
Preparation and Pharmacokinetic Characterization of an Anti-Virulence Compound Nanosuspensions

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of CCG-211790 Nanosuspensions
2.3. Dynamic Light Scattering (DLS)
2.4. Scanning Electron Microscopy
2.5. High-Performance Liquid Chromatography (HPLC) Analysis of Nanosuspensions
2.6. Stability of CCG-211790 Nanosuspensions
2.7. In Vitro Dissolution Study
2.8. Pilot PK Study of CCG-211790 Powder Suspension
2.9. PK Studies of Nanosuspensions
2.10. UPLC-MS/MS Analysis of Plasma Samples
2.11. Statistics
3. Results
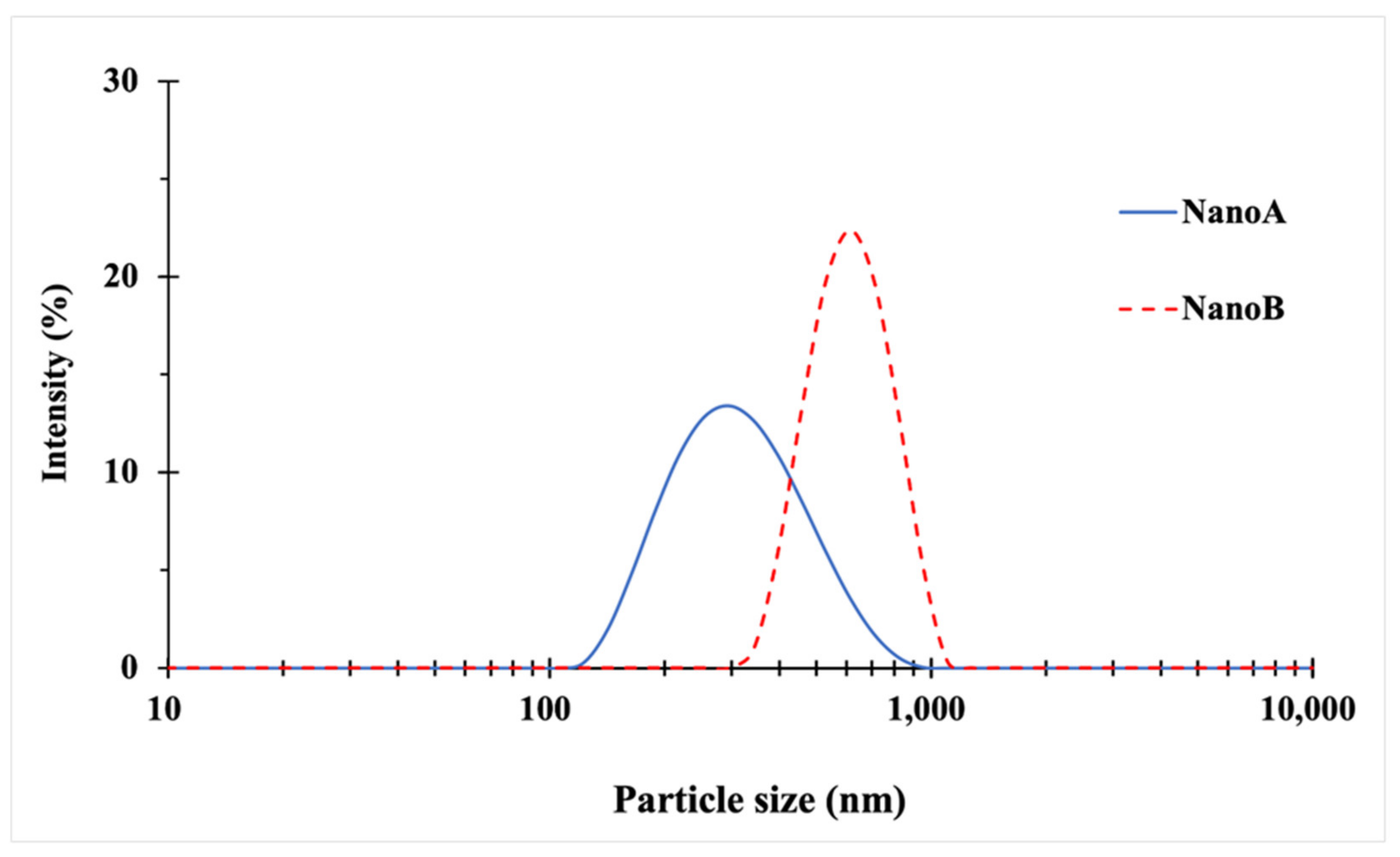
3.1. Particle Size and Morphology
3.2. Storage Stability of Nanosuspensions
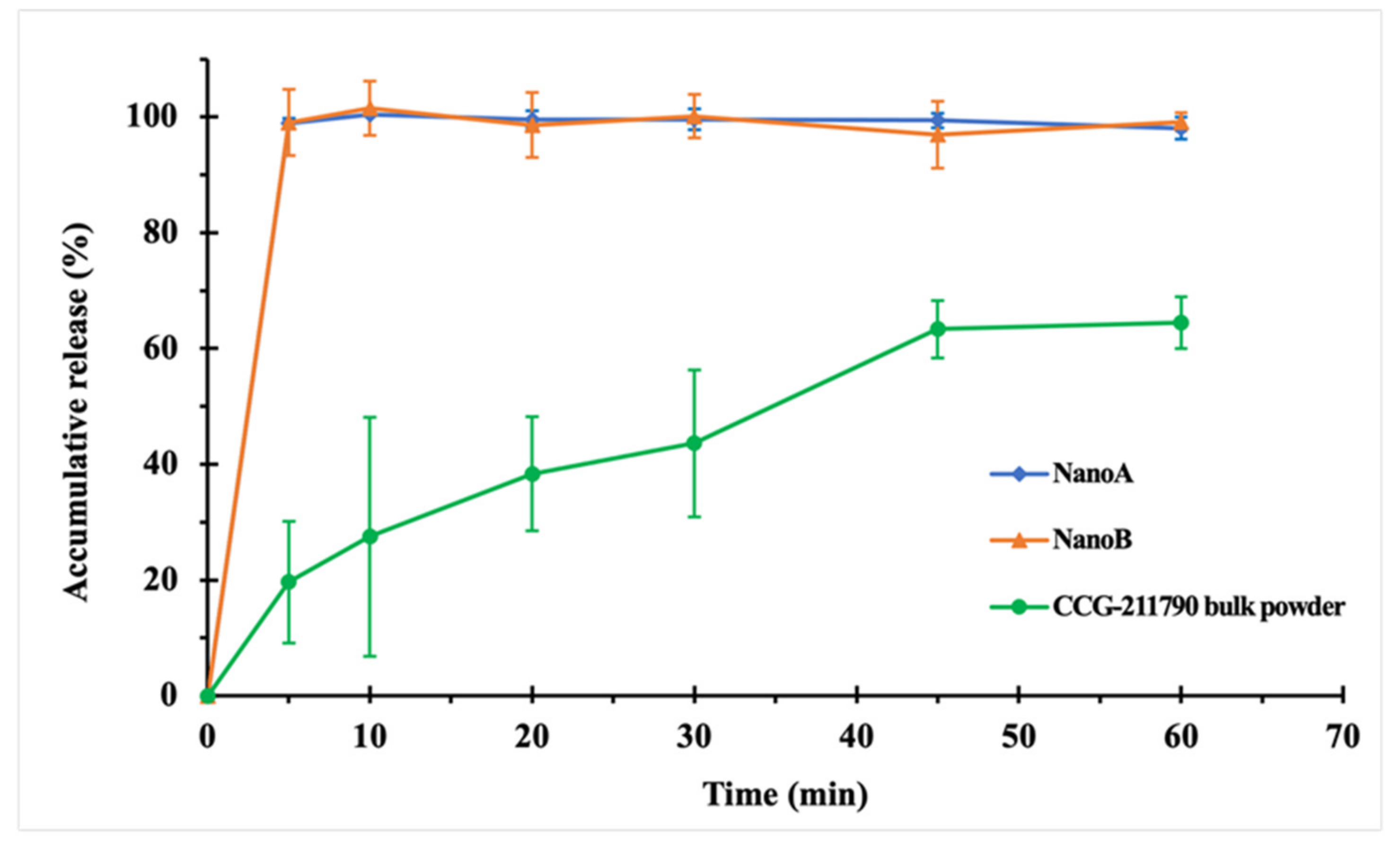
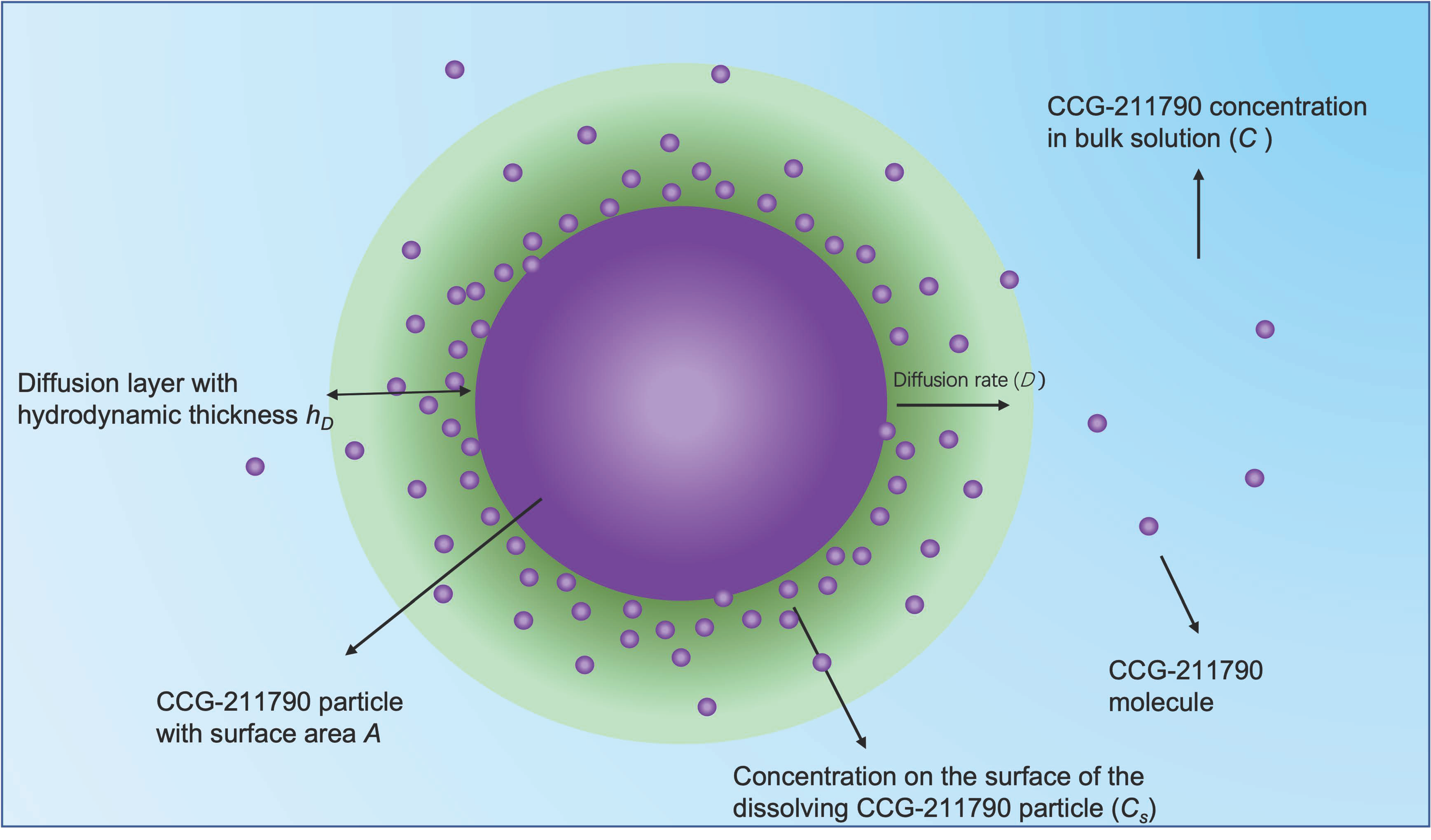
3.3. In Vitro Dissolution Studies
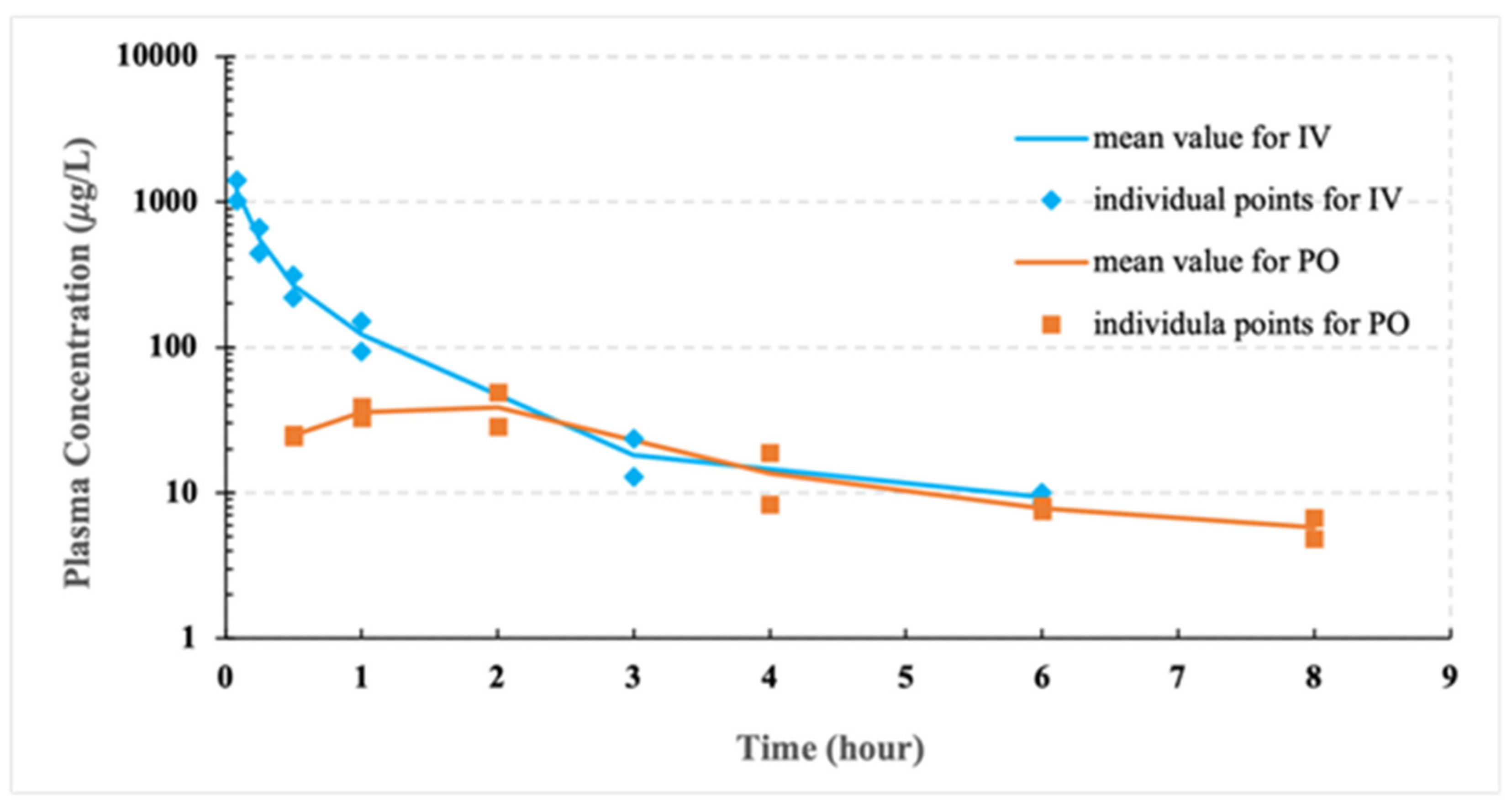
3.4. Pilot PK Studies
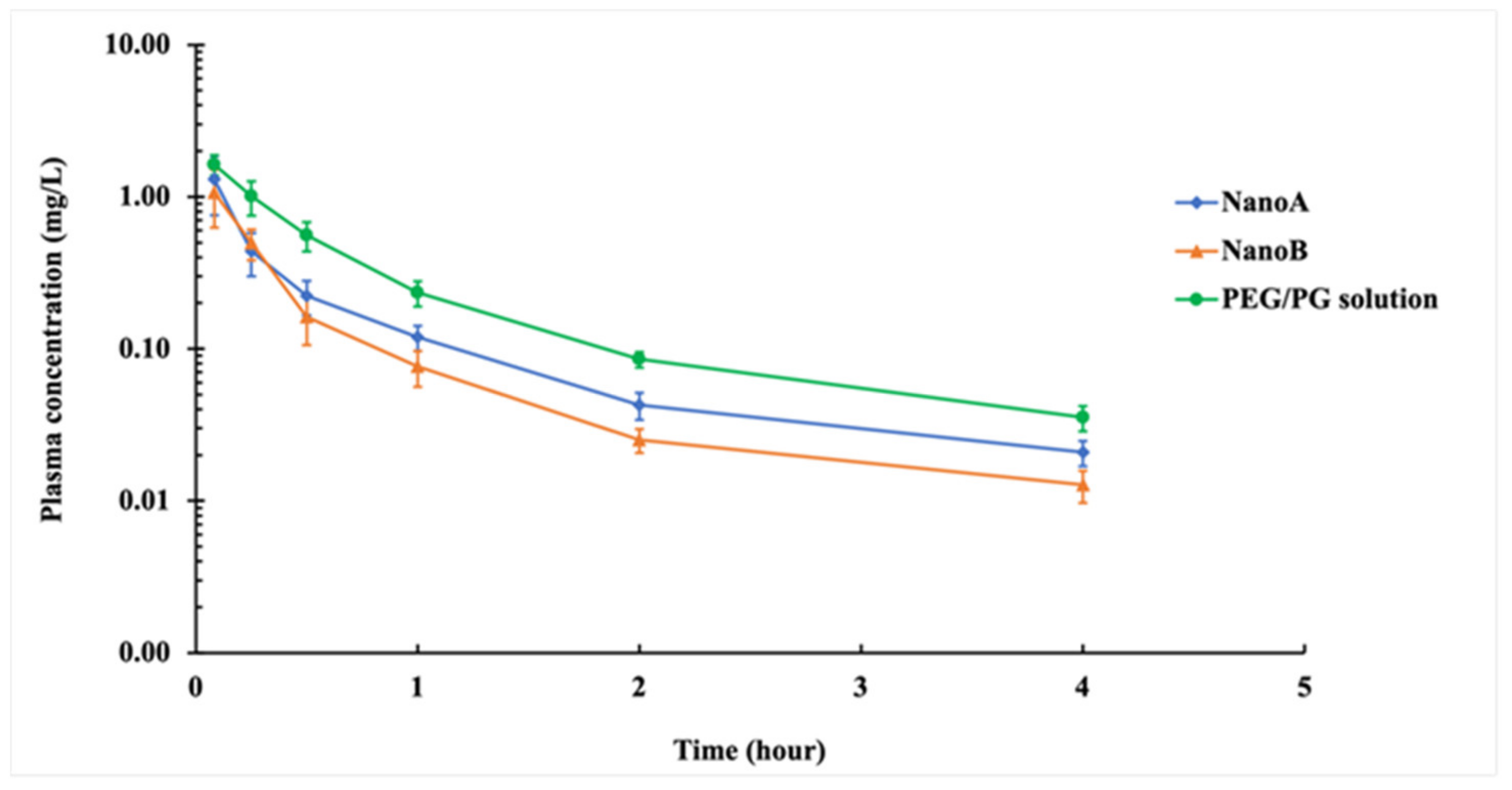
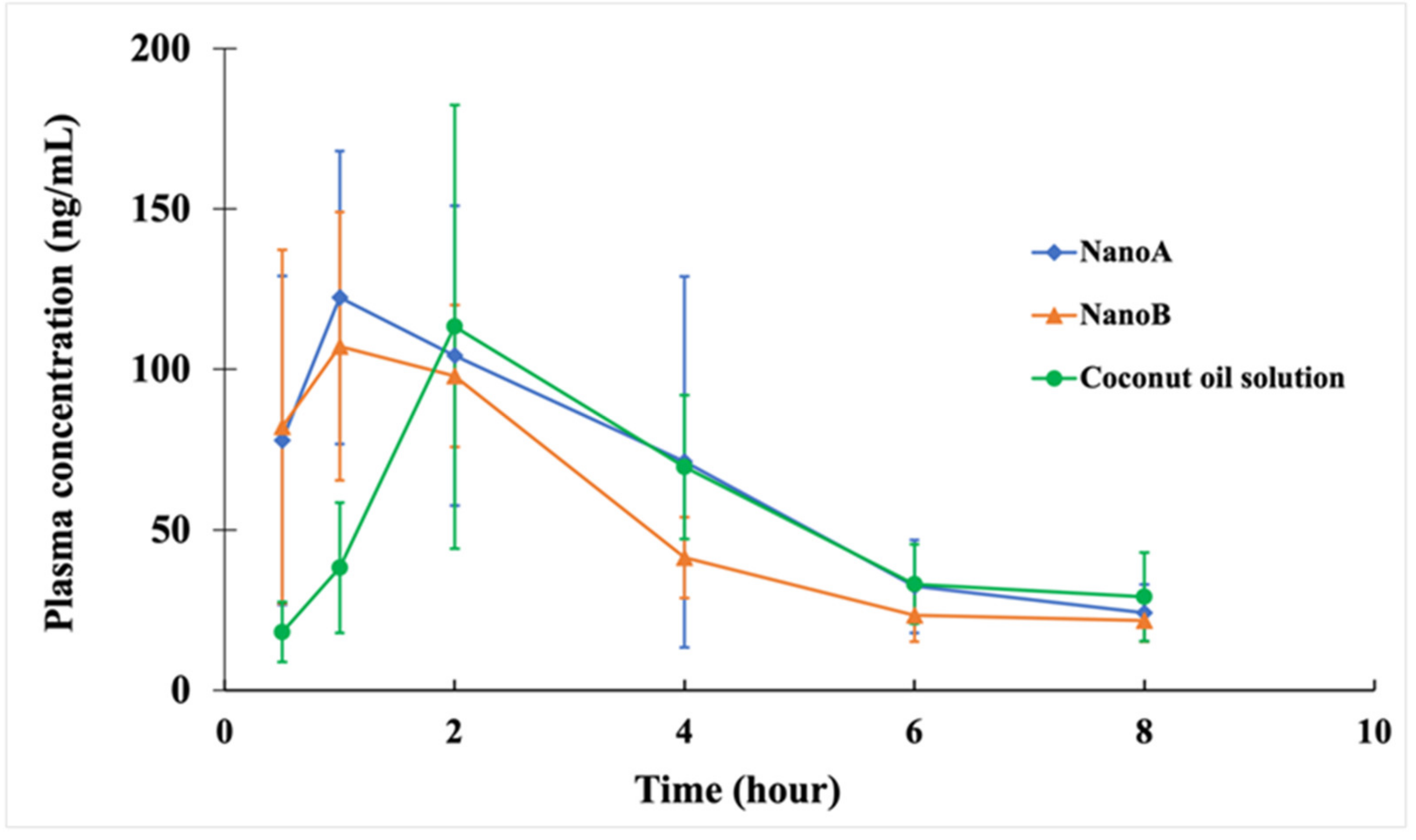
3.5. PK Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bush, K.; Courvalin, P.; Dantas, G.; Davies, J.; Eisenstein, B.; Huovinen, P.; Jacoby, G.A.; Kishony, R.; Kreiswirth, B.N.; Kutter, E.; et al. Tackling antibiotic resistance. Nat. Rev. Genet. 2011, 9, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Fair, R.J.; Tor, Y. Antibiotics and Bacterial Resistance in the 21st Century. Perspect. Med. Chem. 2014, 6, PMC.S14459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Perspect. Med. Chem. 2015, 40, 277–283. [Google Scholar]
- Ventola, C.L. The antibiotic resistance crisis: Part 2: Management strategies and new agents. Pharm. Therap. 2015, 40, 344–352. [Google Scholar]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Ma, Y.; Xu, Y.; Yestrepsky, B.D.; Sorenson, R.J.; Chen, M.; Larsen, S.D.; Sun, H. Novel Inhibitors of Staphylococcus aureus Virulence Gene Expression and Biofilm Formation. PLoS ONE 2012, 7, e47255. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Xu, Y.; Sitkiewicz, I.; Ma, Y.; Wang, X.; Yestrepsky, B.D.; Huang, Y.; Lapadatescu, M.C.; Larsen, M.J.; Larsen, S.D.; et al. Inhibitor of streptokinase gene expression improves survival after group A streptococcus infection in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3469–3474. [Google Scholar] [CrossRef] [Green Version]
- Yestrepsky, B.D.; Xu, Y.; Breen, M.E.; Li, X.; Rajeswaran, W.G.; Ryu, J.G.; Sorenson, R.J.; Tsume, Y.; Wilson, M.W.; Zhang, W.; et al. Novel inhibitors of bacterial virulence: Development of 5,6-dihydrobenzo[h]quinazolin-4(3H)-ones for the inhibition of group A streptococcal streptokinase expression. Bioorg. Med. Chem. 2013, 21, 1880–1897. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Qi, F.; Yu, H.; Yestrepsky, B.D.; Larsen, S.D.; Shi, H.; Ji, J.; Anderson, D.W.; Li, H.; Sun, H. Physicochemical properties and formulation development of a novel compound inhibiting Staphylococcus aureus biofilm formation. PLoS ONE 2021, 16, e0246408. [Google Scholar] [CrossRef]
- Pires, S.; Jacquet, R.; Parker, D. Inducible Costimulator Contributes to Methicillin-Resistant Staphylococcus aureus Pneumonia. J. Infect. Dis. 2018, 218, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Choe, D.; Szubin, R.; Dahesh, S.; Cho, S.; Nizet, V.; Palsson, B.; Cho, B.-K. Genome-scale analysis of Methicillin-resistant Staphylococcus aureus USA300 reveals a tradeoff between pathogenesis and drug resistance. Sci. Rep. 2018, 8, 2215. [Google Scholar] [CrossRef] [Green Version]
- Choo, E.J.; Chambers, H.F. Treatment of Methicillin-Resistant Staphylococcus aureus Bacteremia. Infect. Chemother. 2016, 48, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.L.; Chan, L.; Konopka, C.I.; Burkitt, M.; Moffa, M.A.; Bremmer, D.N.; Murillo, M.A.; Watson, C.; Chan-Tompkins, N.H. Appropriateness of antibiotic management of uncomplicated skin and soft tissue infections in hospitalized adult patients. BMC Infect. Dis. 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Pulakat, L.; Anderson, D.W. Challenges and New Therapeutic Approaches in the Management of Chronic Wounds. Curr. Drug Targets 2020, 21, 1264–1275. [Google Scholar] [CrossRef]
- Metcalf, D.G.; Bowler, P.G. Biofilm delays wound healing: A review of the evidence. Burn. Trauma 2013, 1, 5–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinton, A.; Carter, T. Chronic Wound Biofilms: Pathogenesis and Potential Therapies. Lab. Med. 2015, 46, 277–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bas, S.; Kramer, M.; Stopar, D. Biofilm Surface Density Determines Biocide Effectiveness. Front. Microbiol. 2017, 8, 2443. [Google Scholar] [CrossRef]
- Hall, C.W.; Mah, T.-F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Mah, T.-F.C.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureusBiofilms Prevent Macrophage Phagocytosis and Attenuate Inflammation In Vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef] [Green Version]
- Jesaitis, A.J.; Franklin, M.J.; Berglund, D.; Sasaki, M.; Lord, C.I.; Bleazard, J.B.; Duffy, J.E.; Beyenal, H.; Lewandowski, Z. Compromised Host Defense on Pseudomonas aeruginosa Biofilms: Characterization of Neutrophil and Biofilm Interactions. J. Immunol. 2003, 171, 4329–4339. [Google Scholar] [CrossRef] [Green Version]
- Kumar, T.; Teo, I.; McCormick, B.B. Systemic Toxicity of Intraperitoneal Vancomycin. Case Rep. Nephrol. 2016, 2016, 3968690. [Google Scholar] [CrossRef] [Green Version]
- Cadle, R.M.; Mansouri, M.D.; O Darouiche, R. Vancomycin-Induced Elevation of Liver Enzyme Levels. Ann. Pharmacother. 2006, 40, 1186–1189. [Google Scholar] [CrossRef]
- Choi, Y.C.; Saw, S.; Soliman, D.; Bingham, A.L.; Pontiggia, L.; Hunter, K.; Chuang, L.; Siemianowski, L.A.; Ereshefsky, B.; Hollands, J.M. Intravenous Vancomycin Associated With the Development of Nephrotoxicity in Patients With Class III Obesity. Ann. Pharmacother. 2017, 51, 937–944. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Morakul, B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Russo, J.; Fiegel, J.; Brogden, N. Antibiotic Delivery Strategies to Treat Skin Infections When Innate Antimicrobial Defense Fails. Antibiotics 2020, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs. Pharmaceutics 2018, 10, 74. [Google Scholar] [CrossRef] [Green Version]
- Dizaj, S.M.; Vazifehasl, Z.; Salatin, S.; Adibkia, K.; Javadzadeh, Y. Nanosizing of drugs: Effect on dissolution rate. Res. Pharm. Sci. 2015, 10, 95–108. [Google Scholar]
- Murdande, S.B.; Shah, D.A.; Dave, R.H. Impact of Nanosizing on Solubility and Dissolution Rate of Poorly Soluble Pharmaceuticals. J. Pharm. Sci. 2015, 104, 2094–2102. [Google Scholar] [CrossRef]
- Al-Kassas, R.; Bansal, M.; Shaw, J. Nanosizing techniques for improving bioavailability of drugs. J. Control. Release 2017, 260, 202–212. [Google Scholar] [CrossRef]
- Kanthamneni, N.; Valiveti, S.; Patel, M.; Xia, H.; Tseng, Y.C. Enhanced bioavailability of danazol nanosuspensions by wet milling and high-pressure homogenization. Int. J. Pharm. Investig. 2016, 6, 218–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Zhang, Q.; Li, H.; Sun, Y.; Cheng, G.; Zou, M.; Piao, H. Increased bioavailability of efonidipine hydrochloride nanosuspensions by the wet-milling method. Eur. J. Pharm. Biopharm. 2018, 130, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Junghanns, J.U.A.H.; Muller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar]
- Shegokar, R.; Singh, K.K.; Mueller, R.H. Nevirapine nanosuspension: Comparative investigation of production methods. Nanotechnol. Dev. 2011, 1, e4. [Google Scholar] [CrossRef]
- Rabinow, B.E. Nanosuspensions in drug delivery. Nat. Rev. Drug Discov. 2004, 3, 785–796. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Y.; Wu, W. Injected nanocrystals for targeted drug delivery. Acta Pharm. Sin. B 2016, 6, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Salem, H.F. Sustained-release progesterone nanosuspension following intramuscular injection in ovariectomized rats. Int. J. Nanomed. 2010, 5, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Shi, Y.; Yan, Z.; Hao, H.; Zhang, Y.; Zhong, J.; Hou, H. Dosage Form Developments of Nanosuspension Drug Delivery System for Oral Administration Route. Curr. Pharm. Des. 2015, 21, 4355–4365. [Google Scholar] [CrossRef]
- Djebli, N.; Khier, S.; Griguer, F.; Coutant, A.-L.; Tavernier, A.; Fabre, G.; Leriche, C.; Fabre, D. Ocular Drug Distribution After Topical Administration: Population Pharmacokinetic Model in Rabbits. Eur. J. Drug Metab. Pharm. 2017, 42, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Rossi, I.; Sonvico, F.; McConville, J.T.; Rossi, F.; Fröhlich, E.; Zellnitz, S.; Rossi, A.; Del Favero, E.; Bettini, R.; Buttini, F. Nebulized coenzyme Q 10 nanosuspensions: A versatile approach for pulmonary antioxidant therapy. Eur. J. Pharm. Sci. 2018, 113, 159–170. [Google Scholar] [CrossRef]
- Du, J.; Li, X.; Zhao, H.; Zhou, Y.; Wang, L.; Tian, S.; Wang, Y. Nanosuspensions of poorly water-soluble drugs prepared by bottom-up technologies. Int. J. Pharm. 2015, 495, 738–749. [Google Scholar] [CrossRef]
- Sinha, B.; Müller, R.H.; Möschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef]
- Van Eerdenbrugh, B.; Van den Mooter, G.; Augustijns, P. Top-down production of drug nanocrystals: Nanosuspension stabilization, miniaturization and transformation into solid products. Int. J. Pharm. 2008, 364, 64–75. [Google Scholar] [CrossRef]
- Keck, C.M.; Müller, R.H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur. J. Pharm. Biopharm. 2006, 62, 3–16. [Google Scholar] [CrossRef]
- Thomas, J.C. The determination of log normal particle size distributions by dynamic light scattering. J. Colloid Interface Sci. 1987, 117, 187–192. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Kaszuba, M.; McKnight, D.; Connah, M.T.; McNeil-Watson, F.K.; Nobbmann, U. Measuring sub nanometre sizes using dynamic light scattering. J. Nanopart. Res. 2008, 10, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hollis, C.P.; Zhang, Q.; Li, T. Preparation and antitumor study of camptothecin nanocrystals. Int. J. Pharm. 2011, 415, 293–300. [Google Scholar] [CrossRef]
- Arunkumar, N.; Deecaraman, M.; Rani, C. Nanosuspension technology and its applications in drug delivery. Asian J. Pharm. Free. 2014, 3, 168–173. [Google Scholar] [CrossRef]
- Shaal, L.A.; Mishra, P.R.; Muller, R.H.; Keck, C.M. Nanosuspensions of hesperetin: Preparation and characterization. Die Pharm. Int. J. Pharm. Sci. 2014, 69, 173–182. [Google Scholar] [CrossRef]
- Buxton, I.L.; Benet, L.Z. Pharmacokinetics: The dynamics of drug absorption, distribution, metabolism and elimination. In Good-Man & Gilman’s the Pharmacological Basis of Therapeutic, 12th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Abo-El-Sooud, K. Absolute and Relative Bioavailability. In Drug Discovery and Evaluation: Methods in Clinical Pharmacology; Hock, F.J., Gralinski, M.R., Eds.; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2018; pp. 1–7. [Google Scholar]
- Guo, Z.; Zhang, M.; Li, H.; Wang, J.; Kougoulos, E. Effect of ultrasound on anti-solvent crystallization process. J. Cryst. Growth 2005, 273, 555–563. [Google Scholar] [CrossRef]
- Hielscher, T. Ultrasonic production of nano-size dispersions and emulsions. arXiv 2007, arXiv:0708.1831. [Google Scholar]
- Na Kim, H.; Suslick, K.S. The Effects of Ultrasound on Crystals: Sonocrystallization and Sonofragmentation. Crystals 2018, 8, 280. [Google Scholar] [CrossRef] [Green Version]
- Dokoumetzidis, A.; Macheras, P. A century of dissolution research: From Noyes and Whitney to the Biopharmaceutics Classification System. Int. J. Pharm. 2006, 321, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Uchegbu, I. Emulsions and Nanosuspensions for the Formulation of Poorly Soluble Drugs, Edited by R.H. Muller, S. Benita, B. Bohm, Medpharm Scientific Publishers, Stuttgart, ISBN 3 88763 069. Int. J. Pharm. 2001, 212, 143–144. [Google Scholar] [CrossRef]
- Sugano, K. Theoretical comparison of hydrodynamic diffusion layer models used for dissolution simulation in drug discovery and development. Int. J. Pharm. 2008, 363, 73–77. [Google Scholar] [CrossRef]
- Plakkot, S.; de Matas, M.; York, P.; Saunders, M.; Sulaiman, B. Comminution of ibuprofen to produce nano-particles for rapid dissolution. Int. J. Pharm. 2011, 415, 307–314. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, D.; Jiao, Y.; Guo, H.; Zheng, D.; Jia, L.; Duan, C.; Liu, Y.; Tian, X.; Shen, J.; et al. In vitro and in vivo evaluation of riccardin D nanosuspensions with different particle size. Colloids Surf. B Biointerfaces 2013, 102, 620–626. [Google Scholar] [CrossRef]
- Yadav, V.; Yadav, A. Improvement of solubility and dissolution of indomethacin by liquisolid and compaction granulation technique. J. Pharm. Sci. Res. 2009, 1, 44. [Google Scholar]
- Gao, L.; Zhang, D.; Chen, M.; Duan, C.; Dai, W.; Jia, L.; Zhao, W. Studies on pharmacokinetics and tissue distribution of oridonin nanosuspensions. Int. J. Pharm. 2008, 355, 321–327. [Google Scholar] [CrossRef]
- Schmidt, L.E.; Dalhoff, K. Food-Drug Interactions. Drugs 2002, 62, 1481–1502. [Google Scholar] [CrossRef]
- Singh, B.N. Effects of Food on Clinical Pharmacokinetics. Clin. Pharm. 1999, 37, 213–255. [Google Scholar] [CrossRef]
- Welling, P.G. Effects of Food on Drug Absorption. Annu. Rev. Nutr. 1996, 16, 383–415. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle uptake: The phagocyte problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J. Molecular Structure of Tight Junctions and Their Role in Epithelial Transport. Physiology 2001, 16, 126–130. [Google Scholar] [CrossRef]
- Avdeef, A. Absorption and Drug Development: Solubility, Permeability, and Charge State; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Wang, H.; Li, Q.; Reyes, S.; Zhang, J.; Xie, L.; Melendez, V.; Hickman, M.; Kozar, M.P. Formulation and Particle Size Reduction Improve Bioavailability of Poorly Water-Soluble Compounds with Antimalarial Activity. Malar. Res. Treat. 2013, 2013, 769234. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Liu, G.; Ma, J.; Wang, X.; Zhou, L.; Li, X. Drug nanocrystals: In vivo performances. J. Control. Release 2012, 160, 418–430. [Google Scholar] [CrossRef]
- Patravale, V.B.; Date, A.; Kulkarni, R.M. Nanosuspensions: A promising drug delivery strategy. J. Pharm. Pharmacol. 2004, 56, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tan, T.; Gao, L.; Zhao, W.; Wang, P. Preparation of Azithromycin Nanosuspensions by High Pressure Homogenization and its Physicochemical Characteristics Studies. Drug Dev. Ind. Pharm. 2007, 33, 569–575. [Google Scholar] [CrossRef]
- Yadollahi, R.; Vasilev, K.; Simovic, S. Nanosuspension Technologies for Delivery of Poorly Soluble Drugs. J. Nanomater. 2015, 2015, 1. [Google Scholar] [CrossRef]
- Santos, A.C.; Pattekari, P.; Jesus, S.; Veiga, F.; Lvov, Y.; Ribeiro, A. Sonication-Assisted Layer-by-Layer Assembly for Low Solubility Drug Nanoformulation. ACS Appl. Mater. Interfaces 2015, 7, 11972–11983. [Google Scholar] [CrossRef]
- Nguyen, D.N.; Clasen, C.; Mooter, G.V.D. Encapsulating darunavir nanocrystals within Eudragit L100 using coaxial electrospraying. Eur. J. Pharm. Biopharm. 2017, 113, 50–59. [Google Scholar] [CrossRef]
- Kar, M.; Chourasiya, Y.; Maheshwari, R.; Tekade, R.K. Chapter 2-Current Developments in Excipient Science: Implication of Quantitative Selection of Each Excipient in Product Development. In Basic Fundamentals of Drug Delivery; Tekade, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 29–83. [Google Scholar]
- Mozafari, M. Nanotechnology in Wound Care: One Step Closer to the Clinic. Mol. Ther. 2018, 26, 2085–2086. [Google Scholar] [CrossRef] [Green Version]
- Pelgrift, R.Y.; Friedman, A.J. Nanotechnology as a therapeutic tool to combat microbial resistance. Adv. Drug Deliv. Rev. 2013, 65, 1803–1815. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Duration (Week) | Z-Average (nm) | Polydispersity Index | Zeta Potential (mV) | |||
|---|---|---|---|---|---|---|
| 4 °C | RT | 4 °C | RT | 4 °C | RT | |
| 0 | 315 ± 6 | 315 ± 6 | 0.18 ± 0.03 | 0.18 ± 0.03 | −42.3 ± 4.2 | −42.3 ± 4.2 |
| 1 | 311 ± 1 | 310 ± 3 | 0.21 ± 0.01 | 0.22 ± 0.01 | −49.3 ± 0.5 | −48.4 ± 0.3 |
| 2 | 309 ± 3 | 316 ± 7 | 0.20 ± 0.03 | 0.20 ± 0.03 | −49.4 ± 1.3 | −49.5 ± 0.9 |
| 4 | 314 ± 2 | 322 ± 6 | 0.21 ± 0.01 | 0.23 ± 0.01 | −46.1 ± 0.4 | −52.3 ± 0.7 |
| 6 | 315 ± 3 | 319 ± 5 | 0.21 ± 0.01 | 0.20 ± 0.03 | −45.4 ± 0.4 | −51.3 ± 0.3 |
| Duration (Week) | Z-Average (nm) | Polydispersity Index | Zeta Potential (mV) | |||
|---|---|---|---|---|---|---|
| 4 °C | RT | 4 °C | RT | 4 °C | RT | |
| 0 | 915 ± 24 | 915 ± 24 | 0.52 ± 0.03 | 0.52 ± 0.03 | −42.2 ± 0.2 | −42.2 ± 0.2 |
| 1 | 900 ± 99 | 947 ± 60 | 0.58 ± 0.12 | 0.62 ± 0.02 * | −49.5 ± 0.5 * | −50.4 ± 0.7 * |
| 2 | 911 ± 37 | 912 ± 66 | 0.61 ± 0.01 * | 0.62 ± 0.07 | −44.9 ± 0.6 * | −49.1 ± 0.7 * |
| 4 | 966 ± 89 | 878 ± 17 | 0.59 ± 0.03 | 0.54 ± 0.04 | −47.7 ± 1.0 * | −50.2 ± 0.3 * |
| 6 | 856 ± 17 * | 891 ± 24 | 0.57 ± 0.05 | 0.60 ± 0.05 | −46.4 ± 1.4 * | −50.0 ± 1.5 * |
| PK Parameters | Pilot Study for IV |
|---|---|
| Cmax (mg L−1) | 1.21 ± 0.15 |
| AUC0∓∞ (mg L−1 h) | 0.67 ± 0.18 |
| MRT0∓∞ (h) | 0.95 ± 0.07 |
| t1/2β (h) | 1.44 ± 0.15 |
| Vz (L kg−1) | 8.07 ± 2.92 |
| CL (L h−1 kg−1) | 3.85 ± 1.01 |
| PK Parameters | Pilot Study for PO |
|---|---|
| Cmax (mg L−1) | 0.04 ± 0.01 |
| Tmax (h) | 1.50 ± 0.71 |
| AUC0∓∞ (mg L−1 h) | 0.18 ± 0.03 |
| MRT0∓∞ (h) | 4.75 ± 1.20 |
| t1/2β (h) | 3.88 ± 1.68 |
| Vz /F (L kg−1) | 168 ± 101 |
| CL/F (L h−1 kg−1) | 28.8 ± 5.6 |
| FIV | 13.4% |
| PK Parameters | PEG/PG | NanoA | NanoB |
|---|---|---|---|
| Cmax (mg L−1) | 1.63 ± 0.25 | 1.31 ± 0.55 | 1.07 ± 0.44 * |
| AUC0∓∞ (mg L−1 h) | 1.11 ± 0.14 | 0.64 ± 0.19 * | 0.49 ± 0.12 * |
| MRT0∓∞ (h) | 0.98 ± 0.17 | 0.90 ± 0.14 | 0.77 ± 0.21 |
| t1/2β (h) | 1.13 ± 0.27 | 1.08 ± 0.18 | 1.06 ± 0.27 |
| Vz (L kg−1) | 3.66 ± 0.93 | 6.44 ± 1.84 * | 8.37 ± 3.29 * |
| CL (L h−1 kg−1) | 2.28 ± 0.27 | 4.21 ± 1.28 * | 5.28 ± 1.01 * |
| PK Parameters | Coconut Oil | NanoA | NanoB |
|---|---|---|---|
| Cmax (mg L−1) | 0.12 ± 0.07 | 0.13 ± 0.05 | 0.12 ± 0.03 |
| Tmax (h) | 2.33 ± 0.82 | 1.58 ± 1.28 | 1.08 ± 0.49 * |
| AUC0∓∞ (mg L−1 h) | 0.63 ± 0.27 | 0.63 ± 0.17 | 0.50 ± 0.08 |
| MRT0∓∞ (h) | 6.59 ± 3.22 | 5.00 ± 1.14 | 4.34 ± 1.03 |
| t1/2β (h) | 3.82 ± 2.23 | 3.24 ± 1.36 | 2.61 ± 0.45 |
| Vz /F (L kg−1) | 46.1 ± 24.6 | 41.1 ± 21.7 | 38.2 ± 8.9 |
| CL/F (L h−1 kg−1) | 8.99 ± 3.14 | 8.53 ± 2.88 | 10.14 ± 1.64 |
| FPEG/PG | 28.4% | 28.6% | 22.7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Qi, F.; He, X.; Shi, H.; Anderson, D.W.; Li, H.; Sun, H. Preparation and Pharmacokinetic Characterization of an Anti-Virulence Compound Nanosuspensions. Pharmaceutics 2021, 13, 1586. https://doi.org/10.3390/pharmaceutics13101586
Wang N, Qi F, He X, Shi H, Anderson DW, Li H, Sun H. Preparation and Pharmacokinetic Characterization of an Anti-Virulence Compound Nanosuspensions. Pharmaceutics. 2021; 13(10):1586. https://doi.org/10.3390/pharmaceutics13101586
Chicago/Turabian StyleWang, Nan, Feng Qi, Xiaolong He, Honglan Shi, David W. Anderson, Hao Li, and Hongmin Sun. 2021. "Preparation and Pharmacokinetic Characterization of an Anti-Virulence Compound Nanosuspensions" Pharmaceutics 13, no. 10: 1586. https://doi.org/10.3390/pharmaceutics13101586
APA StyleWang, N., Qi, F., He, X., Shi, H., Anderson, D. W., Li, H., & Sun, H. (2021). Preparation and Pharmacokinetic Characterization of an Anti-Virulence Compound Nanosuspensions. Pharmaceutics, 13(10), 1586. https://doi.org/10.3390/pharmaceutics13101586