Uncovering New Drug Properties in Target-Based Drug–Drug Similarity Networks

 ,
,  ,
,  , , and
, , and 
Abstract
:
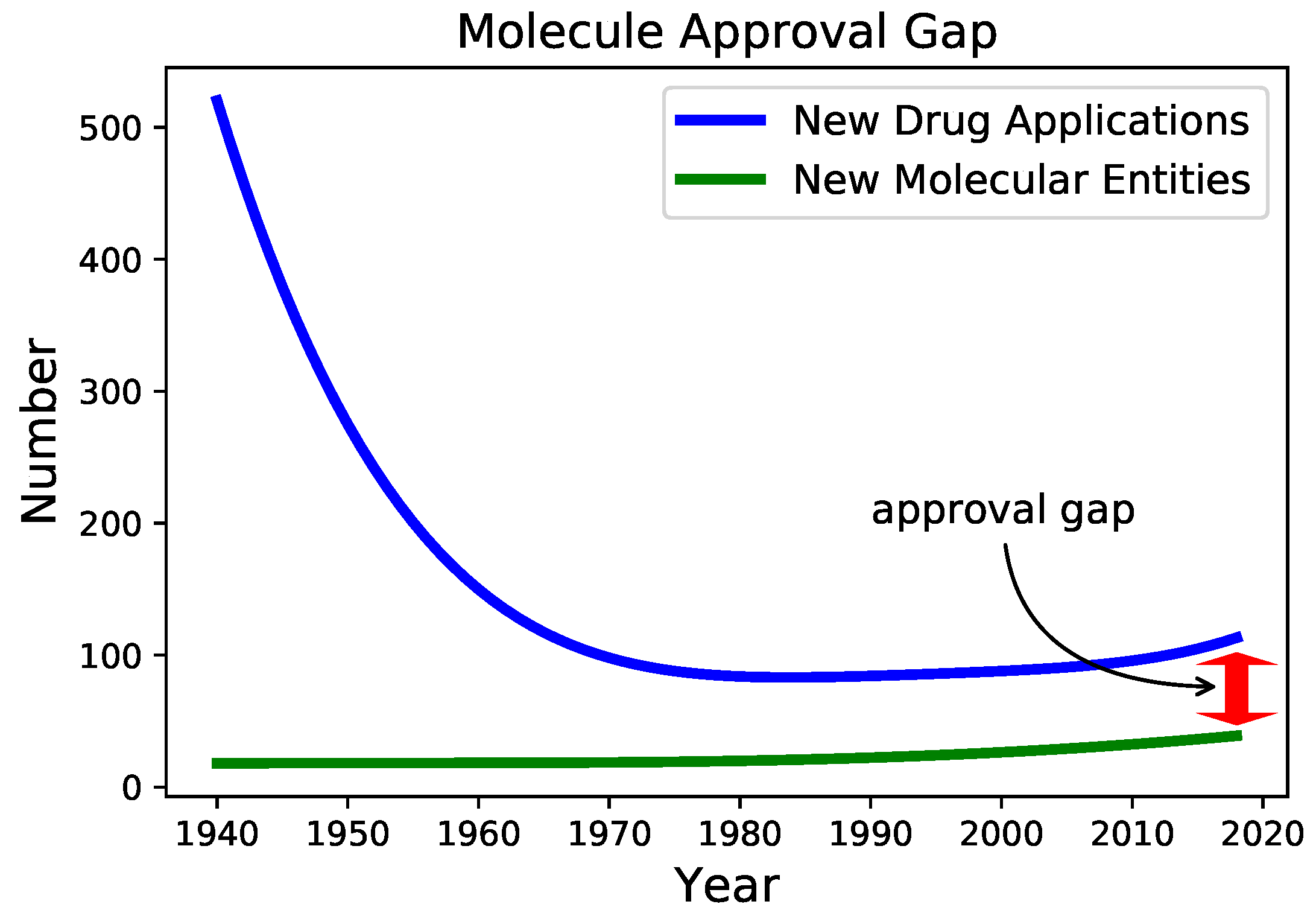
1. Introduction
- Relate each cluster and each community to a pharmacological property or pharmacological action (i.e., label communities and clusters according to the dominant property or pharmacological action), using expert analysis.
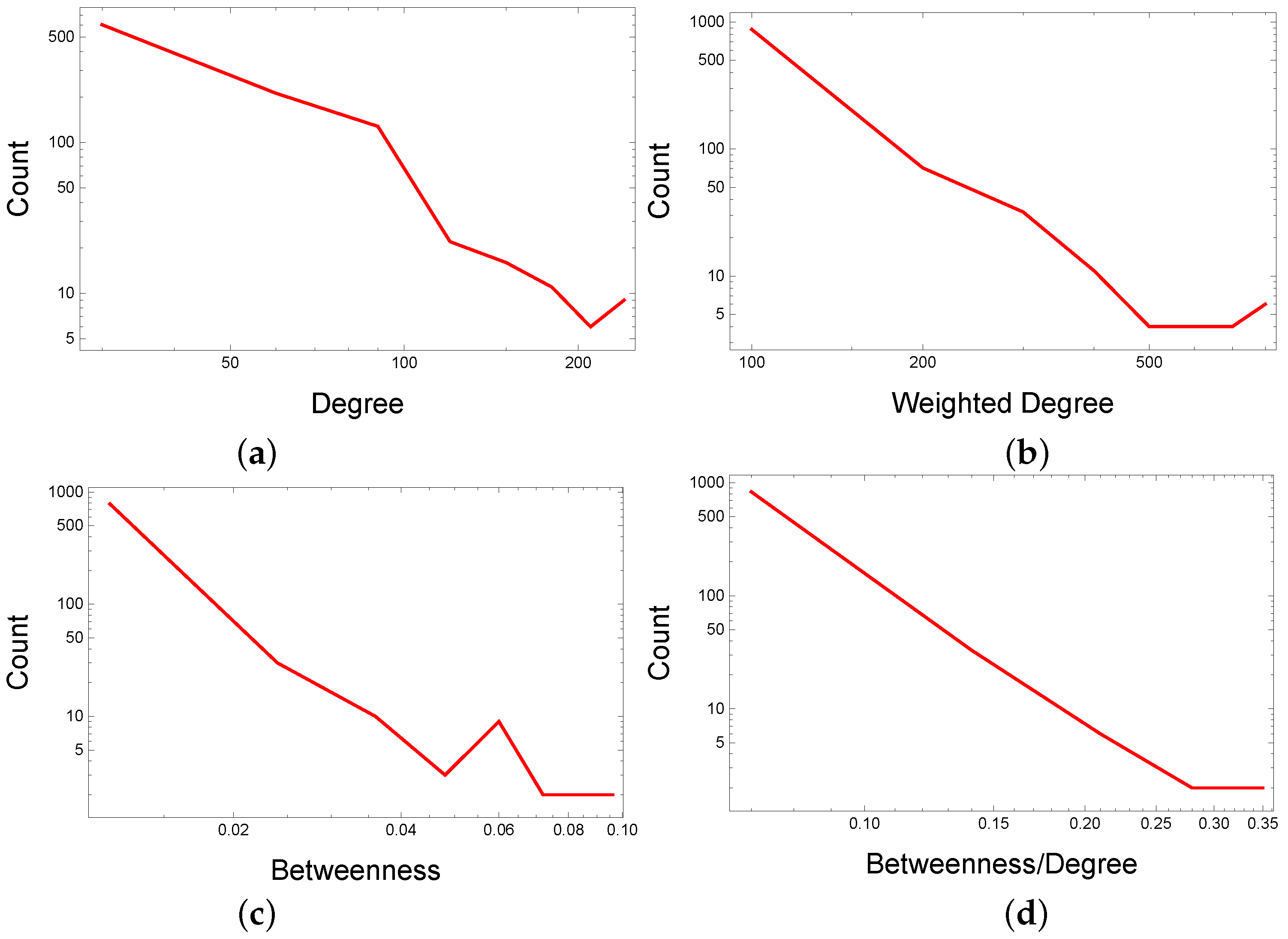
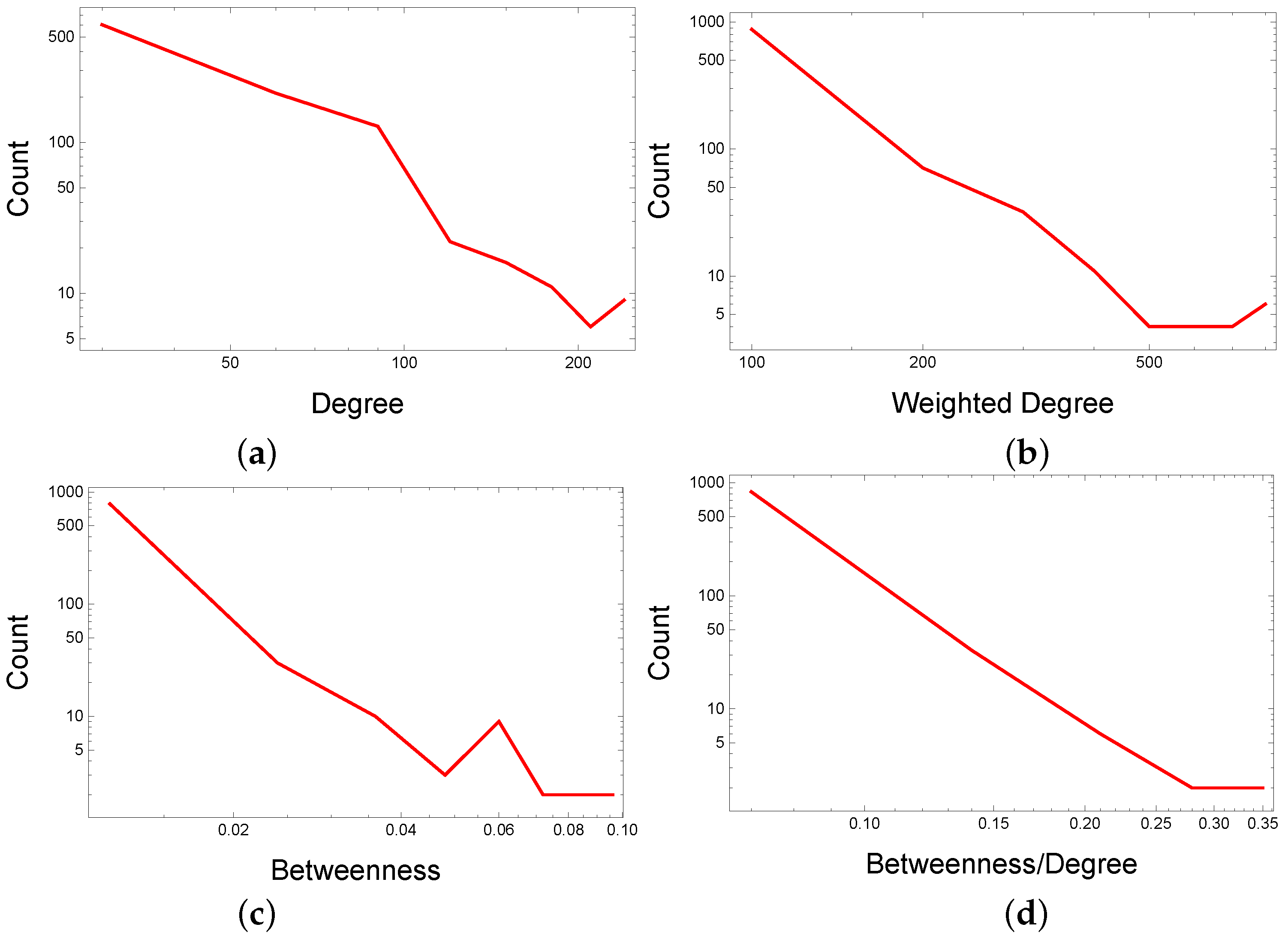
- Identify and select (by betweenness divided by degree, ) within each topological cluster/modularity class community, the top drugs not compliant with the cluster/community label. Network analysis uses centralities to rank nodes (i.e., drugs); we opt for the centrality to find this centrality’s distribution more stable in the DDSN.
- Validate the hinted repositionings by searching the new versions of DrugBank, the electronic records containing the relevant scientific literature (for merely reconstructed repositionings), and by analyzing molecular docking parameters [36] for previously unaccounted repositionings.
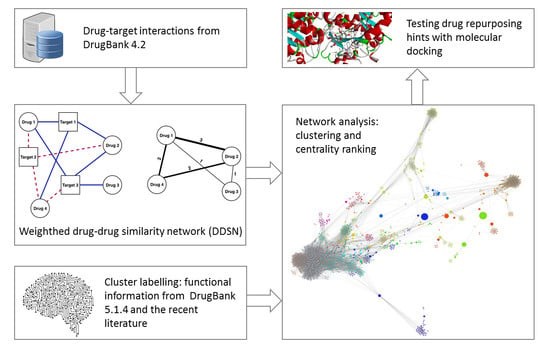
2. Materials and Methods
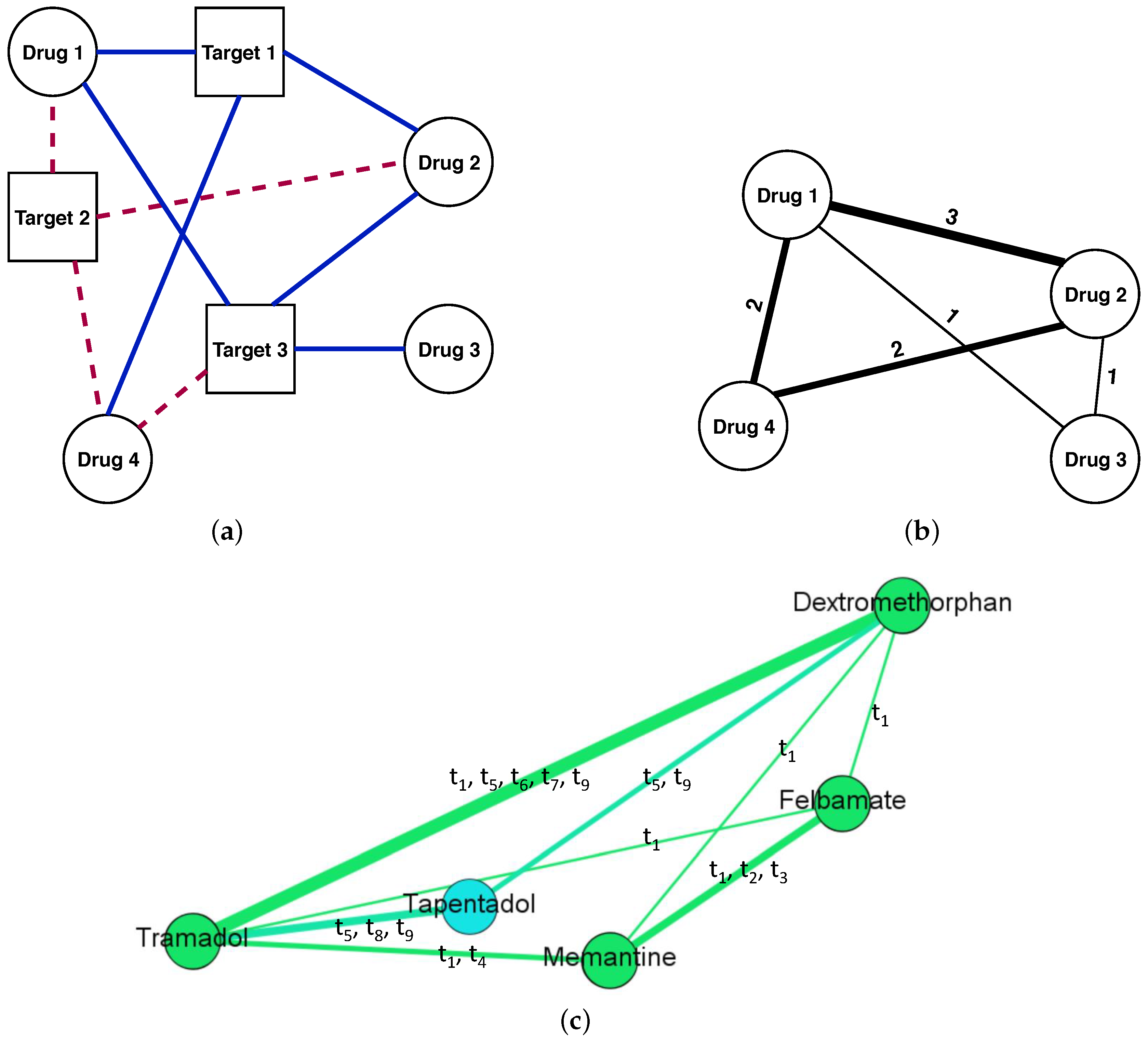
2.1. Building the DDSN
2.2. Network Analysis
2.2.1. Network Clustering
2.2.2. Network Centralities
2.3. Molecular Docking for Repurposing Testing
2.3.1. Testing Procedure
- We define the drug sets to enter the docking process, consisting of drugs hinted as having the pharmacological property (), well-documented drugs with property (reference drugs ), and drugs with little probability of having property (). Our goal is to explore the similarity (in terms of relevant target activity) between the reference drugs and the tested drugs .
- (a)
- consists of the drugs hinted as repurposed for property/properties .
- (b)
- consists of two subsets, reference drugs in the DDSN’s community () and reference drugs not in (), with .
- (c)
- contains typical drugs for other pharmacological properties, with little probability of having property .
- We establish the target sets. Specifically, for pharmacological property , we take into consideration the targets from DrugBank that interact with the drugs in the hinted drug community having property (), and the targets from DrugBank that interact with the drugs with property not included in DDSN’s ().
- For the set of tested drugs , we use molecular docking to check the interactions between all possible drug–target pairs, defined as the Cartesian product of sets and (with ),
- For the set of reference drugs, we apply molecular docking on separately designed drug–target pairs for reference drugs in (), and reference drugs not in () respectively, such that any drug–target pair is well-documented in the literature,and
2.3.2. Ligands and Targets Preparation
2.3.3. Docking Protocol
3. Results
3.1. DDSN Analysis
3.2. Illustrative Examples of Reconstructed Drug Repositionings
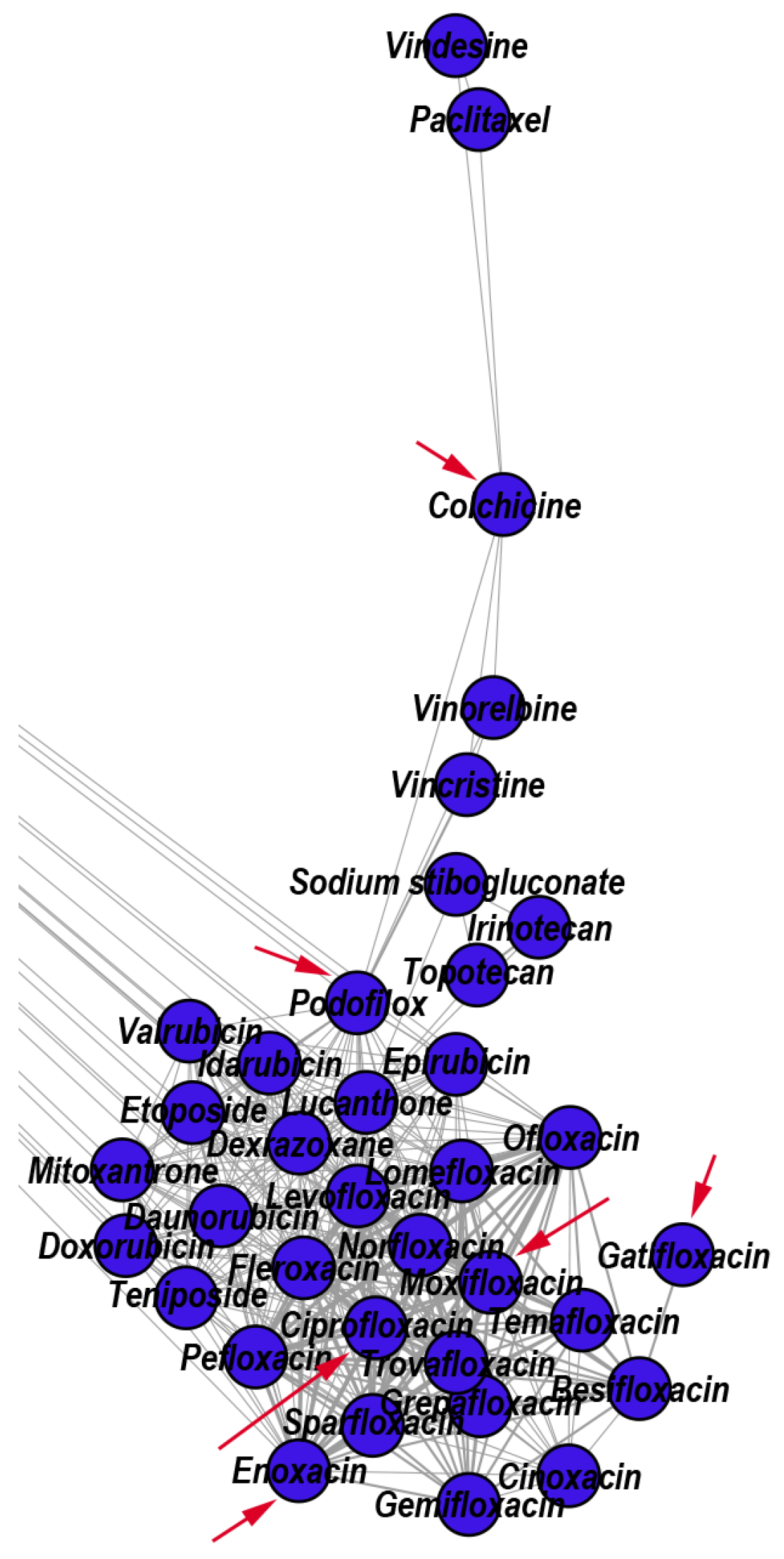
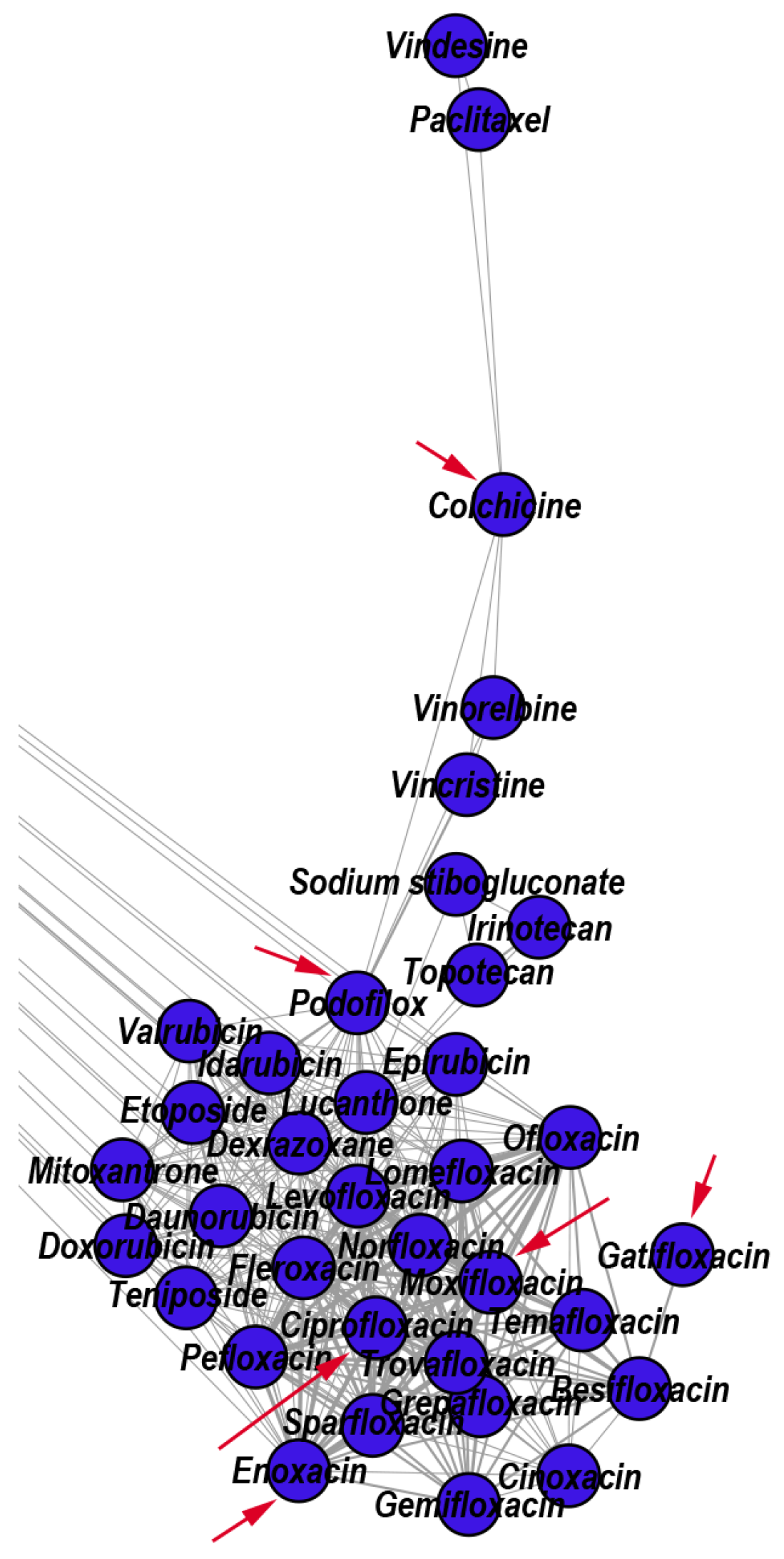
3.2.1. Reconstructed Repurposings as Antineoplastic Agents
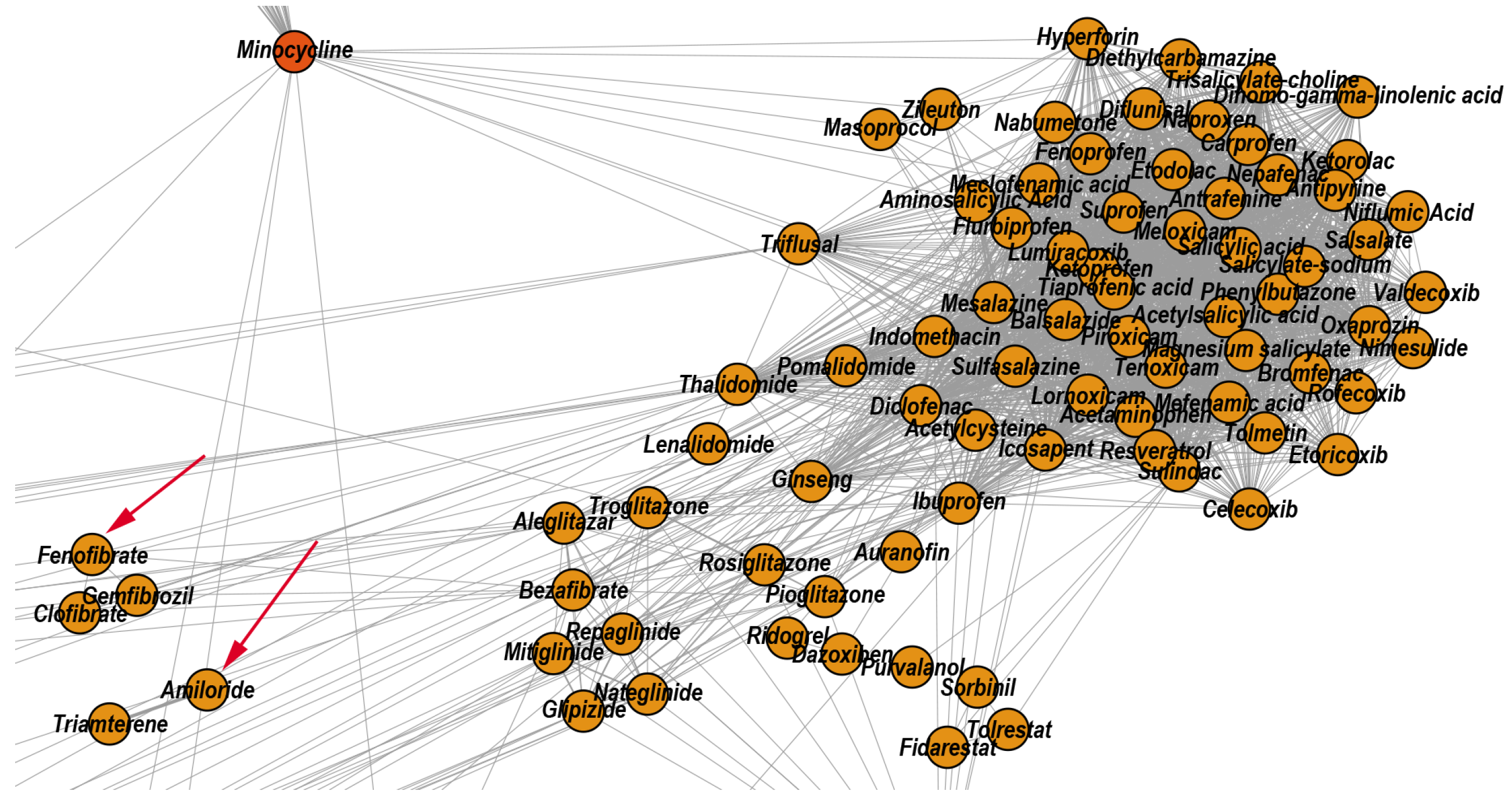
3.2.2. Reconstructed Repurposings as Anti-Inflammatory Drugs
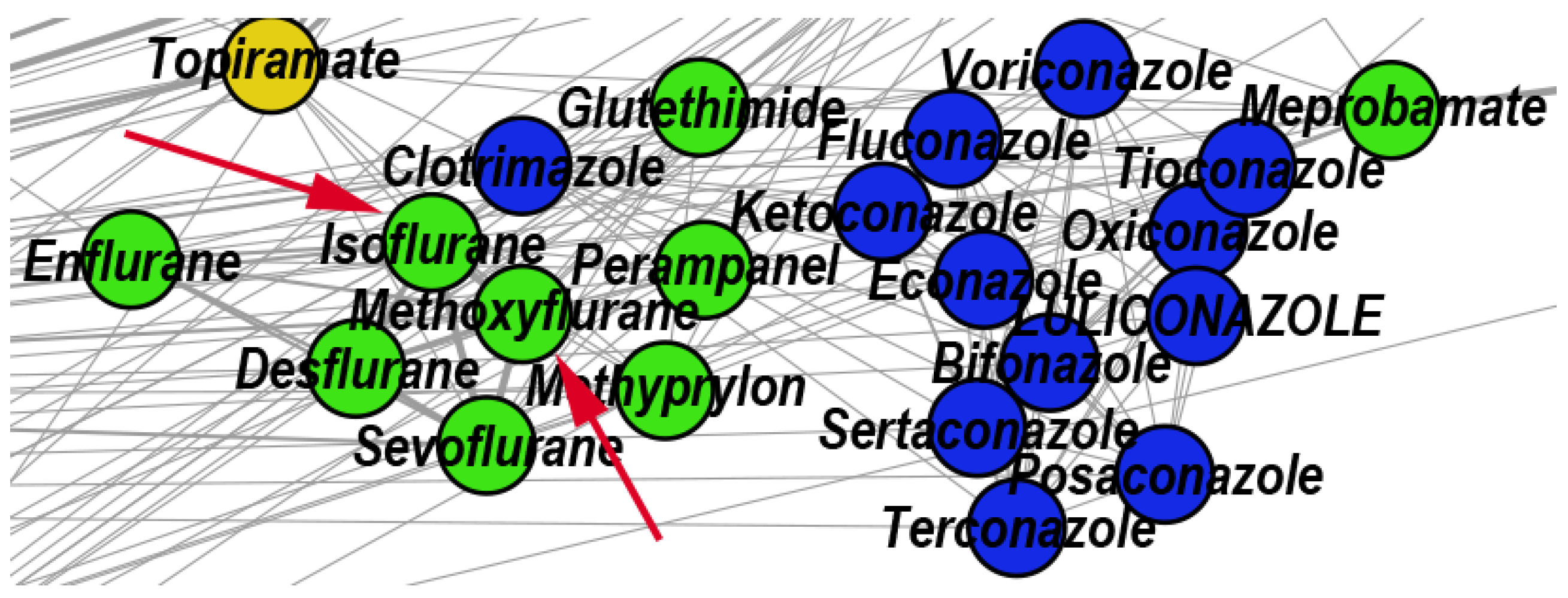
3.2.3. Reconstructed Repurposings as Antifungal Drugs
3.3. Repositioning Hints Prioritization
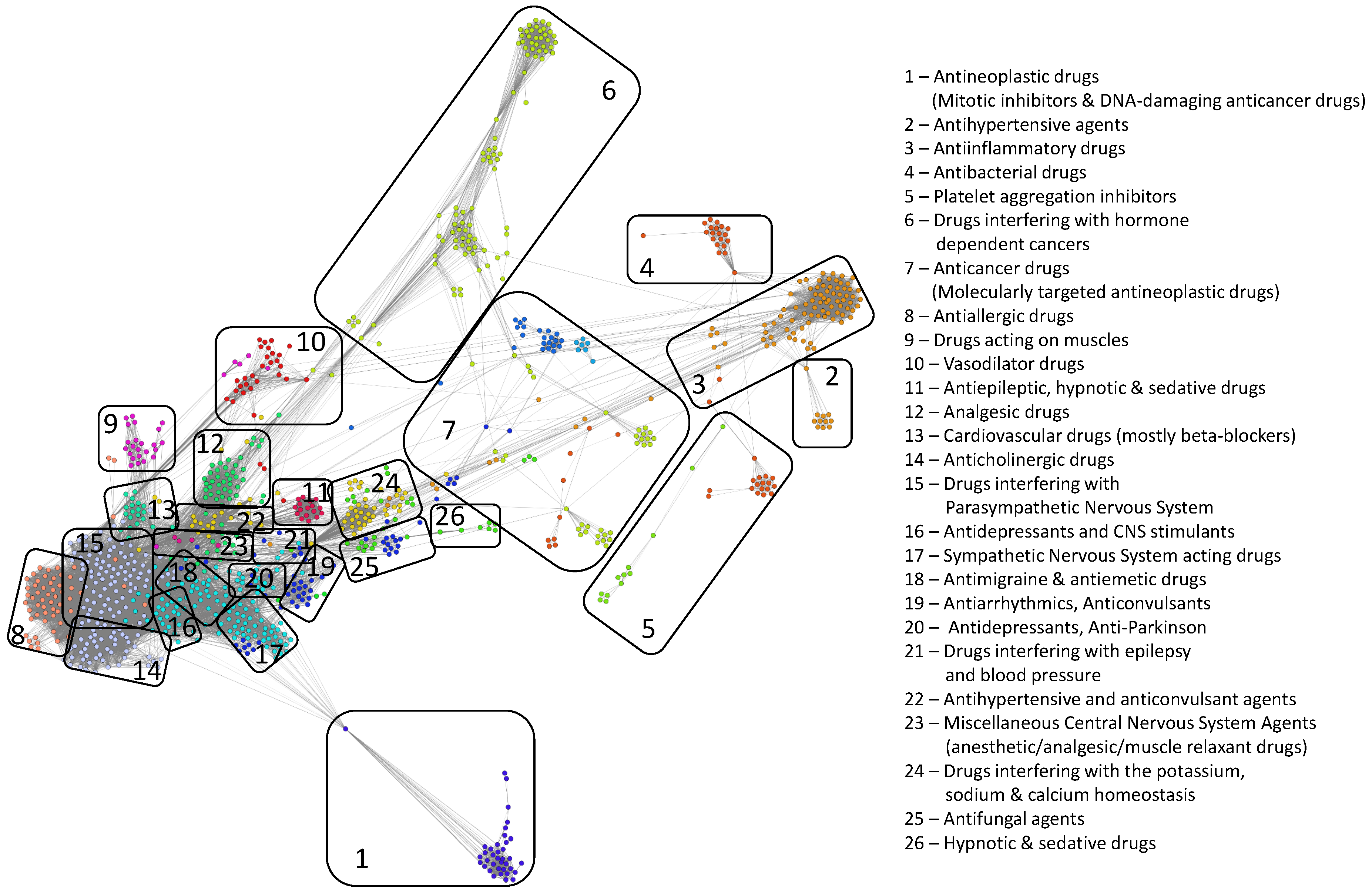
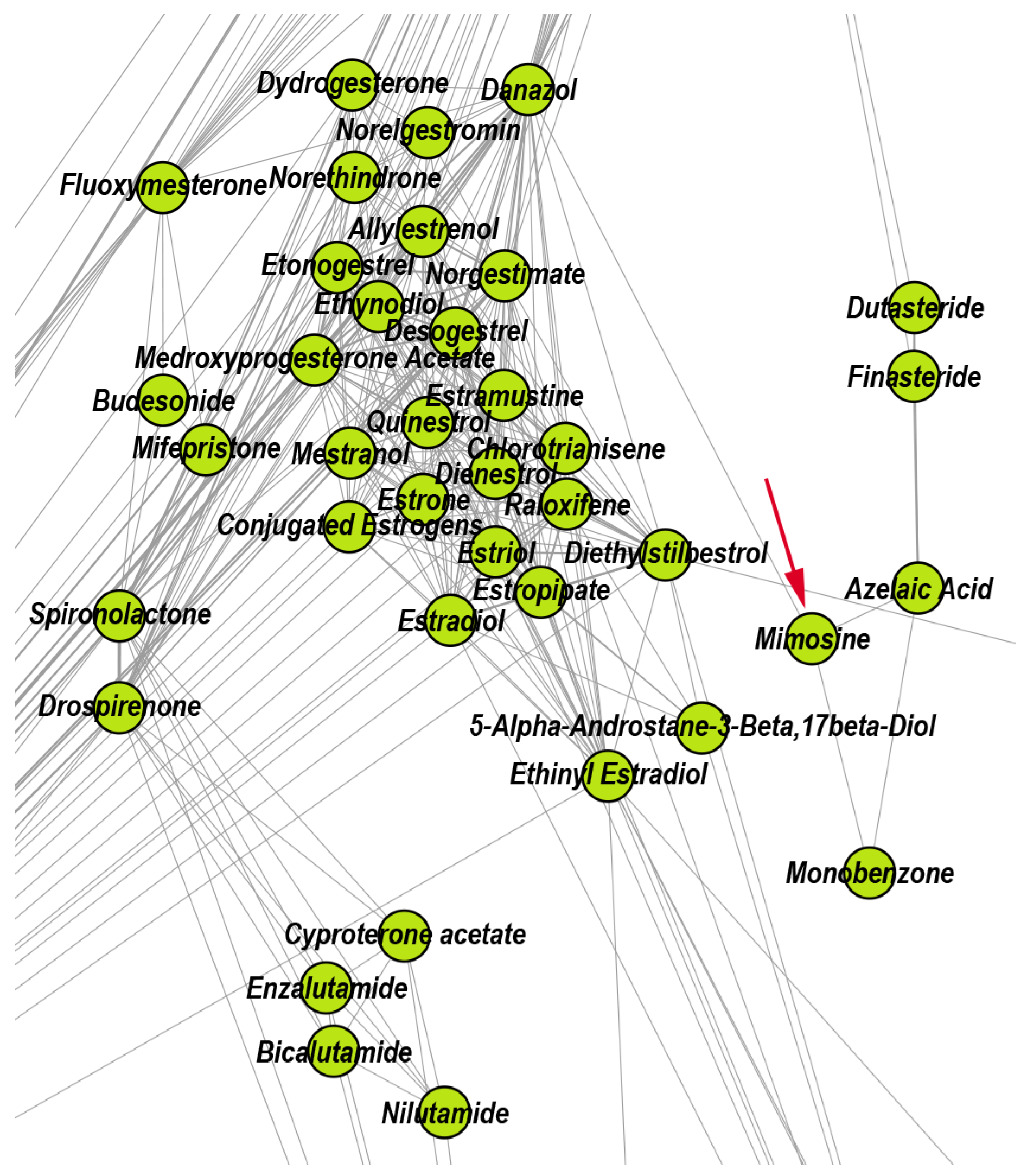
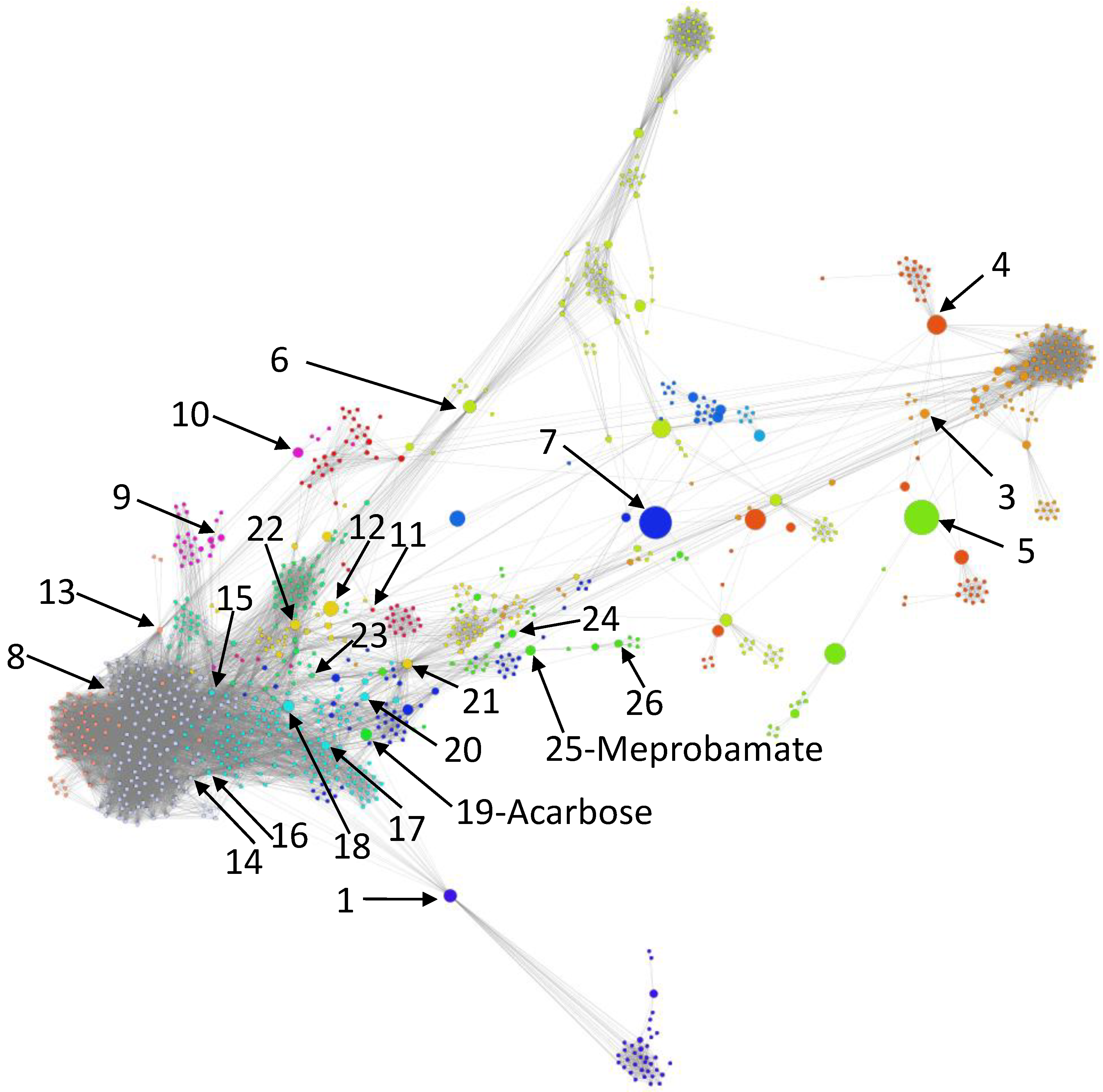
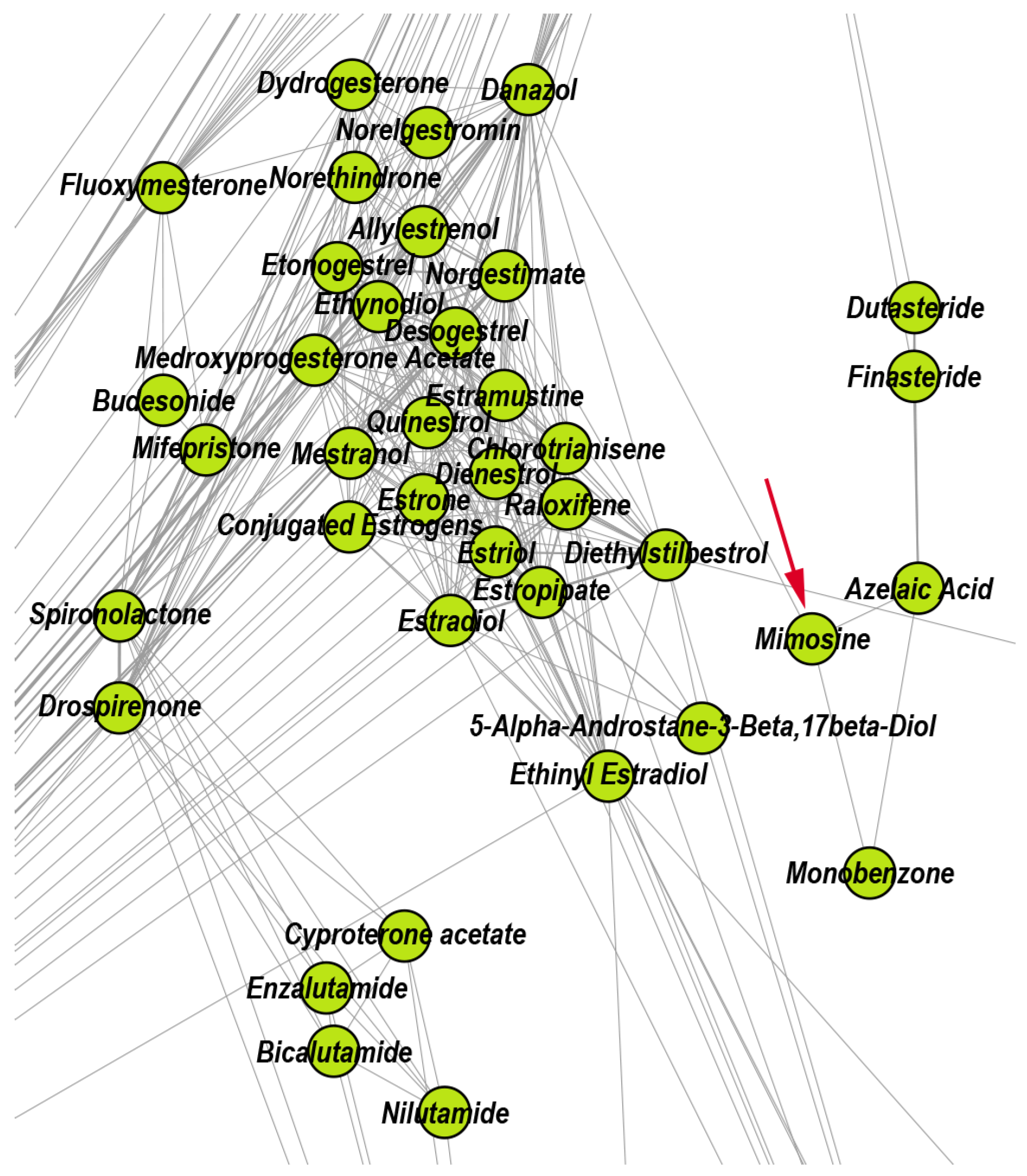
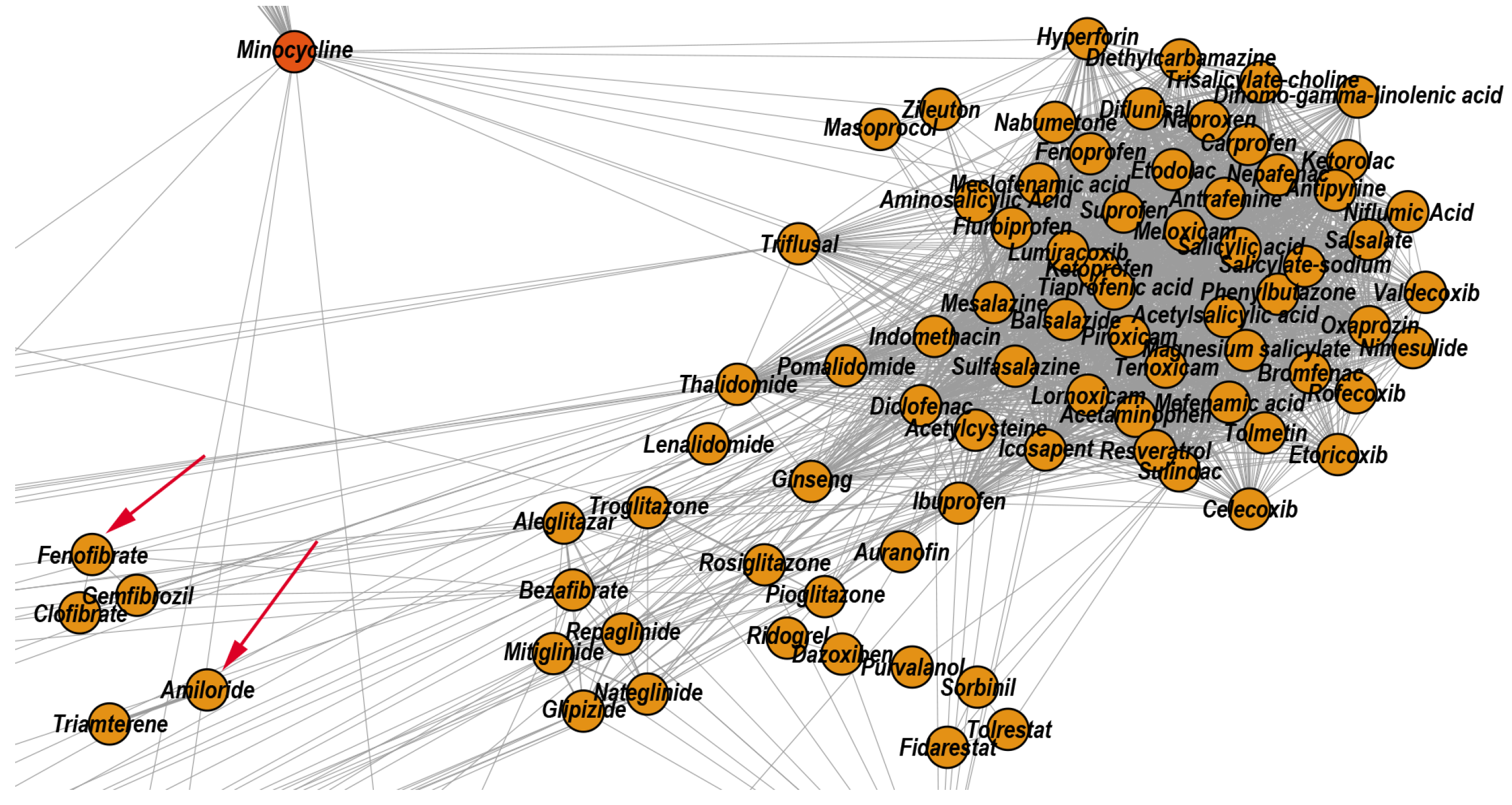
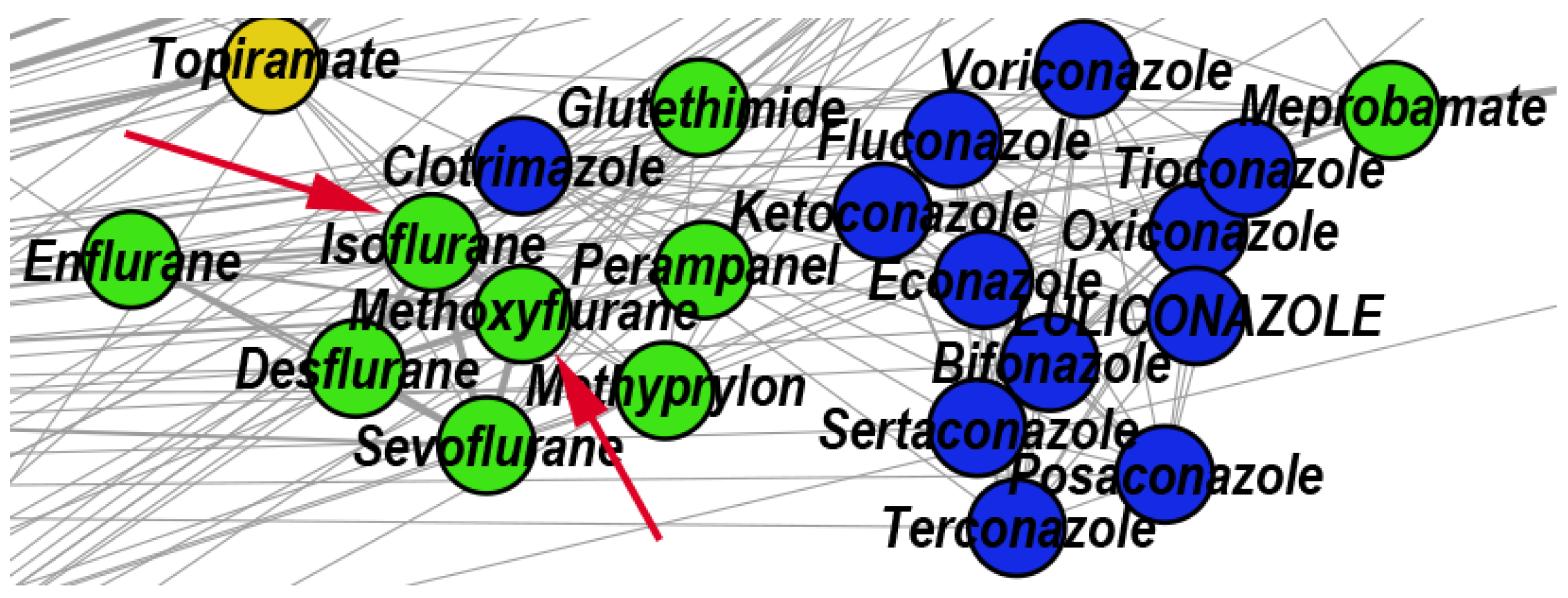
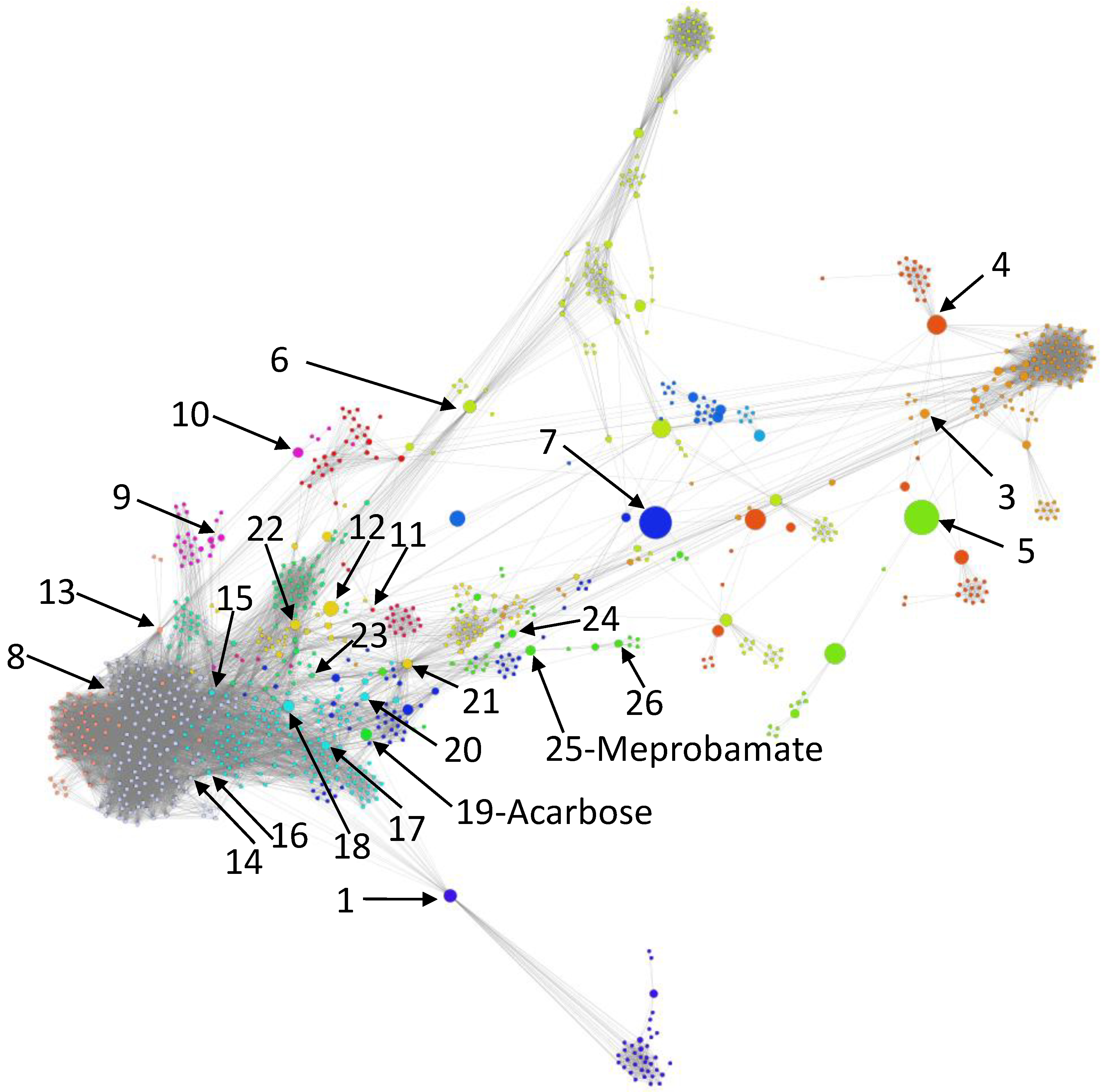
- We uncover the relevant drug properties by generating network communities with ( in our DDSN). Then, using expert analysis, we assign a dominant property to each community. Figure 3 illustrates the 26 DDSN communities as well as their dominant functionality. The dominant community property can be a pharmacological mechanism, a targeted disease, or a targeted organ. For instance, the community 1 () consists of antineoplastic drugs which act as mitotic inhibitors and DNA damaging agents; Community 13 () consists of cardiovascular drugs (antihypertensive, anti-arrhythmic, and anti-angina drugs), mostly beta-blockers.
- In each cluster , we identify the top t drugs according to their values. From these selected drugs, , some stand out by not sharing the community property or properties, and thus, can be repositioned as such. To this end, for eliminated from the drugs whose repurposings were already confirmed (i.e., performed by others and found in the recent literature), thus producing lists of repurposing hints yet to be confirmed by in silico, in vitro, and in vivo experiments, . Table 3 presents the lists of drugs for and (i.e., the top 5 drugs in each community). We chose to provide a reasonable amount of eloquent information in Table 3; we provide the entire sets in the Supplementary file SupplementaryDDSN.
3.4. Repurposing Hints Testing
4. Discussion
4.1. Complex Network Perspective
4.2. Molecular Docking Perspective
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DDIN | Drug–Drug Interaction Network |
| DDSN | Drug–Drug Similarity Network |
| FDA | U.S. Food and Drug Administration |
| NDA | New Drug Applications |
| NME | New Molecular Entities |
References
- Dickson, M.; Gagnon, J.P. The cost of new drug discovery and development. Discov. Med. 2009, 4, 172–179. [Google Scholar]
- Chen, X.Q.; Antman, M.D.; Gesenberg, C.; Gudmundsson, O.S. Discovery pharmaceutics—Challenges and opportunities. Aaps J. 2006, 8, E402–E408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. 2016 FDA drug approvals. Nat. Rev. Drug Discov. 2017, 16, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Graul, A.; Pina, P.; Cruces, E.; Stringer, M. The year’s new drugs & biologics 2016: Part I. Drugs Today 2017, 53, 27. [Google Scholar] [PubMed]
- Pammolli, F.; Magazzini, L.; Riccaboni, M. The productivity crisis in pharmaceutical R&D. Nat. Rev. Drug Discov. 2011, 10, 428–438. [Google Scholar]
- Drug Approvals and Databases. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/resources-information-approved-drugs (accessed on 15 January 2019).
- Csermely, P.; Korcsmáros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41. [Google Scholar] [CrossRef]
- Munos, B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 2009, 8, 959–968. [Google Scholar] [CrossRef]
- Shaughnessy, A.F. Old drugs, new tricks. BMJ 2011, 342, d741. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2015, 17, 2–12. [Google Scholar] [CrossRef] [Green Version]
- Lotfi Shahreza, M.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J.R. A review of network-based approaches to drug repositioning. Brief. Bioinform. 2017, 19, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Nugent, T.; Plachouras, V.; Leidner, J.L. Computational drug repositioning based on side-effects mined from social media. Peerj Comput. Sci. 2016, 2, e46. [Google Scholar] [CrossRef]
- Zhao, M.; Yang, C.C. Mining Online Heterogeneous Healthcare Networks for Drug Repositioning. In Proceedings of the Healthcare Informatics (ICHI), 2016 IEEE International Conference, Chicago, IL, USA, 4–7 October 2016; pp. 106–112. [Google Scholar]
- Shameer, K.; Readhead, B.; T Dudley, J. Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr. Top. Med. Chem. 2015, 15, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Yıldırım, M.A.; Goh, K.I.; Cusick, M.E.; Barabási, A.L.; Vidal, M. Drug—target network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.P.; Kwoh, C.K.; Yang, P.; Li, X.L.; Zheng, J. Drug–target interaction prediction by learning from local information and neighbors. Bioinformatics 2012, 29, 238–245. [Google Scholar] [CrossRef]
- Wang, W.; Yang, S.; Zhang, X.; Li, J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014, 30, 2923–2930. [Google Scholar] [CrossRef]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A Network Integration Approach for Drug-Target Interaction Prediction and Computational Drug Repositioning from Heterogeneous Information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Li, W.; Liu, G.; Tang, Y. Network-based methods for prediction of drug–target interactions. Front. Pharmacol. 2018, 9, 1134. [Google Scholar] [CrossRef] [Green Version]
- Tanoli, Z.; Alam, Z.; Ianevski, A.; Wennerberg, K.; Vähä-Koskela, M.; Aittokallio, T. Interactive visual analysis of drug–target interaction networks using drug target profiler, with applications to precision medicine and drug repurposing. Brief. Bioinform. 2018, 21, 211–220. [Google Scholar] [CrossRef]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Rittman, T.; Ge, H.; Menden, M.; Saez-Rodriguez, J. Transcriptional data: A new gateway to drug repositioning? Drug Discov. Today 2013, 18, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Kovács, I.A.; Barabási, A.L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Cheng, F.; Desai, R.J.; Handy, D.E.; Wang, R.; Schneeweiss, S.; Barabási, A.L.; Loscalzo, J. Network-based approach to prediction and population-based validation of in silico drug repurposing. Nat. Commun. 2018, 9, 2691. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Le, H.; Venkatesh, S. GraphDTA: Prediction of drug–target binding affinity using graph convolutional networks. BioRxiv 2019, 684662. [Google Scholar] [CrossRef] [Green Version]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [Green Version]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.A.; Hochreiter, S. Large-scale comparison of machine learning methods for drug target prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fang, H.; Reagan, K.; Xu, X.; Mendrick, D.L.; Slikker, W.; Tong, W. In silico drug repositioning–what we need to know. Drug Discov. Today 2013, 18, 110–115. [Google Scholar] [CrossRef]
- Kunimoto, R.; Bajorath, J. Design of a tripartite network for the prediction of drug targets. J.-Comput.-Aided Mol. Des. 2018, 32, 321–330. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2007, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Jacomy, M.; Venturini, T.; Heymann, S.; Bastian, M. ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS ONE 2014, 9, e98679. [Google Scholar] [CrossRef] [PubMed]
- Girvan, M.; Newman, M.E. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. Icwsm 2009, 8, 361–362. [Google Scholar]
- Newman, M.E. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.E. Equivalence between modularity optimization and maximum likelihood methods for community detection. Phys. Rev. E 2016, 94, 052315. [Google Scholar] [CrossRef] [Green Version]
- Noack, A. Modularity clustering is force-directed layout. Phys. Rev. E 2009, 79, 026102. [Google Scholar] [CrossRef] [Green Version]
- Barabási, A.L. Network Science; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Topirceanu, A.; Udrescu, M.; Marculescu, R. Weighted betweenness preferential attachment: A new mechanism explaining social network formation and evolution. Sci. Rep. 2018, 8, 10871. [Google Scholar] [CrossRef]
- Protain Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 25 May 2020).
- Zhang Lab. Available online: https://zhanglab.ccmb.med.umich.edu/ModRefiner/ (accessed on 25 May 2020).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grathwohl, W.; Wang, K.C.; Jacobsen, J.H.; Duvenaud, D.; Norouzi, M.; Swersky, K. Your Classifier is Secretly an Energy Based Model and You Should Treat it Like One. arXiv 2019, arXiv:1912.03263. [Google Scholar]
- Udrescu, L.; Sbârcea, L.; Topîrceanu, A.; Iovanovici, A.; Kurunczi, L.; Bogdan, P.; Udrescu, M. Clustering drug–drug interaction networks with energy model layouts: Community analysis and drug repurposing. Sci. Rep. 2016, 6, 32745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Joo, Y.H.; Shin, E.Y.; Park, E.J.; Kim, M.S. Anticancer effects of colchicine on hypopharyngeal cancer. Anticancer Res. 2017, 37, 6269–6280. [Google Scholar]
- Shen, M.; Zhang, Y.; Saba, N.; Austin, C.P.; Wiestner, A.; Auld, D.S. Identification of therapeutic candidates for chronic lymphocytic leukemia from a library of approved drugs. PLoS ONE 2013, 8, e75252. [Google Scholar] [CrossRef]
- Melo, S.; Villanueva, A.; Moutinho, C.; Davalos, V.; Spizzo, R.; Ivan, C.; Rossi, S.; Setien, F.; Casanovas, O.; Simo-Riudalbas, L.; et al. Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proc. Natl. Acad. Sci. USA 2011, 108, 4394–4399. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.; Varshney, P.; Sultana, S.; Yadav, J.; Saini, N. Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation. BMC Cancer 2015, 15, 581. [Google Scholar] [CrossRef] [Green Version]
- Fabian, I.; Reuveni, D.; Levitov, A.; Halperin, D.; Priel, E.; Shalit, I. Moxifloxacin enhances antiproliferative and apoptotic effects of etoposide but inhibits its proinflammatory effects in THP-1 and Jurkat cells. Br. J. Cancer 2006, 95, 1038–1046. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.; Sultana, S.; Yadav, J.; Saini, N. Gatifloxacin induces S and G2-phase cell cycle arrest in pancreatic cancer cells via p21/p27/p53. PLoS ONE 2012, 7, e47796. [Google Scholar] [CrossRef]
- Collaborative Group on Hormonal Factors in Breast Cancer. Type and timing of menopausal hormone therapy and breast cancer risk: Individual participant meta-analysis of the worldwide epidemiological evidence. Lancet 2019, 394, 1159–1168. [Google Scholar] [CrossRef]
- Ma, H.; Bernstein, L.; Pike, M.C.; Ursin, G. Reproductive factors and breast cancer risk according to joint estrogen and progesterone receptor status: A meta-analysis of epidemiological studies. Breast Cancer Res. 2006, 8, R43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, J.C.; Wang, S.M.; Guo, C.H.; Aw, S.E.; Zhao, Y.; Li, J.M.; Hui, K.M.; Lin, V.C. Gene regulation profile reveals consistent anticancer properties of progesterone in hormone-independent breast cancer cells transfected with progesterone receptor. Int. J. Cancer 2005, 117, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Preet, R.; Mohapatra, P.; Mohanty, S.; Sahu, S.K.; Choudhuri, T.; Wyatt, M.D.; Kundu, C.N. Quinacrine has anticancer activity in breast cancer cells through inhibition of topoisomerase activity. Int. J. Cancer 2012, 130, 1660–1670. [Google Scholar] [CrossRef]
- Yap, T.A.; Smith, A.D.; Ferraldeschi, R.; Al-Lazikani, B.; Workman, P.; De Bono, J.S. Drug discovery in advanced prostate cancer: Translating biology into therapy. Nat. Rev. Drug Discov. 2016, 15, 699. [Google Scholar] [CrossRef]
- Chung, L.C.; Tsui, K.H.; Feng, T.H.; Lee, S.L.; Chang, P.L.; Juang, H.H. L-Mimosine blocks cell proliferation via upregulation of B-cell translocation gene 2 and N-myc downstream regulated gene 1 in prostate carcinoma cells. Am. J. Physiol.-Cell Physiol. 2012, 302, C676–C685. [Google Scholar] [CrossRef] [Green Version]
- Belfort, R.; Berria, R.; Cornell, J.; Cusi, K. Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J. Clin. Endocrinol. Metab. 2010, 95, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Goto, M. A comparative study of anti-inflammatory and antidyslipidemic effects of fenofibrate and statins on rheumatoid arthritis. Mod. Rheumatol. 2010, 20, 238–243. [Google Scholar] [CrossRef]
- Barbaro, N.R.; Foss, J.D.; Kryshtal, D.O.; Tsyba, N.; Kumaresan, S.; Xiao, L.; Mernaugh, R.L.; Itani, H.A.; Loperena, R.; Chen, W.; et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017, 21, 1009–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, A.; Parodi, F.; Piacenza, G.; Mantellini, E.; Salio, M.; Cremonte, L.; Grosso, E. Antibacterial and antifungal activity of isoflurane and common anesthetic gases. Minerva Med. 1986, 77, 2007–2010. [Google Scholar]
- Barodka, V.M.; Acheampong, E.; Powell, G.; Lobach, L.; Logan, D.A.; Parveen, Z.; Armstead, V.; Mukhtar, M. Antimicrobial effects of liquid anesthetic isoflurane on Candida albicans. J. Transl. Med. 2006, 4, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauset, A.; Shalizi, C.R.; Newman, M.E. Power-law distributions in empirical data. SIAM Rev. 2009, 51, 661–703. [Google Scholar] [CrossRef] [Green Version]
- Alstott, J.; Bullmore, E.; Plenz, D. powerlaw: A Python package for analysis of heavy-tailed distributions. PLoS ONE 2014, 9, e85777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, R.R.; Fonseca, A.L.d.; Pinto, A.C.D.S.; Maia, E.H.B.; Silva, A.M.D.; Varotti, F.D.P.; Taranto, A.G. Brazilian malaria molecular targets (BraMMT): Selected receptors for virtual high-throughput screening experiments. Mem. Inst. Oswaldo Cruz 2019, 114. [Google Scholar] [CrossRef] [Green Version]
- Udrescu, M.; Udrescu, L. A Drug Repurposing Method Based on Drug–Drug Interaction Networks and Using Energy Model Layouts. In Computational Methods for Drug Repurposing; Springer: Berlin/Heidelberg, Germany, 2019; pp. 185–201. [Google Scholar]
- Mestres, J.; Gregori-Puigjane, E.; Valverde, S.; Sole, R.V. Data completeness—The Achilles heel of drug–target networks. Nat. Biotechnol. 2008, 26, 983–984. [Google Scholar] [CrossRef] [PubMed]
- Borgatti, S.P.; Carley, K.M.; Krackhardt, D. On the robustness of centrality measures under conditions of imperfect data. Soc. Net. 2006, 28, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.; Killingback, T.; Sundaram, B.; Wang, Z. Attack robustness and centrality of complex networks. PLoS ONE 2013, 8, e59613. [Google Scholar] [CrossRef] [Green Version]
- Breathnach, A. Azelaic acid: Potential as a general antitumoural agent. Med. Hypotheses 1999, 52, 221–226. [Google Scholar] [CrossRef]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [Green Version]
- Udrescu, L.; Sbârcea, L.; Fuliaș, A.; Ledeți, I.; Vlase, G.; Barvinschi, P.; Kurunczi, L. Physicochemical analysis and molecular modeling of the Fosinopril β-cyclodextrin inclusion complex. J. Spectrosc. 2014, 2014. [Google Scholar] [CrossRef]
- Chittepu, V.C.; Kalhotra, P.; Osorio-Gallardo, T.; Gallardo-Velázquez, T.; Osorio-Revilla, G. Repurposing of FDA-approved NSAIDs for DPP-4 inhibition as an alternative for diabetes mellitus treatment: Computational and in vitro study. Pharmaceutics 2019, 11, 238. [Google Scholar] [CrossRef] [Green Version]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput.-Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- DesJarlais, R.; Seibel, G.; Kuntz, I.; Furth, P.; Alvarez, J.; De Montellano, P.O.; DeCamp, D.; Babe, L.; Craik, C. Structure-based design of nonpeptide inhibitors specific for the human immunodeficiency virus 1 protease. Proc. Natl. Acad. Sci. USA 1990, 87, 6644–6648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalla, D.; Oerton, E.; Bender, A. Drug repurposing review. Ref. Modul. Chem. Mol. Sci. Chem. Eng. 2017. [Google Scholar] [CrossRef]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef] [Green Version]
- Elokely, K.M.; Doerksen, R.J. Docking challenge: Protein sampling and molecular docking performance. J. Chem. Inf. Model. 2013, 53, 1934–1945. [Google Scholar] [CrossRef] [Green Version]
- Maia, E.H.; Medaglia, L.R.; da Silva, A.M.; Taranto, A.G. Molecular Architect: A User-Friendly Workflow for Virtual Screening. ACS Omega 2020, 5, 6628–6640. [Google Scholar] [CrossRef] [Green Version]
- Martin, Y.C.; Kofron, J.L.; Traphagen, L.M. Do structurally similar molecules have similar biological activity? J. Med. Chem. 2002, 45, 4350–4358. [Google Scholar] [CrossRef]
- Yang, L.; Chen, J.; Shi, L.; Hudock, M.P.; Wang, K.; He, L. Identifying unexpected therapeutic targets via chemical-protein interactome. PLoS ONE 2010, 5, e9568. [Google Scholar] [CrossRef]
- Simon, Z.; Peragovics, Á.; Vigh-Smeller, M.; Csukly, G.; Tombor, L.; Yang, Z.; Zahoránszky-Köhalmi, G.; Végner, L.; Jelinek, B.; Hári, P.; et al. Drug effect prediction by polypharmacology-based interaction profiling. J. Chem. Inf. Model. 2012, 52, 134–145. [Google Scholar] [CrossRef]
- Haupt, V.J.; Schroeder, M. Old friends in new guise: Repositioning of known drugs with structural bioinformatics. Briefings Bioinform. 2011, 12, 312–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
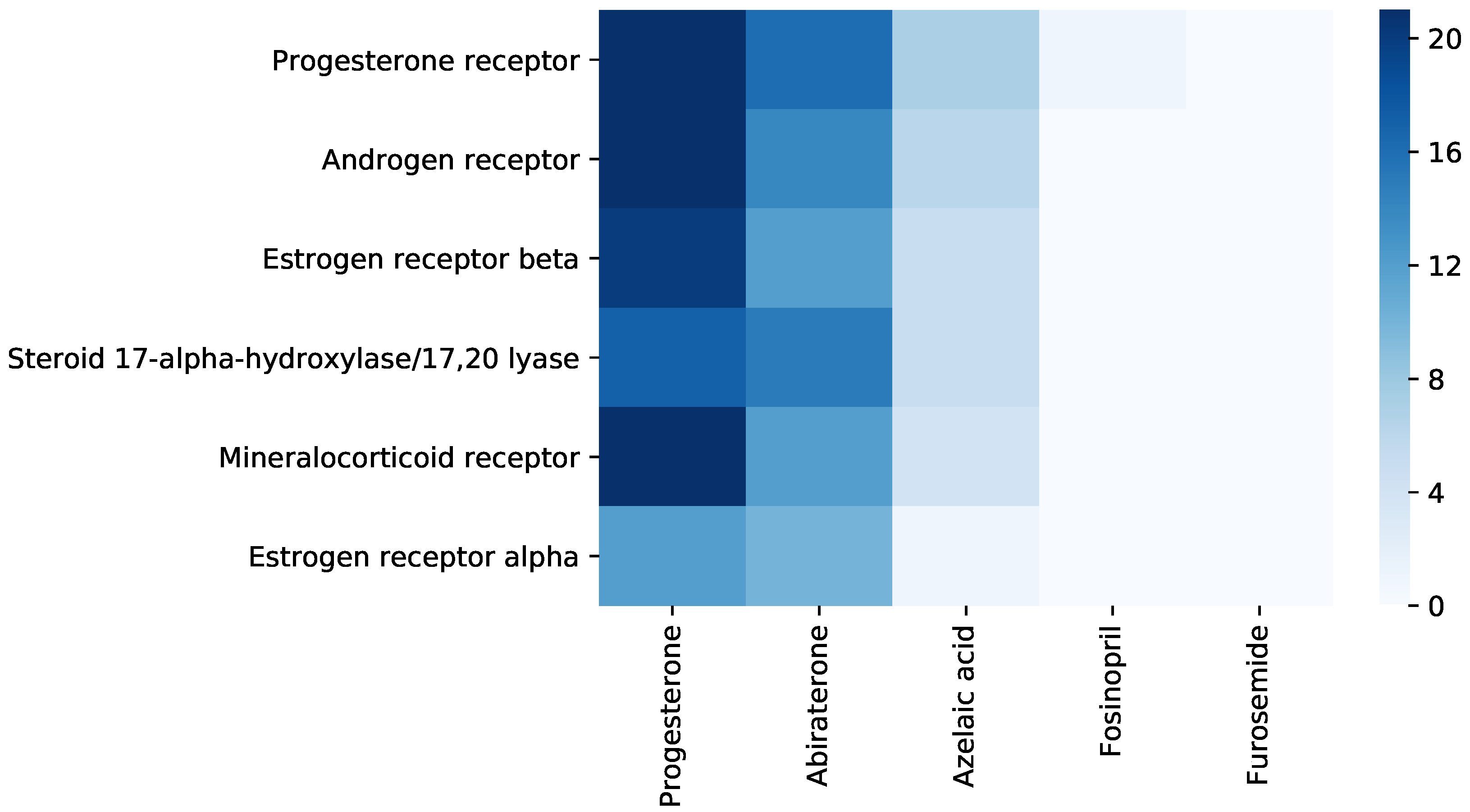
- Hille, U.E.; Hu, Q.; Vock, C.; Negri, M.; Bartels, M.; Müller-Vieira, U.; Lauterbach, T.; Hartmann, R.W. Novel CYP17 inhibitors: Synthesis, biological evaluation, structure–activity relationships and modelling of methoxy-and hydroxy-substituted methyleneimidazolyl biphenyls. Eur. J. Med. Chem. 2009, 44, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- Avendaño, C.; Menendez, J.C. Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- DeVore, N.M.; Scott, E.E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482, 116–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, C.Z.; Man, H.Z.; Saeed, Y.; Chen, D.C.; Wang, L.H.; Jiang, Y.Z. Computational insight into crucial binding features for metabolic specificity of cytochrome P450 17A1. Inform. Med. Unlocked 2019, 15, 100172. [Google Scholar] [CrossRef]
- Micheletti, G.; Calonghi, N.; Farruggia, G.; Strocchi, E.; Palmacci, V.; Telese, D.; Bordoni, S.; Frisco, G.; Boga, C. Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells. Molecules 2020, 25, 404. [Google Scholar] [CrossRef] [Green Version]
- Du, X.J.; Peng, X.J.; Zhao, R.Q.; Zhao, W.G.; Dong, W.L.; Liu, X.H. Design, synthesis and antifungal activity of threoninamide carbamate derivatives via pharmacophore model. J. Enzym. Inhib. Med. Chem. 2020, 35, 682–691. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Nodes [#] | DrugBank [%] | Literature [%] | Not Confirmed [%] | Hints | |
|---|---|---|---|---|---|---|
| 1 | Antineoplastic (mitotic inhibitors and DNA-damaging) | 37 | 40.54 | 37.84 | 21.62 | Besifloxacin Pefloxacin Norfloxacin Ofloxacin |
| 2 | Antihypertensive (sartans) | 10 | 100 | 0 | 0 | – |
| 3 | Anti-inflammatory | 84 | 65.48 | 28.57 | 5.95 | Glipizide |
| 4 | Antibacterial tetracyclines and Aminoglycosides | 20 | 95.00 | 0 | 5.00 | Plerixafor |
| 5 | Platelet aggregation inhibitor | 29 | 10.34 | 82.76 | 6.90 | – |
| 6 | Interfering with hormone-dependent cancers | 93 | 26.88 | 65.59 | 7.53 | Azelaic ac. |
| 7 | Anticancer (molecularly targeted) | 92 | 23.91 | 50.00 | 26.09 | Suramin Acetohydroxamic ac. Glyburide Gliquidone Tolbutamide |
| 8 | Anti-allergic | 51 | 86.27 | 11.76 | 1.96 | Butriptyline |
| 9 | Acting on muscles | 25 | 72.00 | 16.00 | 12.00 | – |
| 10 | Vasodilator | 37 | 48.65 | 24.32 | 27.03 | Tofisopam Mefloquine Oxtriphylline Enprofylline Roflumilast Aminophylline |
| 11 | Antiepileptic, hypnotic, and sedative | 19 | 84.21 | 10.53 | 5.26 | Barbituric ac. deriv. |
| 12 | Analgesic and used in opiate withdrawal & side-effects | 46 | 89.13 | 8.70 | 2.17 | – |
| 13 | Antihypertensive, anti-arrhythmic, anti-angina (mostly beta-blockers) | 26 | 92.31 | 3.85 | 3.85 | – |
| 14 | Anticholinergic | 53 | 100 | 0 | 0 | – |
| 15 | Interfering with Parasympathetic Nervous System | 97 | 42.27 | 42.27 | 15.46 | Doxazosin Terazosin Prazosin Paliperidone Aripiprazole Fenoldopam Dapiprazole Alfuzosin Tamsulosin Silodosin Amisulpiride Carphenazine Acetophenazine |
| Properties | Nodes [#] | DrugBank [%] | Literature [%] | Not Confirmed [%] | Hints | |
|---|---|---|---|---|---|---|
| 16 | Antidepressant and Central Nervous System stimulant | 26 | 92.31 | 7.69 | 0 | – |
| 17 | Sympathetic Nervous System acting | 61 | 85.25 | 8.20 | 6.56 | – |
| 18 | Antimigraine and antiemetic | 26 | 42.31 | 26.92 | 30.77 | Captodiame Ropinirole MDMA Dofetilide Rotigotine L-DOPA |
| 19 | Antiarrhythmic and anticonvulsant | 24 | 66.67 | 12.50 | 20.83 | Acarbose Hexylcaine |
| 20 | Antidepressant and anti-Parkinson | 21 | 57.14 | 14.29 | 28.57 | Quinidine Propafenone Cinchocaine MMDA Aprindine |
| 21 | Interfering with epilepsy and blood pressure | 12 | 41.67 | 25.00 | 33.33 | Miconazole Quinidine barbiturate |
| 22 | Antihypertensive and anticonvulsant | 20 | 80.00 | 15.00 | 5.00 | – |
| 23 | Anesthetic, analgesic, and muscle relaxant | 19 | 73.68 | 5.26 | 21.05 | Halofantrine Ibutilide Pentolinium |
| 24 | Interfering with K, Na, Ca homeostasis | 51 | 50.98 | 13.73 | 35.29 | Progabide Bethanidine Ellagic ac. Vigabatrin Ethinamate |
| 25 | Antifungal | 22 | 59.09 | 9.09 | 31.82 | Meprobamate Enflurane Sevoflurane Desflurane |
| 26 | Hypnotic and sedative | 7 | 100 | 0 | 0 | – |
| All | – | 1008 | 59.52 | 26.98 | 13.49 | – |
| 1 | 2 | 3 | 4 | 5 | ||
|---|---|---|---|---|---|---|
| 1 | Amsacrine | Colchicine | Podofilox | Lucanthone | Besifloxacin | |
| 2 | – | – | – | – | – | |
| 3 | Amiloride | Marimastat | Diclofenac | Thalidomide | Telmisartan | |
| 4 | Minocycline | Framycetin | Amikacin Tobramycin Netilmicin | Doxycycline Clomocycline Oxytetracycline | – | |
| 5 | Treprostinil | Iloprost | Captopril | Bimatoprost | Candoxatril | |
| 6 | Progesterone | Mimosine | Fluticasone propionate | Danazol | Spironolactone | |
| 7 | Vandetanib | Dalteparin | Dehydroepiandrosterone | Amlexanox | Atorvastatin | |
| 8 | Olopatadine | Terfenadine | Flunarizine | Astemizole | Epinastine | |
| 9 | Succinylcholine | Carbachol | Decamethonium | Pilocarpine | Cevimeline | |
| 10 | Nicotine | Melatonin | Amrinone | Dipyridamole | Naloxone | |
| 11 | Quinine | Phenobarbital Secobarbital Pentobarbital | Barbital Hexobarbital Aprobarbital | – | – | |
| 12 | Nimodipine | Adenosine | Drotaverine | Pentazocine | Loperamide | |
| 13 | Ketotifen | Amiodarone | Sotalol | Bevantolol | Penbutolol | |
| 14 | Disopyramide | Scopolamine | Ethopropazine | Paroxetine | Rocuronium | |
| 15 | Minaprine | Amitriptyline | Agomelatine | Orphenadrine | Imipramine | |
| 16 | Cocaine | Chloroprocaine | Procaine | Phenermine | Milnacipran | |
| 17 | Epinephrine | 4-Methoxyamphetamine | Pseudoephedrine | Ephedra | Methamphetamine | |
| 18 | Ginkgo biloba | Captodiame | Cisapride | Bromocriptine | Carteolol | |
| 19 | Acarbose | Lidocaine | Mexiletine | Etomidate | Flecainide | |
| 20 | Phenelzine | Agmatine | Quinidine Propafenone | Ephedrine | Amphetamine | |
| 21 | Zonisamide | Miconazole | Ethanol | Quinidine barbiturate | – | |
| 22 | Felodipine | Bepridil | Verapamil | Dextromethorphan | Amlodipine | |
| 23 | Halothane | Halofantrine | Tramadol | Ibutilide | Tubocurarine | |
| 24 | Thiamylal | Valproic Acid | Progabide | Bethanidine | Topiramate | |
| 25 | Meprobamate | Enflurane | Tioconazole | Clotrimazole | Methoxyflurane Isoflurane Sevoflurane | |
| 26 | Flunitrazepam | Eszopiclone | – | – | – | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udrescu, L.; Bogdan, P.; Chiş, A.; Sîrbu, I.O.; Topîrceanu, A.; Văruţ, R.-M.; Udrescu, M. Uncovering New Drug Properties in Target-Based Drug–Drug Similarity Networks. Pharmaceutics 2020, 12, 879. https://doi.org/10.3390/pharmaceutics12090879
Udrescu L, Bogdan P, Chiş A, Sîrbu IO, Topîrceanu A, Văruţ R-M, Udrescu M. Uncovering New Drug Properties in Target-Based Drug–Drug Similarity Networks. Pharmaceutics. 2020; 12(9):879. https://doi.org/10.3390/pharmaceutics12090879
Chicago/Turabian StyleUdrescu, Lucreţia, Paul Bogdan, Aimée Chiş, Ioan Ovidiu Sîrbu, Alexandru Topîrceanu, Renata-Maria Văruţ, and Mihai Udrescu. 2020. "Uncovering New Drug Properties in Target-Based Drug–Drug Similarity Networks" Pharmaceutics 12, no. 9: 879. https://doi.org/10.3390/pharmaceutics12090879
APA StyleUdrescu, L., Bogdan, P., Chiş, A., Sîrbu, I. O., Topîrceanu, A., Văruţ, R.-M., & Udrescu, M. (2020). Uncovering New Drug Properties in Target-Based Drug–Drug Similarity Networks. Pharmaceutics, 12(9), 879. https://doi.org/10.3390/pharmaceutics12090879