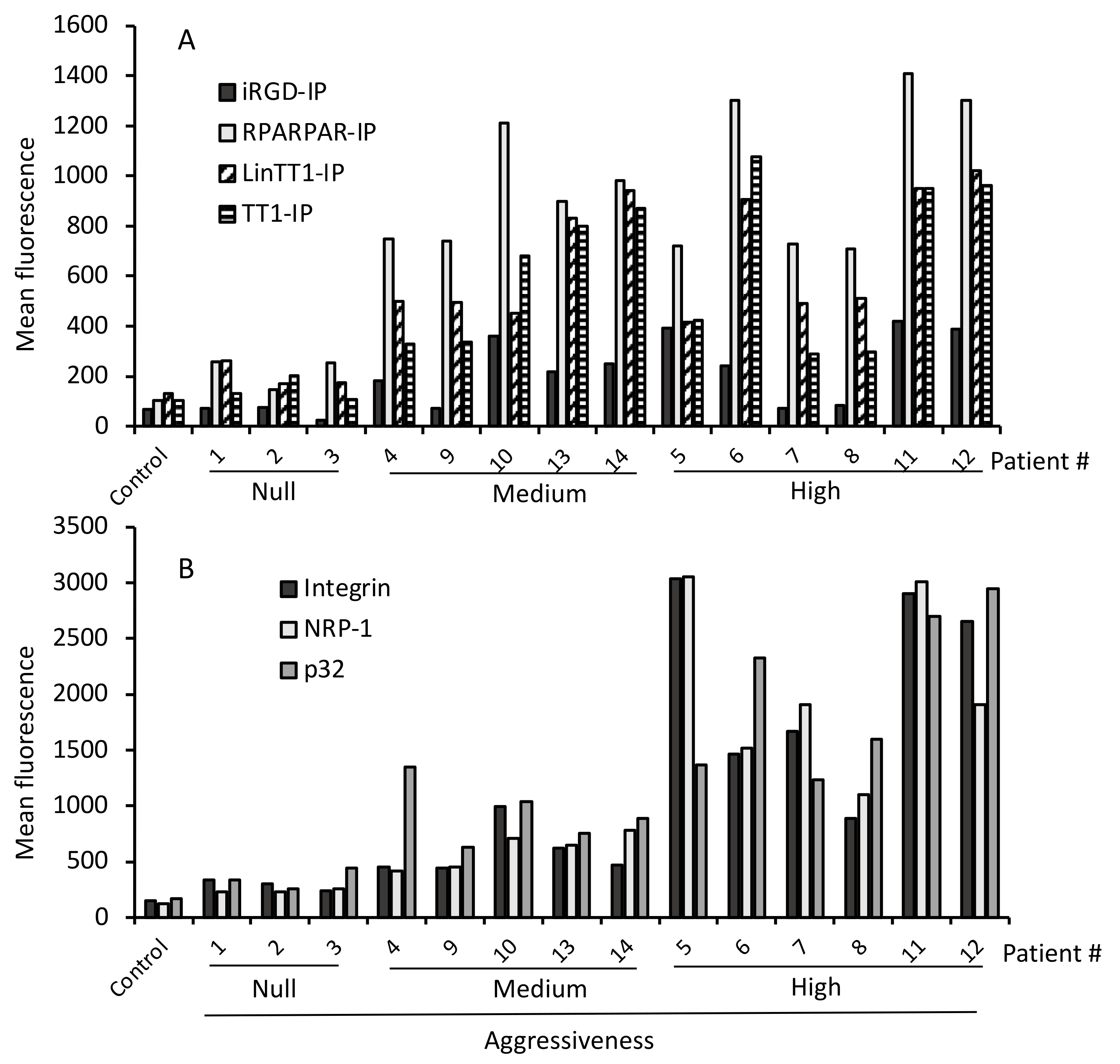
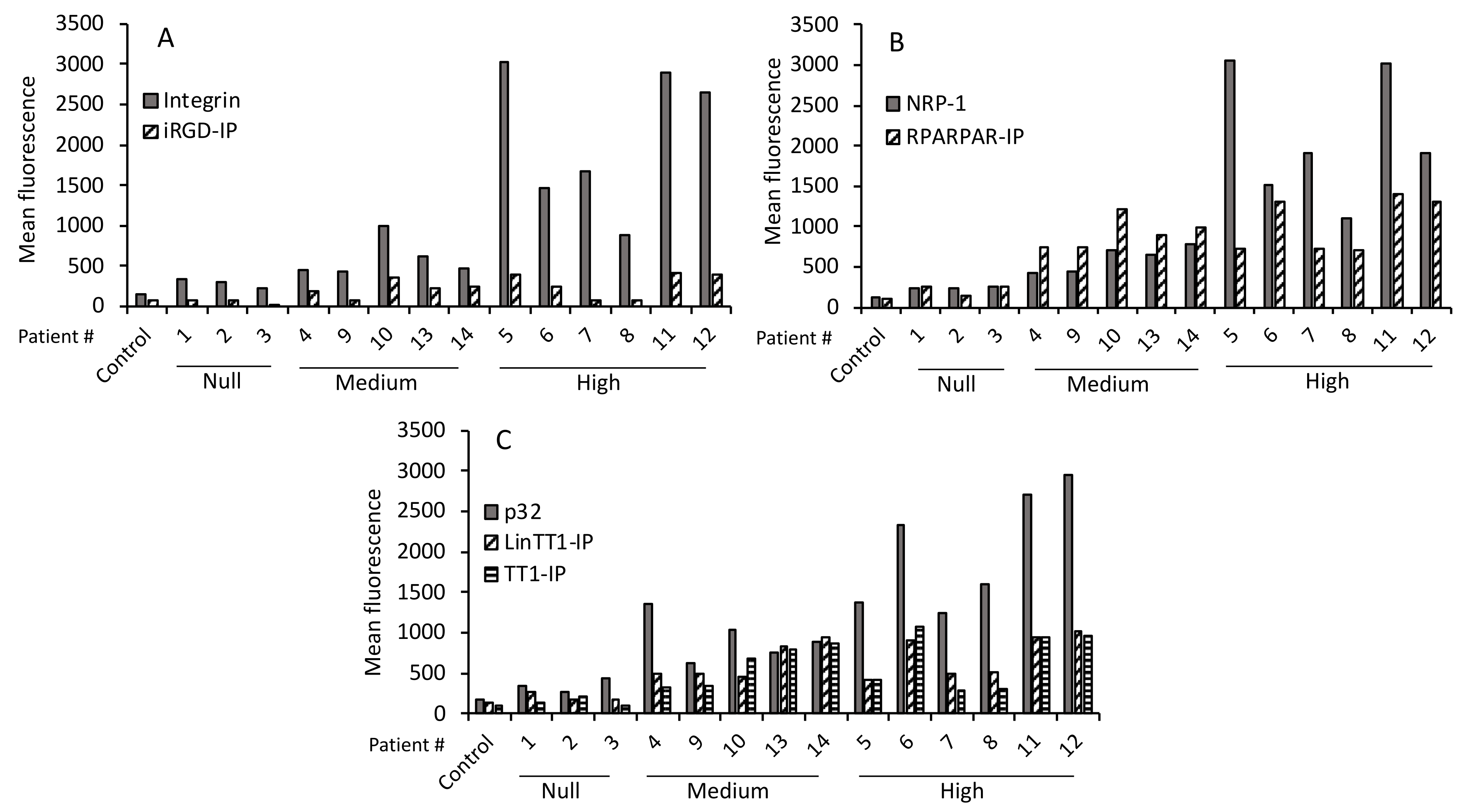
3.4. Internalization of Tumor-Penetrating and Interfering Peptides (TPP-IP) into Primary Tumoral Hepatocytes via Specific Receptors
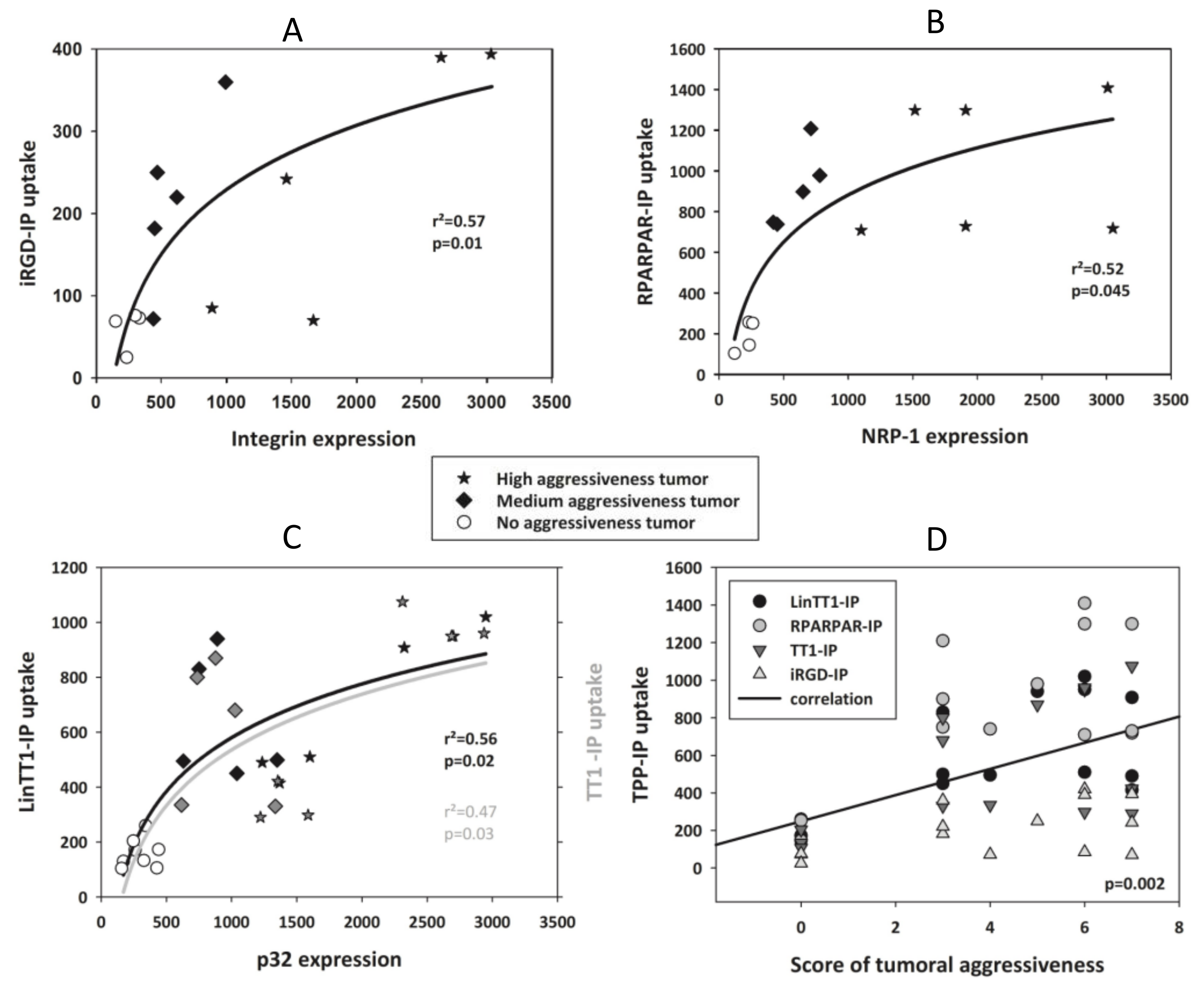
3.5. TPP Internalization and Receptor Expression Correlated with Tumor Aggressiveness
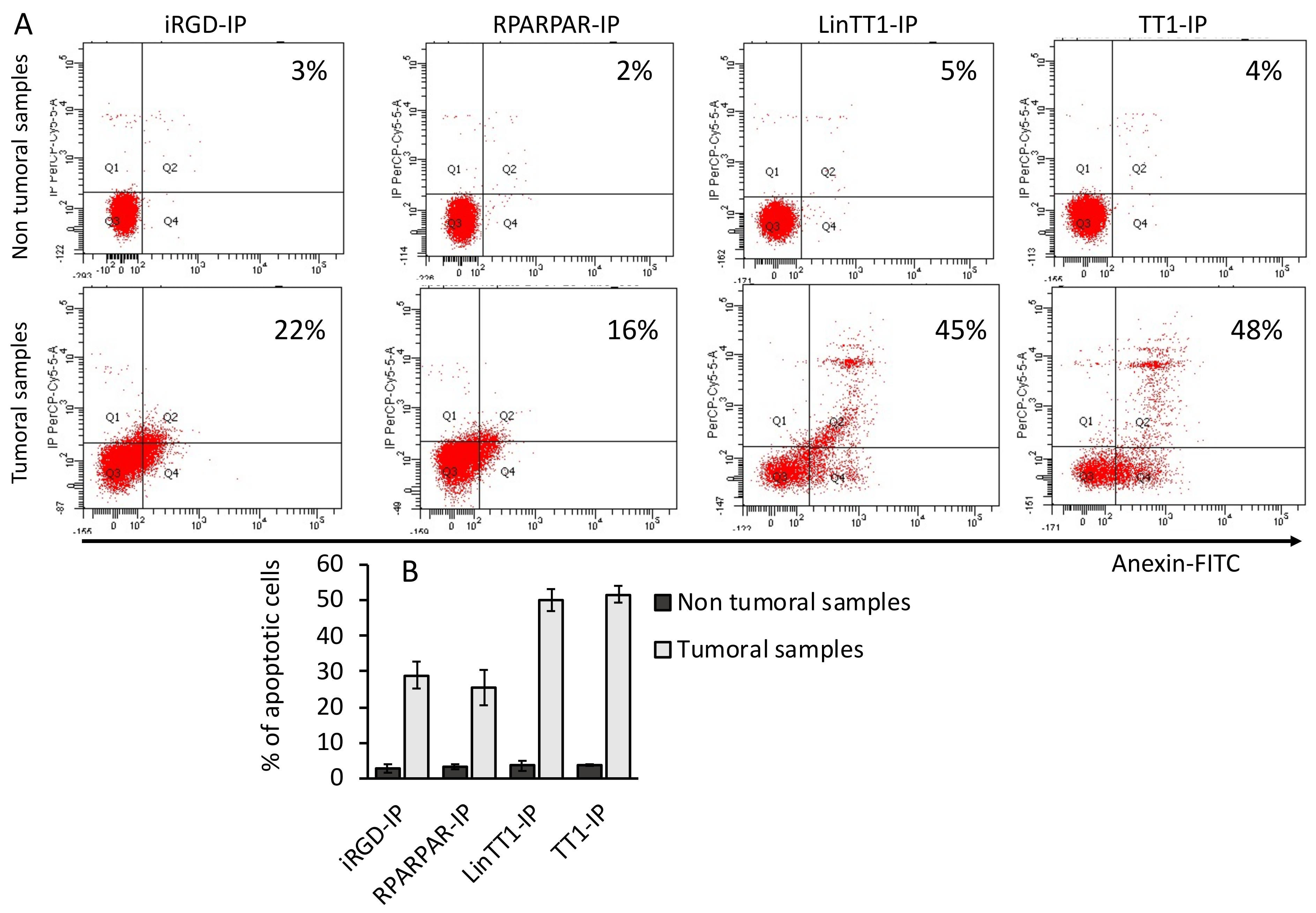
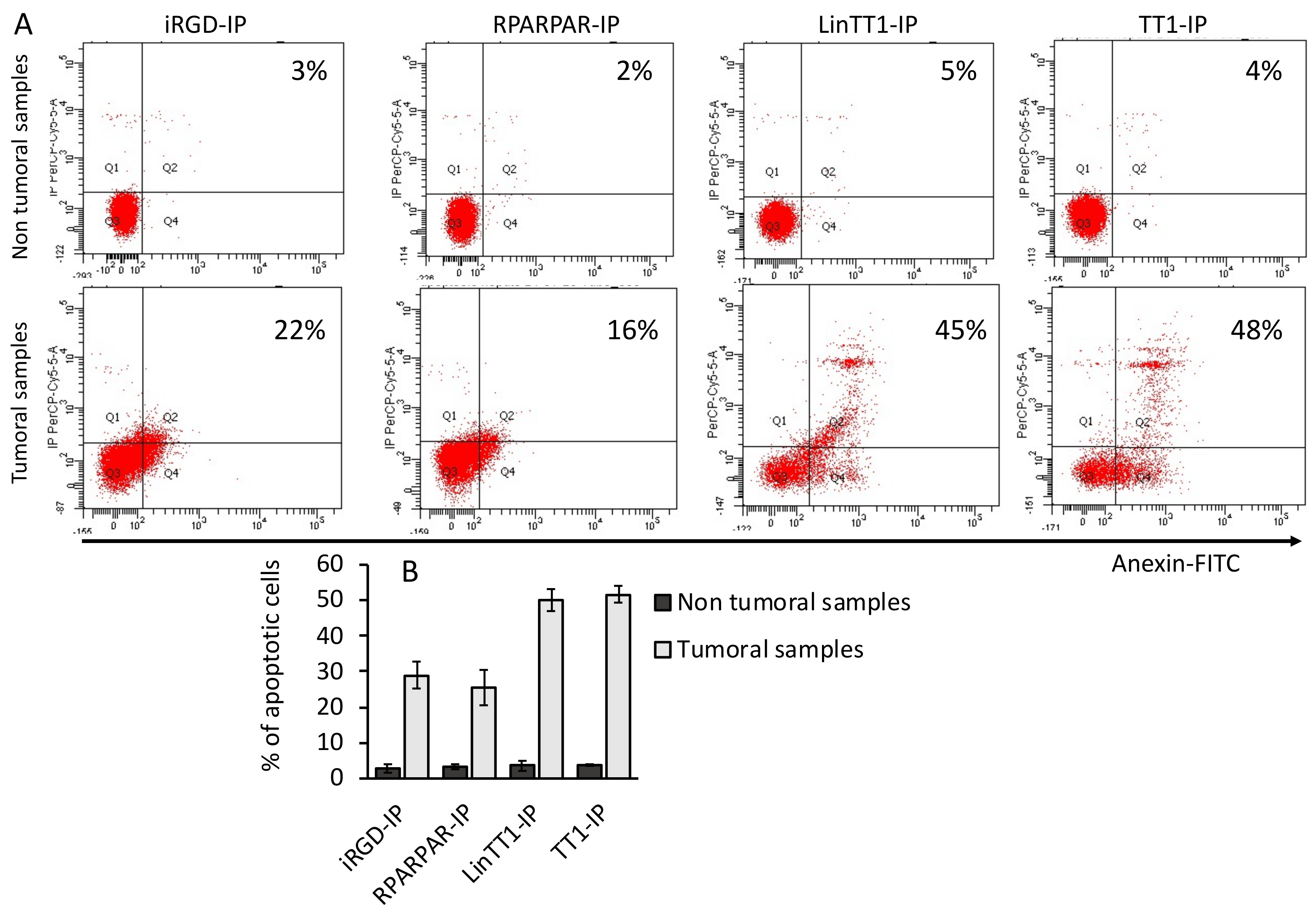
3.6. Apoptotic Effect of TPP-IPs on Tumoral Hepatocytes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xie, H.G.; Frueh, F.W. Pharmacogenomics steps toward personalized medicine. Future Med. 2005, 2, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Watters, J.W.; McLeod, H.L. Cancer pharmacogenomics: Current and future applications. Biochim. Biophys. Acta 2003, 1603, 99–111. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seebacher, N.A.; Stacy, A.E.; Porter, G.M.; Merlot, A.M. Clinical development of targeted and immune based anti-cancer therapies. J. Exp. Clin. Cancer Res. 2019, 38, 156. [Google Scholar] [CrossRef]
- Man, S.; Luo, C.; Yan, M.; Zhao, G.; Ma, L.; Gao, W. Treatment for liver cancer: From sorafenib to natural products. Eur. J. Med. Chem. 2021, 224, 113690. [Google Scholar] [CrossRef]
- Wildner, G. Tumors, tumor therapies, autoimmunity and the eye. Autoimmun. Rev. 2021, 20, 102892. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Ruoslahti, E. Tumor-penetrating peptides. Front. Oncol. 2013, 3, 216. [Google Scholar] [CrossRef] [Green Version]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Zanuy, D.; Kotla, R.; Nussinov, R.; Teesalu, T.; Sugahara, K.N.; Aleman, C.; Haspel, N. Sequence dependence of C-end rule peptides in binding and activation of neuropilin-1 receptor. J. Struct. Biol. 2013, 182, 78–86. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [Green Version]
- Willmore, A.M.; Simon-Gracia, L.; Toome, K.; Paiste, P.; Kotamraju, V.R.; Molder, T.; Sugahara, K.N.; Ruoslahti, E.; Braun, G.B.; Teesalu, T. Targeted silver nanoparticles for ratiometric cell phenotyping. Nanoscale 2016, 8, 9096–9101. [Google Scholar] [CrossRef] [Green Version]
- Wonder, E.; Simon-Gracia, L.; Scodeller, P.; Majzoub, R.N.; Kotamraju, V.R.; Ewert, K.K.; Teesalu, T.; Safinya, C.R. Competition of charge-mediated and specific binding by peptide-tagged cationic liposome-DNA nanoparticles in vitro and in vivo. Biomaterials 2018, 166, 52–63. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Fogal, V.; Zhang, L.; Krajewski, S.; Ruoslahti, E. Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008, 68, 7210–7218. [Google Scholar] [CrossRef] [Green Version]
- Simon-Gracia, L.; Scodeller, P.; Fuentes, S.S.; Vallejo, V.G.; Rios, X.; San Sebastian, E.; Sidorenko, V.; Di Silvio, D.; Suck, M.; De Lorenzi, F.; et al. Application of polymersomes engineered to target p32 protein for detection of small breast tumors in mice. Oncotarget 2018, 9, 18682–18697. [Google Scholar] [CrossRef] [Green Version]
- Paasonen, L.; Sharma, S.; Braun, G.B.; Kotamraju, V.R.; Chung, T.D.; She, Z.G.; Sugahara, K.N.; Yliperttula, M.; Wu, B.; Pellecchia, M.; et al. New p32/gC1qR Ligands for Targeted Tumor Drug Delivery. Chembiochem 2016, 17, 570–575. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Scodeller, P.; Braun, G.B.; de Mendoza, T.H.; Yamazaki, C.M.; Kluger, M.D.; Kitayama, J.; Alvarez, E.; Howell, S.B.; Teesalu, T.; et al. A tumor-penetrating peptide enhances circulation-independent targeting of peritoneal carcinomatosis. J. Control. Release 2015, 212, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Saalik, P.; Lingasamy, P.; Toome, K.; Mastandrea, I.; Rousso-Noori, L.; Tobi, A.; Simon-Gracia, L.; Hunt, H.; Paiste, P.; Kotamraju, V.R.; et al. Peptide-guided nanoparticles for glioblastoma targeting. J. Control. Release 2019, 308, 109–118. [Google Scholar] [CrossRef]
- Hunt, H.; Simon-Gracia, L.; Tobi, A.; Kotamraju, V.R.; Sharma, S.; Nigul, M.; Sugahara, K.N.; Ruoslahti, E.; Teesalu, T. Targeting of p32 in peritoneal carcinomatosis with intraperitoneal linTT1 peptide-guided pro-apoptotic nanoparticles. J. Control. Release 2017, 260, 142–153. [Google Scholar] [CrossRef]
- Simon-Gracia, L.; Hunt, H.; Teesalu, T. Peritoneal Carcinomatosis Targeting with Tumor Homing Peptides. Molecules 2018, 23, 1190. [Google Scholar] [CrossRef] [Green Version]
- Simon-Gracia, L.; Hunt, H.; Scodeller, P.; Gaitzsch, J.; Kotamraju, V.R.; Sugahara, K.N.; Tammik, O.; Ruoslahti, E.; Battaglia, G.; Teesalu, T. iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials 2016, 104, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Diaz Bessone, M.I.; Simon-Gracia, L.; Scodeller, P.; Ramirez, M.L.A.; Lago Huvelle, M.A.; Soler-Illia, G.; Simian, M. iRGD-guided tamoxifen polymersomes inhibit estrogen receptor transcriptional activity and decrease the number of breast cancer cells with self-renewing capacity. J. Nanobiotechnol. 2019, 17, 120. [Google Scholar] [CrossRef]
- Scodeller, P.; Asciutto, E.K. Targeting Tumors Using Peptides. Molecules 2020, 25, 808. [Google Scholar] [CrossRef] [Green Version]
- Simon-Gracia, L.; Sidorenko, V.; Uustare, A.; Ogibalov, I.; Tasa, A.; Tshubrik, O.; Teesalu, T. Novel Anthracycline Utorubicin for Cancer Therapy. Angew. Chem. Int. Ed. Engl. 2021, 60, 17018–17027. [Google Scholar] [CrossRef]
- Ikemoto, H.; Lingasamy, P.; Anton Willmore, A.M.; Hunt, H.; Kurm, K.; Tammik, O.; Scodeller, P.; Simon-Gracia, L.; Kotamraju, V.R.; Lowy, A.M.; et al. Hyaluronan-binding peptide for targeting peritoneal carcinomatosis. Tumor Biol. 2017, 39, 1010428317701628. [Google Scholar] [CrossRef] [Green Version]
- Simon-Gracia, L.; Savier, E.; Parizot, C.; Brossas, J.Y.; Loisel, S.; Teesalu, T.; Conti, F.; Charlotte, F.; Scatton, O.; Aoudjehane, L.; et al. Bifunctional Therapeutic Peptides for Targeting Malignant B Cells and Hepatocytes: Proof of Concept in Chronic Lymphocytic Leukemia. Adv. Ther. 2020, 3, 2000131. [Google Scholar] [CrossRef]
- Lu, X.Y.; Xi, T.; Lau, W.Y.; Dong, H.; Zhu, Z.; Shen, F.; Wu, M.C.; Cong, W.M. Hepatocellular carcinoma expressing cholangiocyte phenotype is a novel subtype with highly aggressive behavior. Ann. Surg. Oncol. 2011, 18, 2210–2217. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Iguchi, T.; Aishima, S.; Sanefuji, K.; Fujita, N.; Sugimachi, K.; Gion, T.; Taketomi, A.; Shirabe, K.; Maehara, Y.; Tsuneyoshi, M. Both fibrous capsule formation and extracapsular penetration are powerful predictors of poor survival in human hepatocellular carcinoma: A histological assessment of 365 patients in Japan. Ann. Surg. Oncol. 2009, 16, 2539–2546. [Google Scholar] [CrossRef]
- Decaens, T.; Roudot-Thoraval, F.; Badran, H.; Wolf, P.; Durand, F.; Adam, R.; Boillot, O.; Vanlemmens, C.; Gugenheim, J.; Dharancy, S.; et al. Impact of tumour differentiation to select patients before liver transplantation for hepatocellular carcinoma. Liver. Int. 2011, 31, 792–801. [Google Scholar] [CrossRef]
- Ziol, M.; Pote, N.; Amaddeo, G.; Laurent, A.; Nault, J.C.; Oberti, F.; Costentin, C.; Michalak, S.; Bouattour, M.; Francoz, C.; et al. Macrotrabecular-massive hepatocellular carcinoma: A distinctive histological subtype with clinical relevance. Hepatology 2018, 68, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Duvoux, C.; Roudot-Thoraval, F.; Decaens, T.; Pessione, F.; Badran, H.; Piardi, T.; Francoz, C.; Compagnon, P.; Vanlemmens, C.; Dumortier, J.; et al. Liver transplantation for hepatocellular carcinoma: A model including alpha-fetoprotein improves the performance of Milan criteria. Gastroenterology 2012, 143, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Akiba, J.; Nakashima, O.; Hattori, S.; Naito, Y.; Kusano, H.; Kondo, R.; Nakayama, M.; Tanikawa, K.; Todoroki, K.; Umeno, Y.; et al. The expression of arginase-1, keratin (K) 8 and K18 in combined hepatocellular-cholangiocarcinoma, subtypes with stem-cell features, intermediate-cell type. J. Clin. Pathol. 2016, 69, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Kataoka, H. Glypican 3-Targeted Therapy in Hepatocellular Carcinoma. Cancers 2019, 11, 1339. [Google Scholar] [CrossRef] [Green Version]
- Sempoux, C.; Chang, C.; Gouw, A.; Chiche, L.; Zucman-Rossi, J.; Balabaud, C.; Bioulac-Sage, P. Benign hepatocellular nodules: What have we learned using the patho-molecular classification. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 322–327. [Google Scholar] [CrossRef]
- Moudi, B.; Heidari, Z.; Mahmoudzadeh-Sagheb, H. Study of liver in HBV-related hepatocellular carcinoma: Stereology shows quantitative differences in liver structure. Eur. J. Histochem. 2018, 62, 2950. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Haber, P.K.; Puigvehi, M.; Castet, F.; Lourdusamy, V.; Montal, R.; Tabrizian, P.; Buckstein, M.; Kim, E.; Villanueva, A.; Schwartz, M.; et al. Evidence-based management of HCC: Systematic review and meta-analysis of randomized controlled trials (2002–2020). Gastroenterology 2021, 161, 879–898. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.J.; McFarlane, T.; Tully, S.; Wong, W.W.L. Lenvatinib Versus Sorafenib as First-Line Treatment of Unresectable Hepatocellular Carcinoma: A Cost-Utility Analysis. Oncologist 2019, 25, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahmad, E.; Rabbani, G.; Haque, S.; Arshad, M.; Khan, R.H. Identification and design of antimicrobial peptides for therapeutic applications. Curr. Protein Pept. Sci. 2012, 13, 211–223. [Google Scholar] [CrossRef]
- Rabbani, G.; Baig, M.H.; Ahmad, K.; Choi, I. Protein-protein Interactions and their Role in Various Diseases and their Prediction Techniques. Curr. Protein Pept. Sci. 2018, 19, 948–957. [Google Scholar] [CrossRef]
- Haesen, D.; Sents, W.; Lemaire, K.; Hoorne, Y.; Janssens, V. The Basic Biology of PP2A in Hematologic Cells and Malignancies. Front. Oncol. 2014, 4, 347. [Google Scholar] [CrossRef] [Green Version]
- Ciccone, M.; Calin, G.A.; Perrotti, D. From the Biology of PP2A to the PADs for Therapy of Hematologic Malignancies. Front. Oncol. 2015, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Kalev, P.; Sablina, A.A. Protein phosphatase 2A as a potential target for anticancer therapy. Anticancer Agents Med. Chem. 2011, 11, 38–46. [Google Scholar] [CrossRef]
- Switzer, C.H.; Cheng, R.Y.; Vitek, T.M.; Christensen, D.J.; Wink, D.A.; Vitek, M.P. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene 2011, 30, 2504–2513. [Google Scholar] [CrossRef] [Green Version]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 2013, 123, 4144–4157. [Google Scholar] [CrossRef]
- Carr, B.I.; Guerra, V. A Hepatocellular Carcinoma Aggressiveness Index and Its Relationship to Liver Enzyme Levels. Oncology 2016, 90, 215–220. [Google Scholar] [CrossRef]
- Carr, B.I.; Guerra, V.; Giannini, E.G.; Farinati, F.; Ciccarese, F.; Rapaccini, G.L.; Di Marco, M.; Benvegnu, L.; Zoli, M.; Borzio, F.; et al. A Liver Index and its Relationship to Indices of HCC Aggressiveness. J. Integr. Oncol. 2016, 5, 178. [Google Scholar] [CrossRef]
- Miao, H.Q.; Lee, P.; Lin, H.; Soker, S.; Klagsbrun, M. Neuropilin-1 expression by tumor cells promotes tumor angiogenesis and progression. FASEB J. 2000, 14, 2532–2539. [Google Scholar] [CrossRef] [Green Version]
- Jubb, A.M.; Strickland, L.A.; Liu, S.D.; Mak, J.; Schmidt, M.; Koeppen, H. Neuropilin-1 expression in cancer and development. J. Pathol. 2012, 226, 50–60. [Google Scholar] [CrossRef]
- Saha, P.; Datta, K. Multi-functional, multicompartmental hyaluronan-binding protein 1 (HABP1/p32/gC1qR): Implication in cancer progression and metastasis. Oncotarget 2018, 9, 10784–10807. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Asghar, U.; Meyer, T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J. Hepatol. 2012, 56, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Bos, C.L.; Kodach, L.L.; van den Brink, G.R.; Diks, S.H.; van Santen, M.M.; Richel, D.J.; Peppelenbosch, M.P.; Hardwick, J.C. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene 2006, 25, 6447–6456. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.; Wang, W.; Chai, K.; Yang, S.; Li, F.; Jiang, K. Combination treatment with triptolide and hydroxycamptothecin synergistically enhances apoptosis in A549 lung adenocarcinoma cells through PP2A-regulated ERK, p38 MAPKs and Akt signaling pathways. Int. J. Oncol. 2015, 46, 1007–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.S. Medical uses of mylabris in ancient China and recent studies. J. Ethnopharmacol. 1989, 26, 147–162. [Google Scholar] [CrossRef]
- Li, W.; Xie, L.; Chen, Z.; Zhu, Y.; Sun, Y.; Miao, Y.; Xu, Z.; Han, X. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 2010, 101, 1226–1233. [Google Scholar] [CrossRef] [PubMed]
- Kinch, M.S. An overview of FDA-approved biologics medicines. Drug Discov. Today 2015, 20, 393–398. [Google Scholar] [CrossRef]
- Carlson, S.G.; Eng, E.; Kim, E.G.; Perlman, E.J.; Copeland, T.D.; Ballermann, B.J. Expression of SET, an inhibitor of protein phosphatase 2A, in renal development and Wilms’ tumor. J. Am. Soc. Nephrol. 1998, 9, 1873–1880. [Google Scholar] [CrossRef]
- Chae, H.; Lim, J.; Kim, M.; Park, J.; Kim, Y.; Han, K.; Lee, S.; Min, W.S. Phenotypic and genetic characterization of adult T-cell acute lymphoblastic leukemia with del(9)(q34);SET-NUP214 rearrangement. Ann. Hematol. 2012, 91, 193–201. [Google Scholar] [CrossRef]
- Cristobal, I.; Garcia-Orti, L.; Cirauqui, C.; Cortes-Lavaud, X.; Garcia-Sanchez, M.A.; Calasanz, M.J.; Odero, M.D. Overexpression of SET is a recurrent event associated with poor outcome and contributes to protein phosphatase 2A inhibition in acute myeloid leukemia. Haematologica 2012, 97, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Cristobal, I.; Rincon, R.; Manso, R.; Carames, C.; Zazo, S.; Madoz-Gurpide, J.; Rojo, F.; Garcia-Foncillas, J. Deregulation of the PP2A inhibitor SET shows promising therapeutic implications and determines poor clinical outcome in patients with metastatic colorectal cancer. Clin. Cancer Res. 2015, 21, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Hung, M.H.; Chen, Y.L.; Chu, P.Y.; Shih, C.T.; Yu, H.C.; Tai, W.T.; Shiau, C.W.; Chen, K.F. Upregulation of the oncoprotein SET determines poor clinical outcomes in hepatocellular carcinoma and shows therapeutic potential. Oncogene 2016, 35, 4891–4902. [Google Scholar] [CrossRef]
- Fukukawa, C.; Shima, H.; Tanuma, N.; Ogawa, K.; Kikuchi, K. Up-regulation of I-2(PP2A)/SET gene expression in rat primary hepatomas and regenerating livers. Cancer Lett. 2000, 161, 89–95. [Google Scholar] [CrossRef]
- Agarwal, A.; MacKenzie, R.J.; Pippa, R.; Eide, C.A.; Oddo, J.; Tyner, J.W.; Sears, R.; Vitek, M.P.; Odero, M.D.; Christensen, D.J.; et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin. Cancer Res. 2014, 20, 2092–2103. [Google Scholar] [CrossRef] [Green Version]
- Hung, M.H.; Wang, C.Y.; Chen, Y.L.; Chu, P.Y.; Hsiao, Y.J.; Tai, W.T.; Chao, T.T.; Yu, H.C.; Shiau, C.W.; Chen, K.F. SET antagonist enhances the chemosensitivity of non-small cell lung cancer cells by reactivating protein phosphatase 2A. Oncotarget 2016, 7, 638–655. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Hung, M.H.; Shih, C.T.; Hsieh, F.S.; Kuo, C.W.; Tsai, M.H.; Chang, S.S.; Hsiao, Y.J.; Chen, L.J.; Chao, T.I.; et al. Antagonizing SET Augments the Effects of Radiation Therapy in Hepatocellular Carcinoma through Reactivation of PP2A-Mediated Akt Downregulation. J. Pharmacol. Exp. Ther. 2018, 366, 410–421. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age | Type Tumor | AFP | Log10 AFP | Partially Encapsulated (0/1) | Satellite Nodule (0/1) | Vascular Invasion (0/1) | Differentiation (1/2/3) 1 | Macrotrabecular (0/1) | Aggressiveness 2 | Aggression Class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 48.5 | Hepatocellular adenoma | 1.6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | Null |
| 2 | M | 65.3 | Necrotic lymph node | 2.6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | Null |
| 3 | F | 53.5 | Angiomyolipoma | 4.9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | Null |
| 4 | M | 56.5 | Microtrabecular and pseudoglandular, Nuclear grade 2 | 7.7 | 0 | 1 | 0 | 0 | 2 | 0 | 3 | Moderate |
| 5 | F | 47.4 | Microtrabecular | 1010 | 3 | 1 | 1 | 0 | 2 | 0 | 7 | High |
| 6 | F | 59.9 | Trabecular, Edmondson grade 2, nuclear grade 2 | 28,000 | 4 | 1 | 0 | 0 | 2 | 0 | 7 | High |
| 7 | M | 76.4 | Microtrabecular and pseudoglandular | 6662 | 3 | 1 | 0 | 0 | 2 | 1 | 7 | High |
| 8 | M | 47.1 | Macro-trabecular, Edmonson grade 3, nuclear grade 3 | 6 | 0 | 1 | 1 | 1 | 2 | 1 | 6 | High |
| 9 | M | 73.4 | Edmondson grade 3, nuclear grade 3 | 1.4 | 0 | 1 | 0 | 0 | 2 | 1 | 4 | Moderate |
| 10 | M | 67.3 | Edmonson grade 2, nuclear grade 2 | 6.4 | 0 | 1 | 0 | 0 | 2 | 0 | 3 | Moderate |
| 11 | M | 57.5 | Macrotrabecular | 5.1 | 0 | 1 | 1 | 1 | 2 | 1 | 6 | High |
| 12 | M | 69.3 | Trabecular, Edmonson grade 2, nuclear grade 2 | 343 | 2 | 1 | 0 | 2 | 2 | 0 | 6 | High |
| 13 | M | 68.7 | Edmonson grade 2 HCC, nuclear grade 2 | 2.5 | 0 | 1 | 0 | 2 | 2 | 0 | 3 | Moderate |
| 14 | M | 78.8 | Trabecular | 341 | 2 | 0 | 0 | 2 | 2 | 0 | 5 | Moderate |
| Peptide ID | Sequence |
|---|---|
| iRGD-IP | FITC -Ahx-ETVTLLVALKVRYRERIT-Ahx-CRGDKGPDC-CONH2 (C-C disulfide bond) |
| RPARPAR-IP | FITC -Ahx-ETVTLLVALKVRYRERIT-Ahx-RPARPAR-OH |
| LinTT1-IP | FITC -Ahx-ETVTLLVALKVRYRERIT-Ahx-AKRGARSTA-CONH2 |
| TT1-IP | FITC -Ahx-ETVTLLVALKVRYRERIT-Ahx-CKRGARSTC-CONH2 (C-C disulfide bond) |
| Patient | CK19 | HepPar | GPC3 | Nuclear β-Catenin | Glutamine Synthetase |
|---|---|---|---|---|---|
| 1 | − | + | − | + | − |
| 2 | necrosis | necrosis | necrosis | necrosis | necrosis |
| 3 | − | − | − | − | − |
| 4 | − | + | + | 10–20% | +++ |
| 5 | − | − | − | 0 | 0 |
| 6 | − | + | − | − | ++ |
| 7 | − | +++ | − | + | +++ |
| 8 | + | +++ | − | − | − |
| 9 | − | + | + | − | − |
| 10 | − | + | − | − | + |
| 11 | − | +++ | + | − | − |
| 12 | − | +++ | + | − | − |
| 13 | − | +++ | − | − | − |
| 14 | − | + | +++ | + | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savier, E.; Simon-Gracia, L.; Charlotte, F.; Tuffery, P.; Teesalu, T.; Scatton, O.; Rebollo, A. Bi-Functional Peptides as a New Therapeutic Tool for Hepatocellular Carcinoma. Pharmaceutics 2021, 13, 1631. https://doi.org/10.3390/pharmaceutics13101631
Savier E, Simon-Gracia L, Charlotte F, Tuffery P, Teesalu T, Scatton O, Rebollo A. Bi-Functional Peptides as a New Therapeutic Tool for Hepatocellular Carcinoma. Pharmaceutics. 2021; 13(10):1631. https://doi.org/10.3390/pharmaceutics13101631
Chicago/Turabian StyleSavier, Eric, Lorena Simon-Gracia, Frederic Charlotte, Pierre Tuffery, Tambet Teesalu, Olivier Scatton, and Angelita Rebollo. 2021. "Bi-Functional Peptides as a New Therapeutic Tool for Hepatocellular Carcinoma" Pharmaceutics 13, no. 10: 1631. https://doi.org/10.3390/pharmaceutics13101631
APA StyleSavier, E., Simon-Gracia, L., Charlotte, F., Tuffery, P., Teesalu, T., Scatton, O., & Rebollo, A. (2021). Bi-Functional Peptides as a New Therapeutic Tool for Hepatocellular Carcinoma. Pharmaceutics, 13(10), 1631. https://doi.org/10.3390/pharmaceutics13101631