
Histological Changes in Renal, Hepatic and Cardiac Tissues of Wistar Rats after 6 Weeks Treatment with Bipyridine Gold (III) Complex with Dithiocarbamate Ligands

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
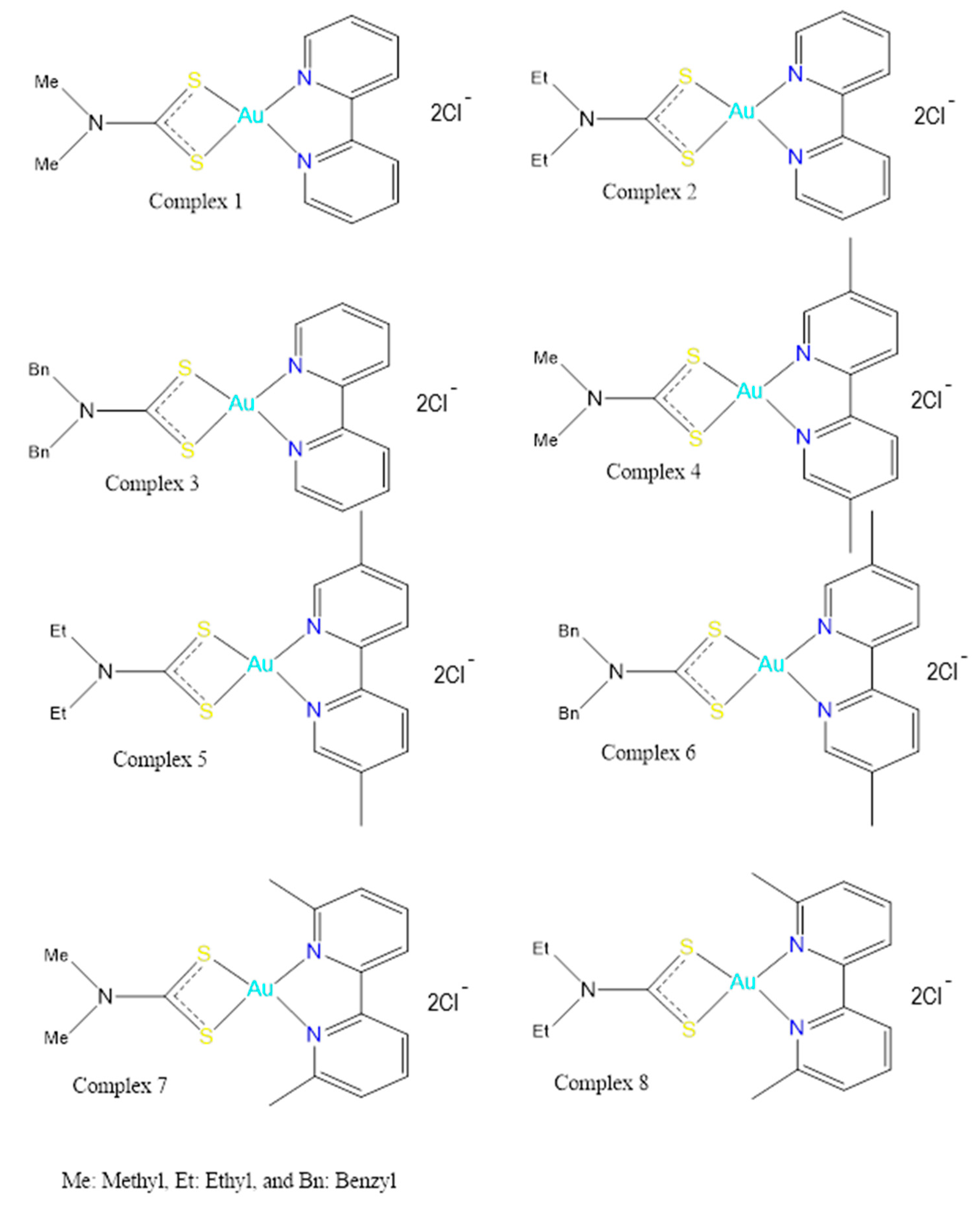
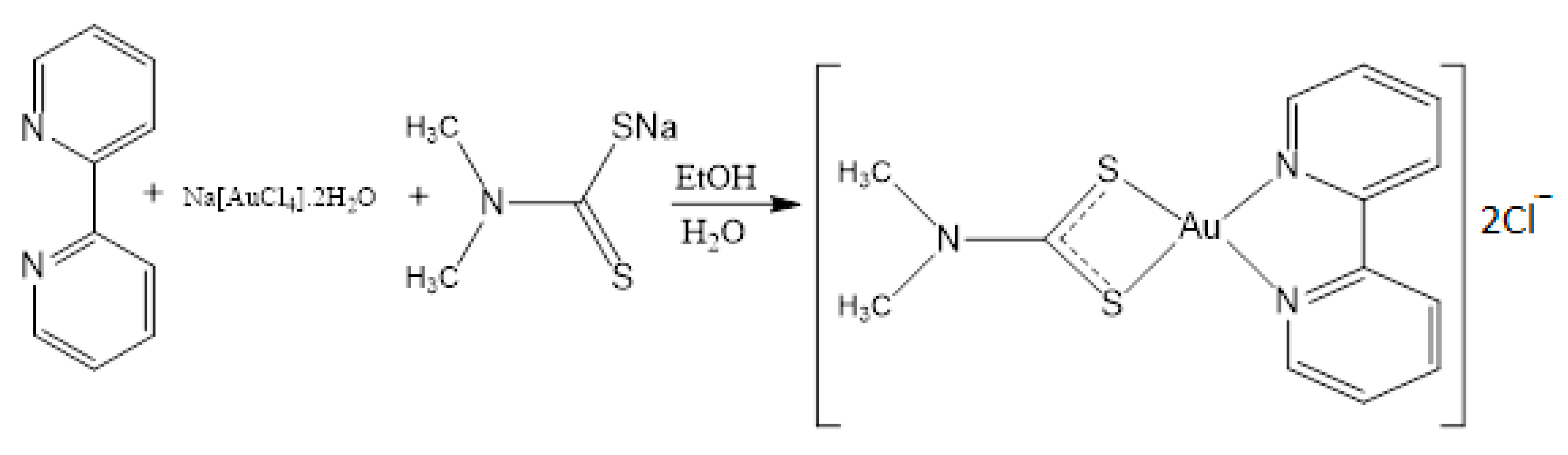
2.1. Syntheis of Complex (1)
2.2. Acute Toxicity Study (Single Dose)
2.3. Subacute Toxicity Study
2.4. Histopathological Evaluation
2.5. Analysis
3. Results
3.1. Acute Toxicity Study
3.2. Sub-Acute Toxicity Study
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kostova, I. Platinum complexes as anticancer agents. Recent Patent Anticancer. Drug Discov. 2006, 1, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, L.; Ghiselli, S.; Guaran, V.; Chicca, M.; Simoni, E.; Olivetto, E.; Lelli, G.; Martini, A. Correlation of adverse effects of cisplatin administration in patients affected by solid tumours: A retrospective evaluation. Oncol. Rep. 2013, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casini, A.; Wai-Yin Sun, R.; Ott, I. Medicinal chemistry of gold anticancer metallodrugs (Chapter-7). In Metallo-Drugs: Development and Action of Anticancer Agents; Sigel, A., Sigel, H., Freisinger, E., Sigel, R., Eds.; De Gruyter: Berlin, Germany; Boston, FL, USA, 2018; pp. 199–218. [Google Scholar] [CrossRef]
- Fernández-Moreira, V.; Herrera, R.P.; Gimeno, M.C. Anticancer properties of gold complexes with biologically relevant ligands. Pure Appl. Chem. 2019, 91, 247–269. [Google Scholar] [CrossRef]
- Yang, Z.; Jiang, G.; Xu, Z.; Zhao, S.; Liu, W. Advances in alkynyl gold complexes for use as potential anticancer agents. Coord. Chem. Rev. 2020, 423, 213492. [Google Scholar] [CrossRef]
- Olszewski, U.; Hamilton, G. A better platinum-based anticancer drug yet to come? Anticancer Agents Med. Chem. 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Janković, S.M.; Djeković, A.; Bugarčić, Z.; Janković, S.V.; Lukić, G.; Folić, M.; Canović, D. Effects of aurothiomalate and gold(III) complexes on spontaneous motility of isolated human oviduct. Biometals 2012, 25, 919–925. [Google Scholar] [CrossRef]
- Kalayda, G.V.; Wagner, C.H.; Jaehde, U. Relevance of copper transporter 1 for cisplatin resistance in human ovarian carcinoma cells. J. Inorg. Biochem. 2012, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buckley, R.G.; Elsome, A.M.; Fricker, S.P.; Henderson, G.R.; Theobald, B.R.; Parish, R.V.; Howe, B.P.; Kelland, L.R. Antitumor properties of some 2-[(dimethylamino)methyl]phenylgold(III) complexes. J. Med. Chem. 1996, 39, 5208–5214. [Google Scholar] [CrossRef]
- Calamai, P.; Carotti, S.; Guerri, A.; Mazzei, T.; Messori, L.; Mini, E.; Orioli, P.; Speroni, G.P. Cytotoxic effects of gold(III) complexes on established human tumor cell lines sensitive and resistant to cisplatin. Anticancer Drug Des. 1998, 13, 67–80. [Google Scholar]
- Ahmed, A.; Al Tamimi, D.M.; Isab, A.A.; Alkhawajah, A.M.; Shawarby, M.A. Histological changes in kidney and liver of rats due to gold (III) compound [Au(en)Cl(2)]Cl. PLoS ONE 2012, 7, e51889. [Google Scholar]
- Marzano, C.; Ronconi, L.; Chiara, F.; Giron, M.C.; Faustinelli, I.; Cristofori, P.; Trevisan, A.; Fregona, D. Gold (III)-dithiocarbamato anticancer agents: Activity, toxicology and histopathological studies in rodents. Int. J. Can. 2011, 129, 487–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altaf, M.; Monim-Ul-Mehboob, M.; Kawde, A.N.; Corona, G.; Larcher, R.; Ogasawara, M.; Casagrande, N.; Celegato, M.; Borghese, C.; Siddik, Z.H.; et al. New bipyridine gold(III) dithiocarbamate-containing complexes exerted a potent anticancer activity against cisplatin-resistant cancer cells independent of p53 status. Oncotarget 2017, 8, 490–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OECD Series on Principles of Good Laboratory Practice (GLP) and Compliance Monitoring. Available online: http://www.oecd.org/chemicalsafety/testing/oecdseriesonprinciplesofgoodlaboratorypracticeglpandcompliancemonitoring.htm (accessed on 2 October 2019).
- Ghosh, M.N. Satistical Analysis, Fundamentals of Experimental Pharmacology, 2nd ed.; Scientific Book Agency: Calcutta, India, 1984; p. 189. [Google Scholar]
- Miller, L.C.; Tainter, M.L. Estimation of ED50 and its error by means of log-probit graph paper. Proc. Soc. Exp. Biol. Med. 1944, 57, 261. [Google Scholar] [CrossRef]
- Underwood, J.C.E. Histochemistry. Theoretical and Applied. Vol. 2: Analytical Technology Pearse AGE, 4th ed.; Churchill Livingstone: Edinburgh, UK, 1985. [Google Scholar]
- Zhang, J.; Brown, R.P.; Shaw, M.; Vaidya, V.S.; Zhou, Y.; Espandiari, P.; Sadrieh, N.; Stratmeyer, M.; Keenan, J.; Kilty, C.G.; et al. Immunolocalization of Kim-1, RPA-1, and RPA-2 in Kidney of Gentamicin-, Mercury-, or Chromium-treated Rats: Relationship to Renal Distributions of iNOS and Nitrotyrosine. Toxicol. Pathol. 2008, 36, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Kakar, S. Histological patterns in drug-induced liver disease. J. Clin. Pathol. 2009, 62, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.Y.; Segarra, I.; Chakravarthi, S.; Akram, S.; Judson, J.P. Histopathology and biochemistry analysis of the interaction between sunitinib and paracetamol in mice. BMC Pharmacol. 2010, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Faqi, A.S. A Comprehensive Guide to Toxicology in Preclinical Drug Development; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Izzedine, H.; Perazella, M.A. Anticancer Drug-Induced Acute Kidney Injury. Kidney Int. Rep. 2017, 2, 504–514. [Google Scholar] [CrossRef] [Green Version]
- Arany, I.; Safirstein, R.L. Cisplatin nephrotoxicity. Semin. Nephrol. 2003, 23, 460–464. [Google Scholar] [CrossRef]
- Joybari, A.Y.; Sarbaz, S.; Azadeh, P.; Mirafsharieh, S.A.; Rahbari, A.; Farasatinasab, M.; Mokhtari, M. Oxaliplatin-induced renal tubular vacuolization. Ann. Pharmacother. 2014, 48, 796–800. [Google Scholar] [CrossRef]
- Laftavi, M.R.; Weber-Shrikant, E.; Kohli, R.; Patel, S.; Feng, L.; Said, M.; Dayton, M.; Pankewycz, O. Sirolimus-induced isometric tubular vacuolization: A new sirolimus histopathologic manifestation. Transplant. Proc. 2010, 42, 2547–2550. [Google Scholar] [CrossRef]
- Naesens, M.; Kuypers, D.R.J.; Sarwal, M. Calcineurin inhibitor nephrotoxicity. Clin. J. Am. Soc. Nephrol. 2009, 4, 481–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, E. Adverse effects of drugs on the kidney. Eur. J. Intern. Med. 2016, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.; Uetrecht, J. How Reactive Metabolites Induce an Immune Response That Sometimes Leads to an Idiosyncratic Drug Reaction. Chem. Res. Toxicol. 2017, 30, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [Green Version]
- Björnsson, E.S.; Bergmann, O.M.; Björnsson, H.K.; Kvaran, R.B.; Olafsson, S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 2013, 144, 1419–1425. [Google Scholar] [CrossRef]
- El-Sayyad, H.I.; Ismail, M.F.; Shalaby, F.M.; Abou-El-Magd, R.F.; Gaur, R.L.; Fernando, A.; Raj, M.H.G.; Ouhtit, A. Histopathological effects of cisplatin, doxorubicin and 5-flurouracil (5-FU) on the liver of male albino rats. Int. J. Biol. Sci. 2009, 5, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Grieco, A.; Forgione, A.; Miele, L.; Vero, V.; Greco, A.V.; Gasbarrini, A.; Gasbarrini, G. Fatty liver and drugs. Eur. Rev. Med. Pharmacol. Sci. 2005, 9, 261–263. [Google Scholar]
- Zhao, F.; Xie, P.; Jiang, J.; Zhang, L.; An, W.; Zhan, Y. The effect and mechanism of tamoxifen-induced hepatocyte steatosis in vitro. Int. J. Mol. Sci. 2014, 15, 4019–4030. [Google Scholar] [CrossRef] [Green Version]
- Vitins, A.P.; Kienhuis, A.S.; Speksnijder, E.N.; Roodbergen, M.; Luijten, M.; Van der Ven, L.T.M. Mechanisms of amiodarone and valproic acid induced liver steatosis in mouse in vivo act as a template for other hepatotoxicity models. Arch. Toxicol. 2014, 88, 1573–1588. [Google Scholar] [CrossRef]
- Rabinowich, L.; Shibolet, O. Drug Induced Steatohepatitis: An Uncommon Culprit of a Common Disease. Biomed. Res. Int. 2015, 2015, 168905. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, D.E. The histopathological evaluation of drug-induced liver injury. Histopathology 2017, 70, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell. Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef] [PubMed]
- Henics, T.; Wheatley, D.N. Cytoplasmic vacuolation, adaptation and cell death: A view on new perspectives and features. Biol. Cell. 1999, 91, 485–498. [Google Scholar] [CrossRef]
- Aki, T.; Nara, A.; Uemura, K. Cytoplasmic vacuolization during exposure to drugs and other substances. Cell. Biol. Toxicol. 2012, 28, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Shubin, A.V.; Demidyuk, I.V.; Komissarov, A.A.; Rafieva, L.M.; Kostrov, S.V. Cytoplasmic vacuolization in cell death and survival. Oncotarget 2016, 7, 55863–55889. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Dose (mg/kg) | Log Dose | % Dead | Probits |
|---|---|---|---|---|
| 1 | 50 | 3.912 | 50 | 0.585 |
| 2 | 100 | 4.605 | 100 | 0.783 |
| 3 | 200 | 5.298 | 75 | 0.912 |
| 4 | 400 | 5.991 | 100 | 0.973 |
| 5 | 800 | 6.685 | 100 | 0.994 |
| Control | 0 | 0 | 0 | 0 |
| Animal No. | Treatment | Fate (Upto 42 Days) |
|---|---|---|
| A1–A6 | Distilled water | All survived, sacrificed on day 42, tissues preserved for histopathology |
| B3 | 3.8 mg/kg BW (1/10 of LD50) | Died on day 22, tissues preserved for histopathology |
| B9 | Died on day 32, tissues preserved for histopathology | |
| B1, B2, B4–B8, B10 | Survived up to day 42, sacrificed, tissues preserved for histopathology | |
| C1 | 7.6 mg/kg BW (1/5 of LD50) | Sacrificed on day 37 due to animal distress, tissues preserved for histopathology |
| C2 | Sacrificed on day 37 due to animal distress, tissues preserved for histopathology | |
| C3 | Died on day 35, tissues preserved for histopathology | |
| C4 | Died on day 36, tissues preserved for histopathology |
| Groups (n = 4 in Each Group) | Dose mg/kg | Renal Tubular Necrosis Grade | Renal Tubular Vacuolization | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 4 | 3 | 2 | 1 | 0 | None | Less Than 50% | More Than 50% | ||
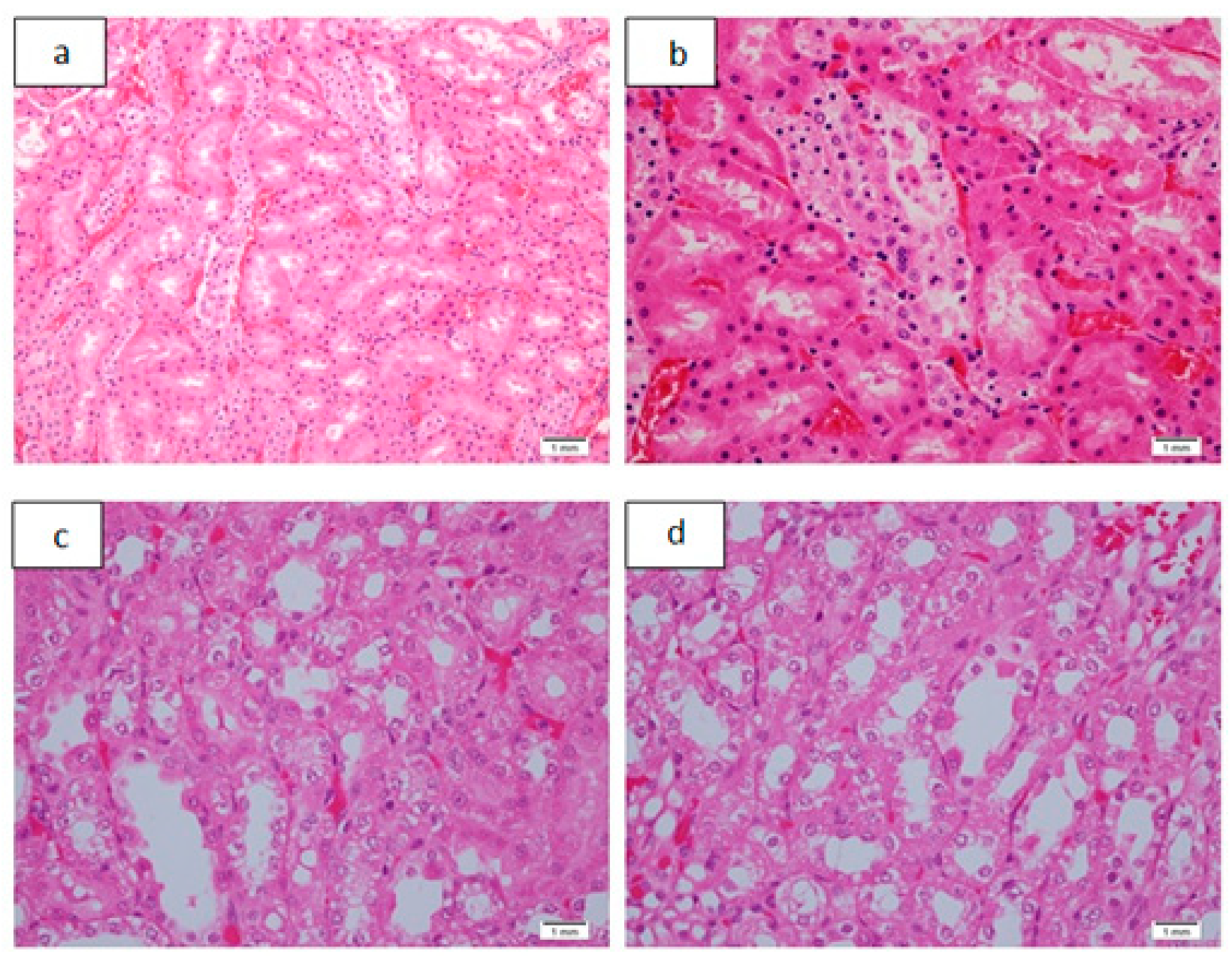
| 1 | 50 | 0 | 0 | 0 | 0 | 25% (n = 1) Figure 3a,b | 75% # (n = 3) | 100% # (n = 4) | 0 | 0 |
| 2 | 100 | 0 | 0 | 0 | 0 | 50% (n = 2) | 50% (n = 2) | 75% # (n = 3) | 25% (n = 1) Figure 3c,d | 0 |
| 3 | 200 | 0 | 0 | 0 | 0 | 25% (n = 1) | 75% # (n = 3) | 50% (n = 2) | 50% (n = 2) | 0 |
| 4 | 400 | 0 | 0 | 0 | 0 | 50% (n = 2) | 50% (n = 2) | 25% (n = 1) | 75% # (n = 3) | 0 |
| 5 | 800 | 0 | 0 | 0 | 0 | 25% (n = 1)) | 75% # (n = 3) | 25% (n = 1) | 75% # (n = 3) | 0 |
| 6 (control) | - | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Groups (n = 4 in Each Group) | Dose mg/kg | Pyelitis/Interstitial Inflammation % | Congestion % | ||||
|---|---|---|---|---|---|---|---|
| None | Mild | Mod/ Marked | None | Mild | Mod/ Marked | ||
| 1 | 50 | 75% # (n = 3) | 25% (n = 1) | 0 | 0 | 50% (n = 2) | 50% (n = 2) |
| 2 | 100 | 75% # (n = 3) | 25% (n = 1) | 0 | 0 | 0 | 100% # (n = 4) |
| 3 | 200 | 0 | 75% # (n = 3) | 25% (n = 1) Figure 4a–d | 0 | 25% (n = 1) | 75% # (n = 3) |
| 4 | 400 | 25% (n = 1) | 25% (n = 1) | 50% (n = 2) | 0 | 0 | 100% # (n = 4) |
| 5 | 800 | 0 | 25% (n = 1) | 75% # (n = 3) | 0 | 25% (n = 1) | 75% # (n = 3) |
| 6 (control) | - | 0 | 0 | 0 | 0 | 0 | 0 |
| Groups (n = 4 in Each Group | Dose mg/kg | Steatosis | Hepatocellular Necrosis/Degeneration | Congestion | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| None | Mild | Moderate Marked | None | Individual Cell Degeneration | Necrosis with Inflammation | None | Mild | Mod/ Marked | ||
| 1 | 50 | 75% # (n = 3) | 25% (n = 1) Figure 5a,b | 0 | 100% # (n = 4) | 0 | 0 | 0 | 75% # (n = 3) | 25% (n = 1) |
| 2 | 100 | 75% # (n = 3) | 25% (n = 1) | 0 | 75% # (n = 3) | 25% (n = 1) | 0 | 25% (n = 1) | 75% # (n = 3)) | 0 |
| 3 | 200 | 75% # (n = 3) | 25% (n = 1) | 0 | 100% # (n = 4) | 0 | 0 | 0 | 50% (n = 2) | 50% (n = 2) |
| 4 | 400 | 75% # (n = 3) | 25% (n = 1) | 0 | 25% (n = 1) | 75% # (n = 3) | 0 | 0 | 0 | 100% # (n = 4) |
| 5 | 800 | 0 | 100% # (n = 4) | 0 | 0 | 100% # (n = 4) | 0 | 0 | 25% (n = 1) | 75 # (n = 3) |
| 6 (control) | - | 100% # (n = 4) | 0 | 0 | 100% # (n = 4) | 0 | 0 | 25% (n = 1) | 75 # (n = 3) | 0 |
| Groups (n = 4 in Each Group) | Dose mg/kg | Inflammation Portal/Lobular | Capsular Acute Inflammation | Cytoplasmic Vacuolization | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| None | Mild | Moderate/ Marked | None | Mild | Mod/ Marked | None | Mild | Moderate/ Marked | ||
| 1 | 50 | 75% # (n = 3) | 25% (n = 1) | 0 | 25% (n = 1) | 75% # (n = 3) Figure 5c | 0 | 25% (n = 1) | 75% # (n = 3) Figure 5d | 0 |
| 2 | 100 | 50% (n = 2) | 50% (n = 2) | 0 | 50% (n = 2) | 50% (n = 2) | 0 | 25% (n = 1) | 75% # (n = 3) | 0 |
| 3 | 200 | 75% # (n = 3) | 25% (n = 1) | 0 | 50% (n = 2) | 25% (n = 1) | 25% (n = 1) | 0 | 50% (n = 2) | 50% (n = 2) |
| 4 | 400 | 25% (n = 1) | 75% # (n = 3) | 0 | 0 | 100% # (n = 4) | 0 | 0 | 75% # (n = 3) | 25% (n = 1) |
| 5 | 800 | 0 | 100% (n = 4) | 0 | 50% (n = 2) | 50% (n = 2) | 0 | 0 | 75% # (n = 3) | 25% (n = 1) |
| 6 (control) | - | 75% # (n = 3) | 25% (n = 1) | 0 | 50% (n = 2) | 50% (n = 2) | 0 | 75% (n = 3) | 25% (n = 1) | 0 |
| Groups (n = 4 in Each Group) | Dose mg/kg | Cytoplasmic Vacuolization | Hyalinization | Congestion | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| None | Mild | Mod | Marked | None | Mild | Mod | Marked | None | Mild | Mod/ marked | ||
| 1 | 50 | 75% # (n = 3) | 25% (n = 1) | 0 | 0 | 75% # (n = 3) | 25% (n = 1) | 0 | 0 | 0 | 50% (n = 2) | 50% (n = 2)) |
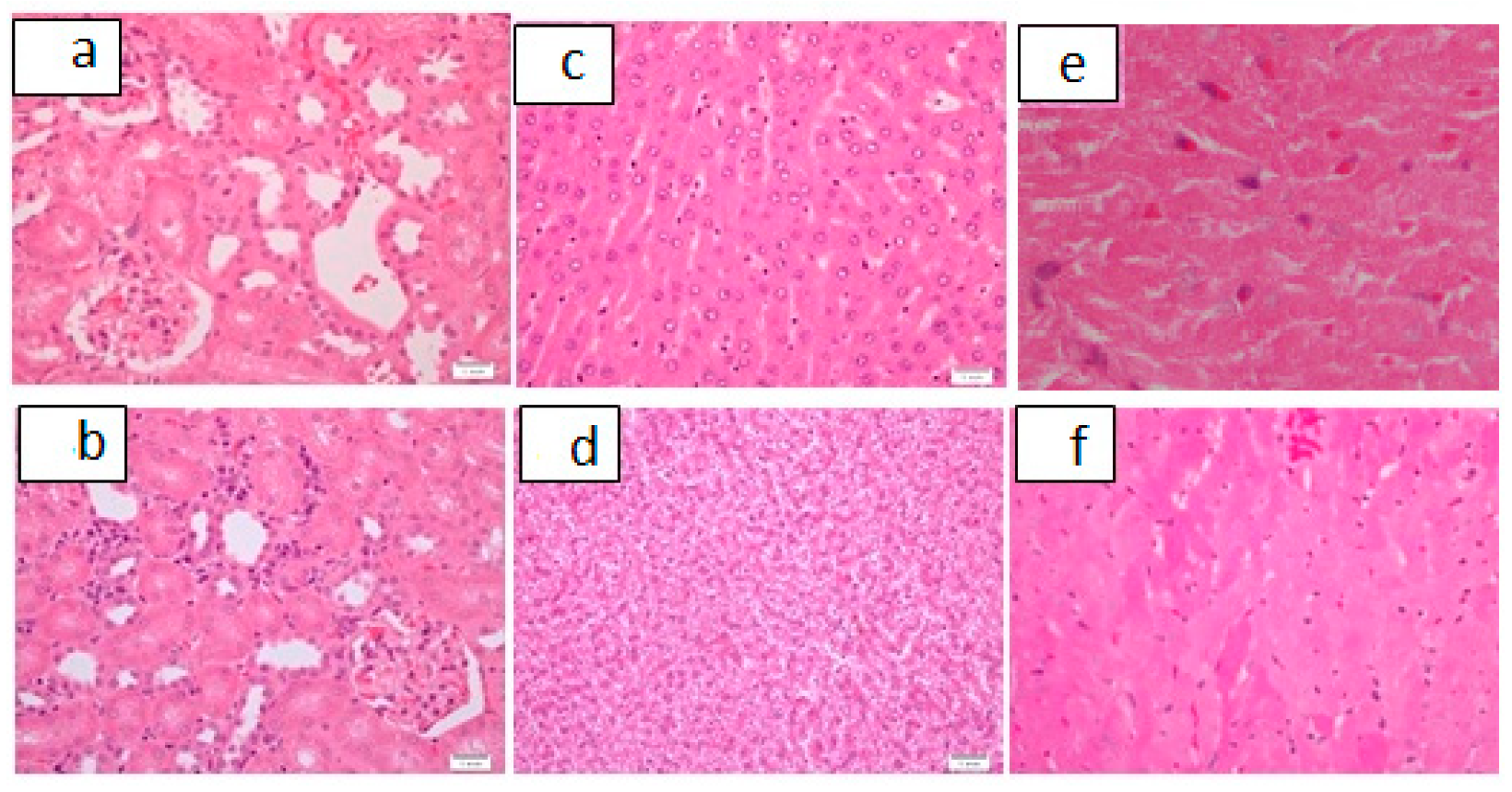
| 2 | 100 | 25% (n = 1) | 75% # (n = 3) | 0 | 0 | 25% (n = 1) | 75% # (n = 3) | 25% (n = 1) Figure 6a,b | 0 | 0 | 0 | 100% # (n = 4) |
| 3 | 200 | 50% (n = 2) | 50% (n = 2)) | 0 | 0 | 75% # (n = 3) | 0 | 0 | 0 | 50% (n = 2) | 50% (n = 2) Figure 6c | |
| 4 | 400 | 75% # (n = 3) | 25% (n = 1) | 0 | 0 | 75% # (n = 3) | 25% (n = 1) | 75 (n = 3) | 0 | 0 | 0 | 100% # (n = 4) |
| 5 | 800 | 50% (n = 2) | 50% Figure 6d | 0 | 0 | 100% # (n = 4) | 0 | 0 | 0 | 25% (n = 1) | 75% # (n = 3) | |
| 6 (control) | - | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 25% (n = 1) | 75% # (n = 3) | 0 |
| Animal Groups | Dose | RENAL | HEPATIC | CARDIAC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal | Necrosis | Interstitial Inflammation | Normal | Portal/Lobular Inflammation | Mild Capsular Inflammation *** | Mild Cytoplasmic Vacuolization *** | Normal | Mild Focal Cardiac Fiber Hyalinization <10% of Tissue Examined | ||||
| Mild | Mod | Mild | Mod | |||||||||
| A Control (n = 6) | 0 | 100%# (n = 6) Figure 7a | 0 | 0 | 0 | 67% # (n = 4) Figure 7b | 33% (n = 2) | 0 | 16% (n = 1) | 0 | 100% # (n = 6) Figure 7c | 0 |
| B * (n = 10) | 3.8 mg/kg | 70% # (n = 7) | 0 | 20% (n = 2) Figure 8a,b | 10% (n = 1) | 70% # (n = 7) | 20% (n = 2) | 10% (n = 1) | 20% (n = 2) | 10% (n = 1) Figure 8c,d | 80% # (n = 8) | 20% (n = 2) Figure 8e,f |
| C ** (n = 4) | 7.6 mg/kg | 50% # (n = 2) | 0 | 25% (n = 1) | 25% (n = 1) | 50% (n = 2) | 50% (n = 2) | 0 | 0 | 50% (n = 2) | 50% (n = 2) | 50% (n = 2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badar, A.; Ahmed, A.; Al-Tamimi, D.M.; Isab, A.A.; Altaf, M.; Ahmed, S. Histological Changes in Renal, Hepatic and Cardiac Tissues of Wistar Rats after 6 Weeks Treatment with Bipyridine Gold (III) Complex with Dithiocarbamate Ligands. Pharmaceutics 2021, 13, 1530. https://doi.org/10.3390/pharmaceutics13101530
Badar A, Ahmed A, Al-Tamimi DM, Isab AA, Altaf M, Ahmed S. Histological Changes in Renal, Hepatic and Cardiac Tissues of Wistar Rats after 6 Weeks Treatment with Bipyridine Gold (III) Complex with Dithiocarbamate Ligands. Pharmaceutics. 2021; 13(10):1530. https://doi.org/10.3390/pharmaceutics13101530
Chicago/Turabian StyleBadar, Ahmed, Ayesha Ahmed, Dalal M. Al-Tamimi, Anvarhusein A. Isab, Muhammad Altaf, and Sania Ahmed. 2021. "Histological Changes in Renal, Hepatic and Cardiac Tissues of Wistar Rats after 6 Weeks Treatment with Bipyridine Gold (III) Complex with Dithiocarbamate Ligands" Pharmaceutics 13, no. 10: 1530. https://doi.org/10.3390/pharmaceutics13101530
APA StyleBadar, A., Ahmed, A., Al-Tamimi, D. M., Isab, A. A., Altaf, M., & Ahmed, S. (2021). Histological Changes in Renal, Hepatic and Cardiac Tissues of Wistar Rats after 6 Weeks Treatment with Bipyridine Gold (III) Complex with Dithiocarbamate Ligands. Pharmaceutics, 13(10), 1530. https://doi.org/10.3390/pharmaceutics13101530