Drugs Modulating CD4+ T Cells Blood–Brain Barrier Interaction in Alzheimer’s Disease

 , , and
, , and
Abstract

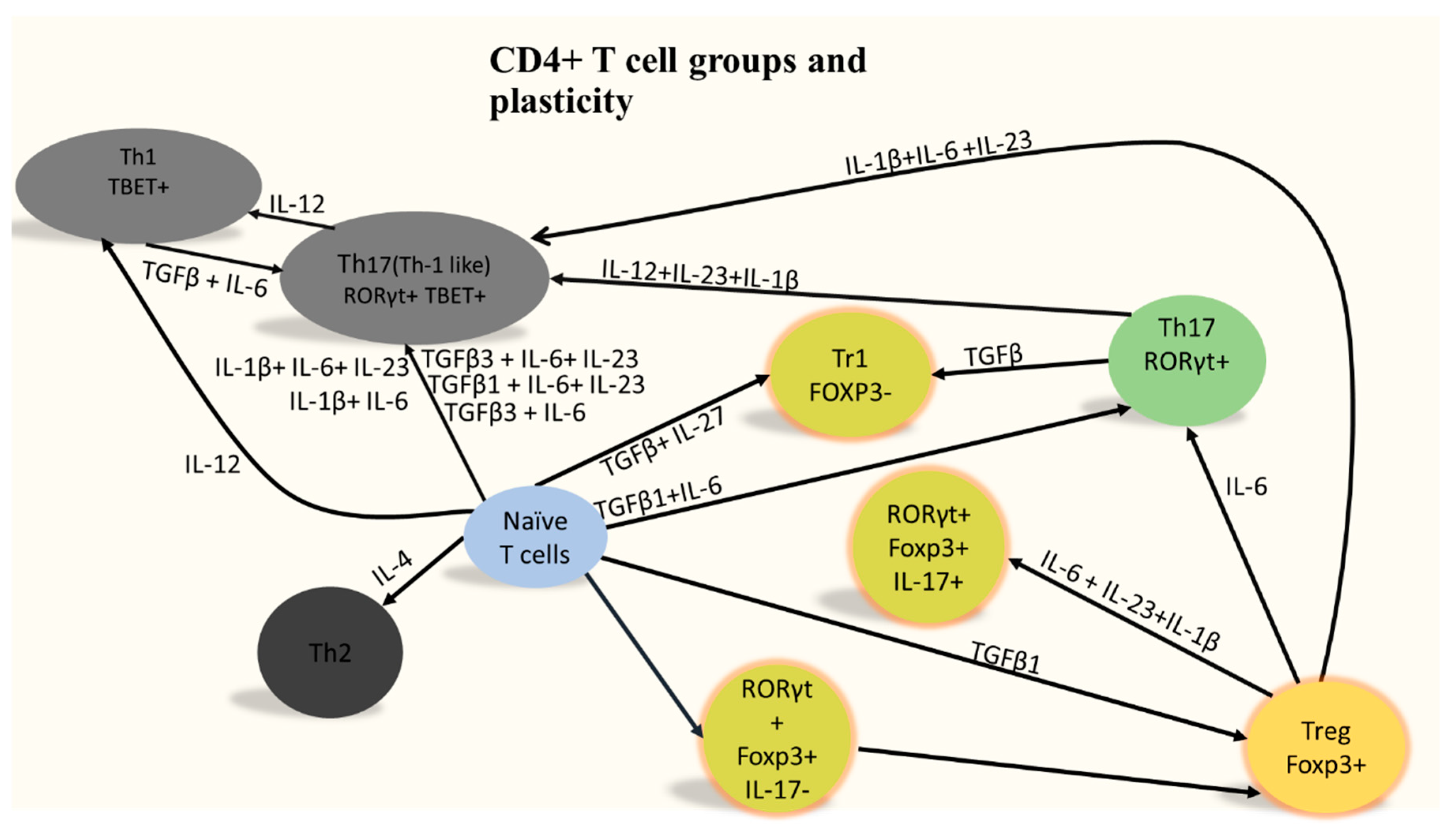
1. Who Are CD4+ T Cells?
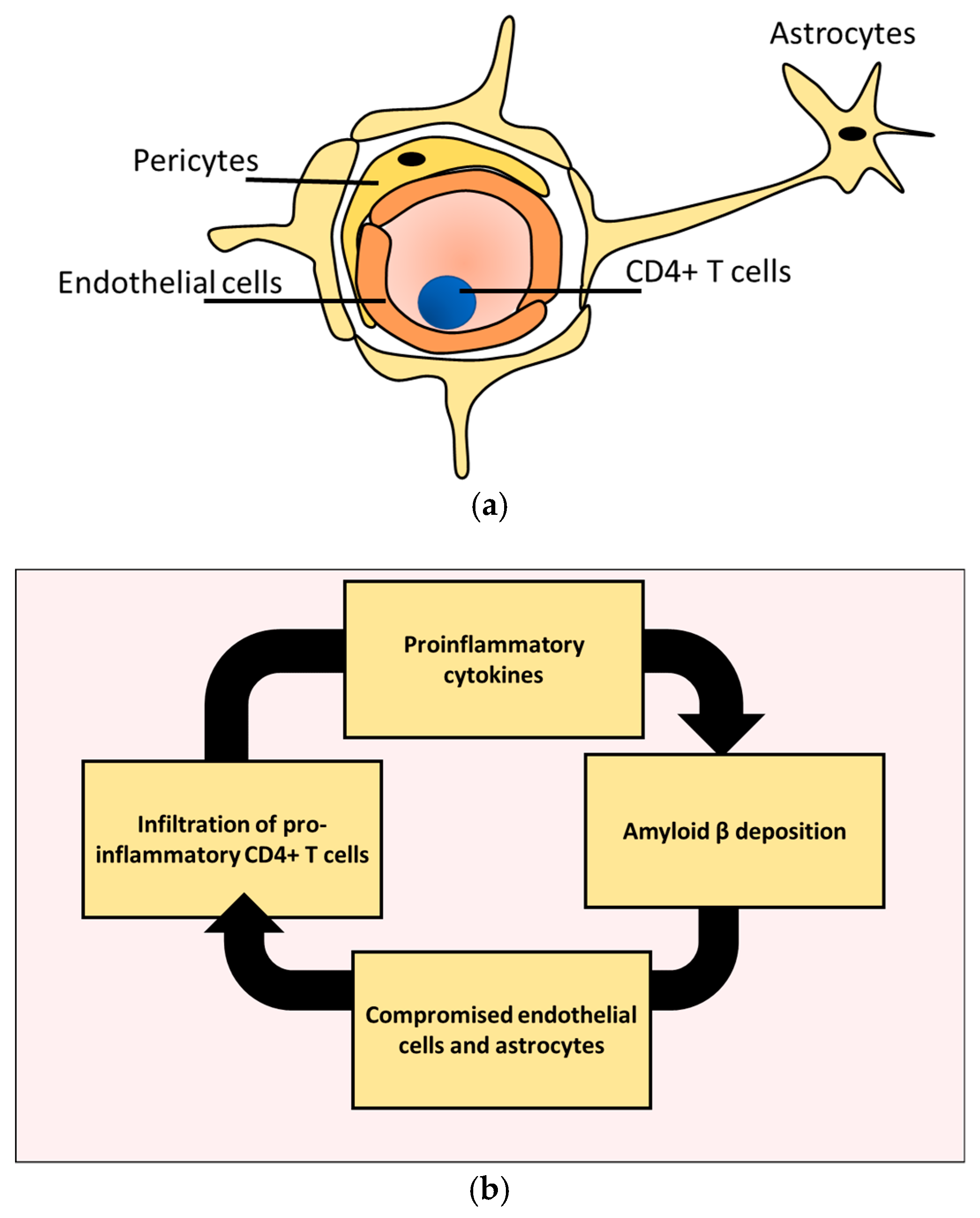
2. CD4+ T Cells Infiltration Affects Alzheimer’s Disease Prognosis
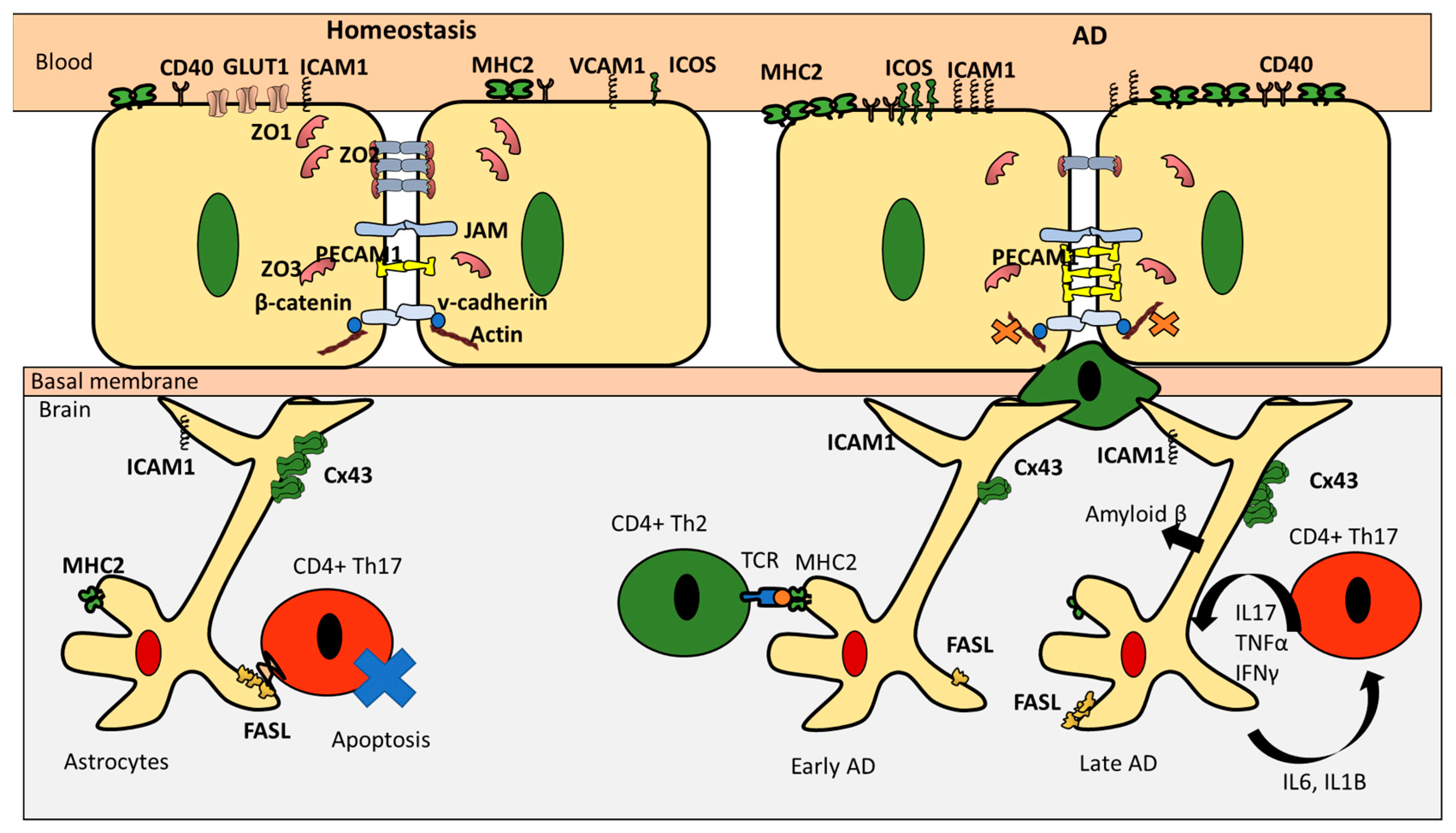
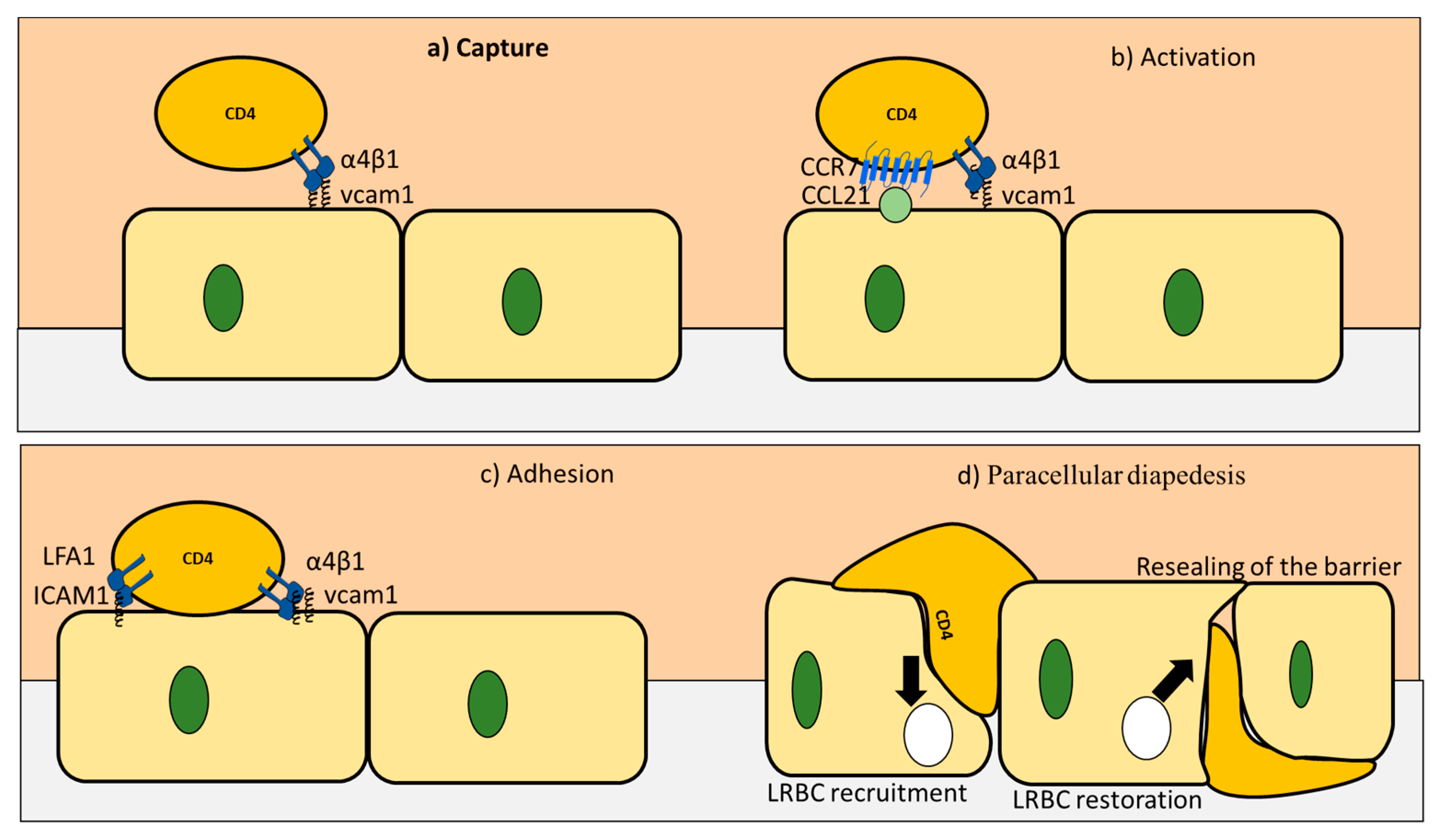
3. CD4+ T Cells Interaction with Endothelial Cells under Homeostasis
4. CD4+ T Cell Interactions with Astrocytes under Homeostasis
5. CD4+ T Cells Endothelial Interactions in AD
6. CD4+ T Cells and Astrocytes in AD
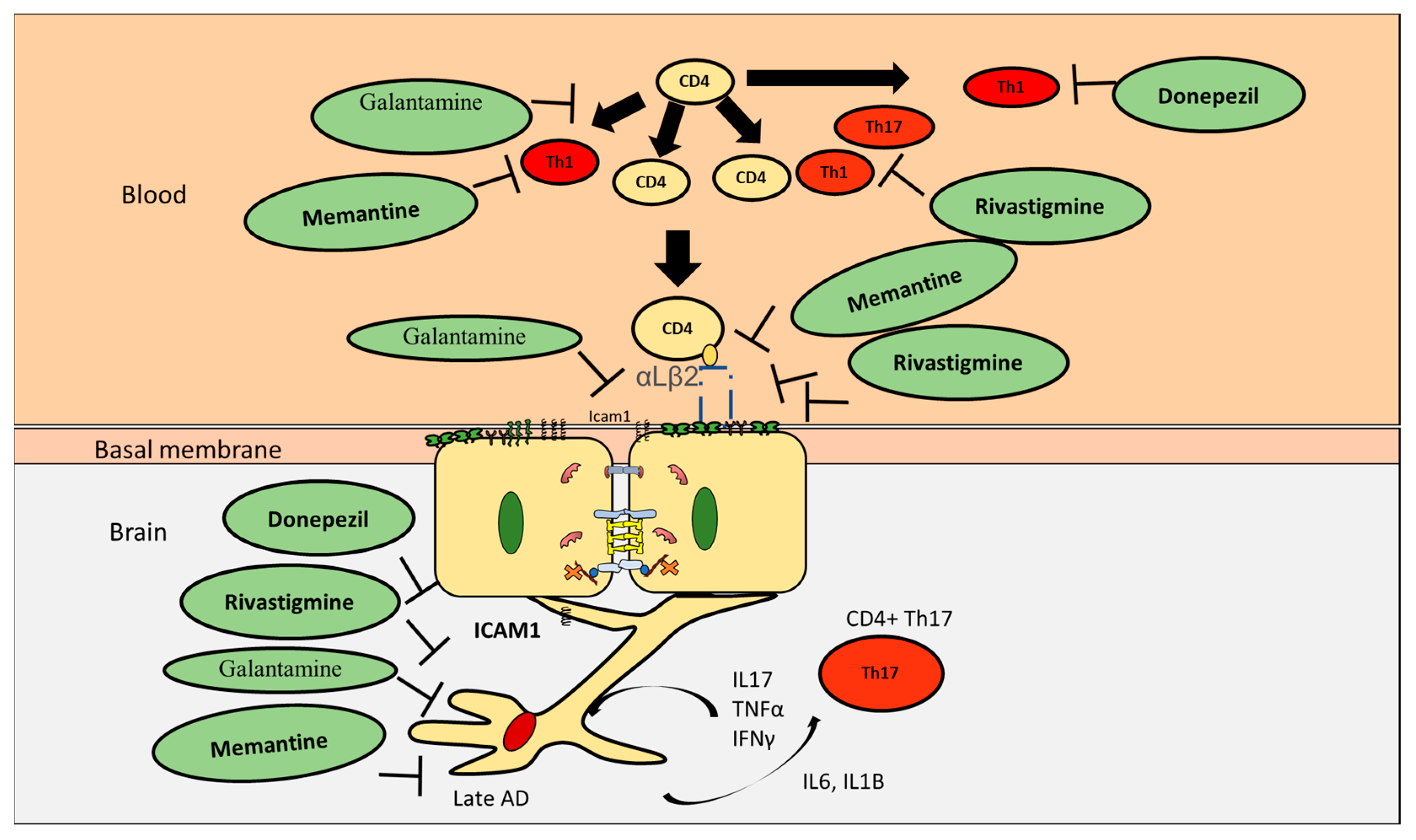
7. Effect of AD Drugs on CD4+ T Cells Proliferation and Differentiation
8. Effect of Current AD Drugs on Endothelial Cells
9. Effect of AD Drugs on CD4+ T Cell Subpopulations Interactions with Astrocytes in AD
10. Alternatives for Existing Classical AD Drugs
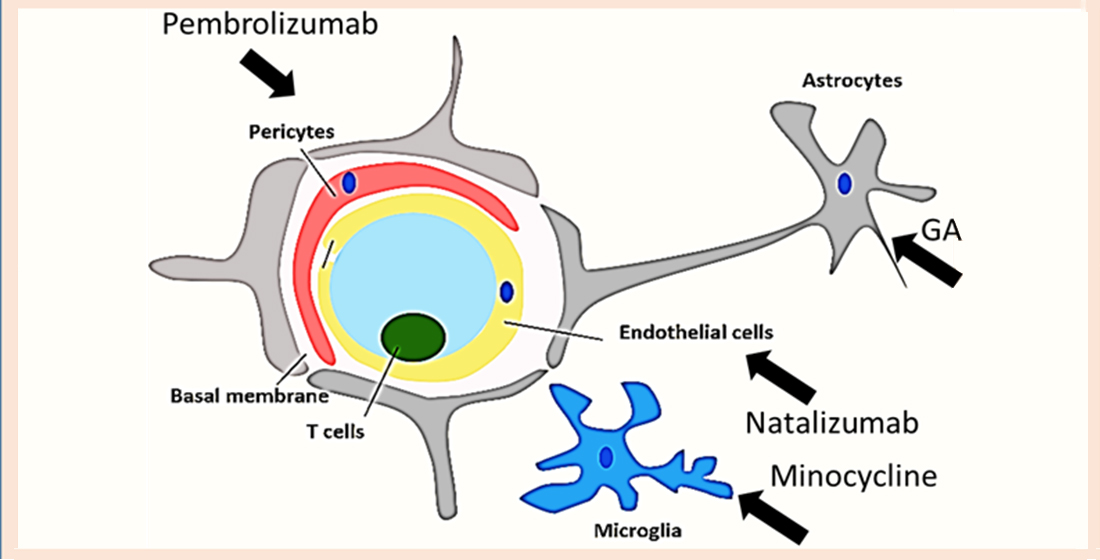
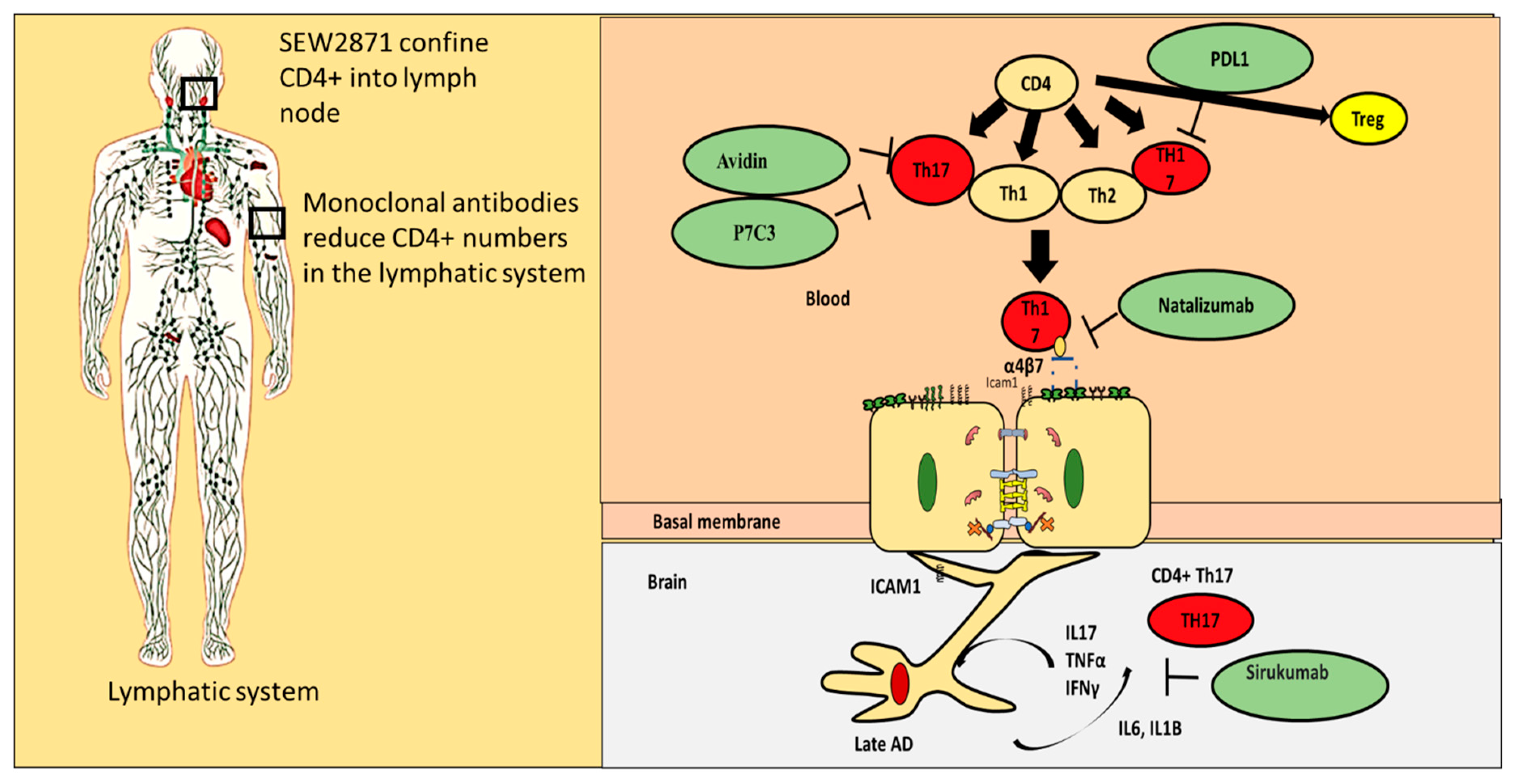
11. Repurposing Drug Strategies That Can Regulate Proinflammatory CD4+ T Cells Interaction with the BBB
- (i)
- Blocking proinflammatory CD4+ T cell subpopulations (e.g., Th1 and Th17) from entering the brain could reduce deposition and enhance cognitive function. Natalizumab is a monoclonal antibody against α4β7 and α4β1 integrins that are expressed on CD4+ T cells. This drug has proven potential in other neurogenerative diseases such as multiple sclerosis. It is important to note that one of the targets of Natalizumab (i.e., α4β7) is predominately expressed on proinflammatory CD4+ T cells and is less expressed on anti-inflammatory CD4+ T cells such as Tregs. [88]. Thus, Natalizumab is more selective for proinflammatory CD4+ T cells. Slavonic acid B has been reported to specifically inhibit Th1 infiltration of the brain in MS [89].
- (ii)
- Confining CD4+ T cells to lymph nodes is an alternative strategy, which could be achieved through employing peripheral modulators such as Sphingosine-1-Phosphate receptor (S1PR). S1PR limits lymphocytes traffic and decreases their peripheral count, mainly by confining them into lymph nodes [90]. Several S1PR agonists (ponesimod, siponimod, amiselimod, and ozanimod) are currently tested in MS clinical trials. SEW2871 administration prevented cognitive abilities in Alzheimer’s rat model, indicating the S1P1R signaling pathway could be a new therapeutic target.
- (iii)
- Monoclonal antibodies such as alemtuzumab decreased peripheral T cell count of both CD4+ T cells. However, its effect on AD patients is not yet known.
- (iv)
- Other compounds that are capable of inhibiting Th17 include Avidin, Curcumin, Naringin, and P7C3
- (v)
- One of the innovative approaches toward exploiting CD4+ T cells in treating AD is controlling CD4+ T cell fate; administration of PDL1 could help CD4+ T cells differentiate into Tregs and not into pathogenic Th17, which could be beneficial especially during the late stages of the disease.
- (vi)
- Inhibiting IL-6 production by astrocytes production could lead to decrease in Th17 differentiation and recruitment; currently available anti-IL-6 drugs are sirukumab, olokizumab, elsilimomab, and siltuximab.
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APC | Antigen presenting cells |
| BBB | Blood brain barrier |
| BEC | Brain Endothelial Cells |
| CNS | central nervous system |
| COX2 | Cyclooxygenase-2 |
| FoxP3 | Forkhead Box P3 |
| ICAM1 | Intercellular Adhesion Molecule 1 |
| IFNγ | Interferon gamma |
| IL-2 | Interleukin 2 |
| Il-6 | Interleukin 6 |
| NLRP3 | PYD domains-containing protein 3 |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| PD-1 | Programmed cell death protein 1 |
| PECAM1 | Platelet endothelial cell adhesion molecule 1 |
| S1PR | Sphingosine-1-Phosphate receptor |
| TGF-β | Transforming growth factor beta |
| TJ | tight junctions |
| TNFα | Tumor necrosis factor alpha |
| VCAM1 | Vascular cell adhesion protein 1 |
| AChE | Acetylcholinesterase |
| ZO1 | Zonula occludens-1 |
| BuChE | Butyrylcholinesterase |
| GLUT1 | Glucose transporter 1 |
| MLCK | Myosin light-chain kinase |
| VE-cadherin | Vascular endothelial cell-specific cadherin |
References
- Mickael, M.E.; Bhaumik, S.; Basu, R. Retinoid-related orphan receptor RORgammat in CD4(+) T-Cell-mediated intestinal homeostasis and inflammation. Am. J. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Wynn, T.A. Evolution of Th2 immunity: A rapid repair response to tissue destructive pathogens. PLoS Pathog. 2011, 7, e1002003. [Google Scholar] [CrossRef] [PubMed]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; Mace, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkhurst, C.N.; Muratet, M.; et al. A validated regulatory network for Th17 cell specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, S.; Basu, R. Cellular and molecular dynamics of Th17 differentiation and its developmental plasticity in the intestinal immune response. Front. Immunol. 2017, 8, 254. [Google Scholar] [CrossRef]
- Saresella, M.; Calabrese, E.; Marventano, I.; Piancone, F.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain Behav. Immun. 2011, 25, 539–547. [Google Scholar] [CrossRef]
- Richartz-Salzburger, E.; Batra, A.; Stransky, E.; Laske, C.; Kohler, N.; Bartels, M.; Buchkremer, G.; Schott, K. Altered lymphocyte distribution in Alzheimer’s disease. J. Psychiatr. Res. 2007, 41, 174–178. [Google Scholar] [CrossRef]
- Ferretti, M.T.; Merlini, M.; Spani, C.; Gericke, C.; Schweizer, N.; Enzmann, G.; Engelhardt, B.; Kulic, L.; Suter, T.; Nitsch, R.M. T-cell brain infiltration and immature antigen-presenting cells in transgenic models of Alzheimer’s disease-like cerebral amyloidosis. Brain Behav. Immun. 2016, 54, 211–225. [Google Scholar] [CrossRef]
- Cai, Z.; Qiao, P.F.; Wan, C.Q.; Cai, M.; Zhou, N.K.; Li, Q. Role of blood-brain barrier in Alzheimer’s disease. J. Alzheimers Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef]
- Tousi, B. The emerging role of bexarotene in the treatment of Alzheimer’s disease: Current evidence. Neuropsychiatr. Dis. Treat. 2015, 11, 311–315. [Google Scholar] [CrossRef]
- Alves, S.; Churlaud, G.; Audrain, M.; Michaelsen-Preusse, K.; Fol, R.; Souchet, B.; Braudeau, J.; Korte, M.; Klatzmann, D.; Cartier, N. Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer’s disease mice. Brain 2017, 140, 826–842. [Google Scholar] [CrossRef] [PubMed]
- Bryson, K.J.; Lynch, M.A. Linking T cells to Alzheimer’s disease: From neurodegeneration to neurorepair. Curr. Opin. Pharmacol. 2016, 26, 67–73. [Google Scholar] [CrossRef]
- Mietelska-Porowska, A.; Wojda, U. T Lymphocytes and inflammatory mediators in the interplay between brain and blood in Alzheimer’s disease: Potential Pools of New Biomarkers. J. Immunol. Res. 2017, 2017, 4626540. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Rosenzweig, N.; Kertser, A.; Deczkowska, A.; Sharif, A.M.; Spinrad, A.; Tsitsou-Kampeli, A.; Sarel, A.; Cahalon, L.; Schwartz, M. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 2015, 6, 7967. [Google Scholar] [CrossRef] [PubMed]
- Haddad-Tovolli, R.; Dragano, N.R.V.; Ramalho, A.F.S.; Velloso, L.A. Development and function of the blood-brain barrier in the context of metabolic control. Front. Neurosci. 2017, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 74. [Google Scholar] [CrossRef] [PubMed]
- Browne, T.C.; McQuillan, K.; McManus, R.M.; O’Reilly, J.A.; Mills, K.H.; Lynch, M.A. IFN-gamma Production by amyloid beta-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J. Immunol. 2013, 190, 2241–2251. [Google Scholar] [CrossRef]
- Cristiano, C.; Volpicelli, F.; Lippiello, P.; Buono, B.; Raucci, F.; Piccolo, M.; Iqbal, A.J.; Irace, C.; Miniaci, M.C.; Perrone Capano, C.; et al. Neutralization of IL-17 rescues amyloid-beta-induced neuroinflammation and memory impairment. Br. J. Pharmacol. 2019, 176, 3544–3557. [Google Scholar] [CrossRef] [PubMed]
- Dansokho, C.; Ait Ahmed, D.; Aid, S.; Toly-Ndour, C.; Chaigneau, T.; Calle, V.; Cagnard, N.; Holzenberger, M.; Piaggio, E.; Aucouturier, P.; et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 2016, 139, 1237–1251. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kanekiyo, T. Blood-brain barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef]
- Alasmari, F.; Alshammari, M.A.; Alasmari, A.F.; Alanazi, W.A.; Alhazzani, K. Neuroinflammatory cytokines induce amyloid beta neurotoxicity through modulating amyloid precursor protein levels/metabolism. Biomed. Res. Int. 2018, 2018, 3087475. [Google Scholar] [CrossRef] [PubMed]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane proteins of the tight junctions at the blood-brain barrier: Structural and functional aspects. Semin. Cell Dev. Biol. 2015, 38, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.; Campbell, M. Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 2016, 4, e1138017. [Google Scholar] [CrossRef] [PubMed]
- Sonar, S.A.; Lal, G. Blood-brain barrier and its function during inflammation and autoimmunity. J. Leukoc. Biol. 2018, 103, 839–853. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Baena, V.; Ge, S.; Jiang, X.; Jellison, E.R.; Kiprono, T.; Agalliu, D.; Pachter, J.S. Appearance of claudin-5(+) leukocytes in the central nervous system during neuroinflammation: A novel role for endothelial-derived extracellular vesicles. J. Neuroinflamm. 2016, 13, 292. [Google Scholar] [CrossRef] [PubMed]
- Cummins, P.M. Occludin: One protein, many forms. Mol. Cell Biol. 2012, 32, 242–250. [Google Scholar] [CrossRef]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef]
- Banh, C.; Fugere, C.; Brossay, L. Immunoregulatory functions of KLRG1 cadherin interactions are dependent on forward and reverse signaling. Blood 2009, 114, 5299–5306. [Google Scholar] [CrossRef]
- Ma, J.; Wang, R.; Fang, X.; Sun, Z. β-catenin/TCF-1 pathway in T cell development and differentiation. J. Neuroimmune Pharmacol. 2012, 7, 750–762. [Google Scholar] [CrossRef]
- Fernandez-Riejos, P.; Najib, S.; Santos-Alvarez, J.; Martin-Romero, C.; Perez-Perez, A.; Gonzalez-Yanes, C.; Sanchez-Margalet, V. Role of leptin in the activation of immune cells. Mediat. Inflamm. 2010, 2010, 568343. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.W.; Gu, C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Wheway, J.; Obeid, S.; Couraud, P.O.; Combes, V.; Grau, G.E. The brain microvascular endothelium supports T cell proliferation and has potential for alloantigen presentation. PLoS ONE 2013, 8, e52586. [Google Scholar] [CrossRef]
- Nishihara, H.; Soldati, S.; Mossu, A.; Rosito, M.; Rudolph, H.; Muller, W.A.; Latorre, D.; Sallusto, F.; Sospedra, M.; Martin, R.; et al. Human CD4(+) T cell subsets differ in their abilities to cross endothelial and epithelial brain barriers in vitro. Fluids Barriers CNS 2020, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J. Neural Transm. 2006, 113, 477–485. [Google Scholar] [CrossRef]
- Noor, S.; Wilson, E.H. Role of C-C chemokine receptor type 7 and its ligands during neuroinflammation. J. Neuroinflammation 2012, 9, 77. [Google Scholar] [CrossRef]
- Beurel, E.; Harrington, L.E.; Buchser, W.; Lemmon, V.; Jope, R.S. Astrocytes modulate the polarization of CD4+ T cells to Th1 cells. PLoS ONE 2014, 9, e86257. [Google Scholar] [CrossRef]
- Xie, L.; Yang, S.H. Interaction of astrocytes and T cells in physiological and pathological conditions. Brain Res. 2015, 1623, 63–73. [Google Scholar] [CrossRef]
- Boulay, A.C.; Mazeraud, A.; Cisternino, S.; Saubamea, B.; Mailly, P.; Jourdren, L.; Blugeon, C.; Mignon, V.; Smirnova, M.; Cavallo, A.; et al. Immune quiescence of the brain is set by astroglial connexin 43. J. Neurosci. 2015, 35, 4427–4439. [Google Scholar] [CrossRef]
- Frohman, E.M.; Frohman, T.C.; Dustin, M.L.; Vayuvegula, B.; Choi, B.; Gupta, A.; van den Noort, S.; Gupta, S. The induction of intercellular adhesion molecule 1 (ICAM-1) expression on human fetal astrocytes by interferon-λ, tumor necrosis factor α, lymphotoxin, and interleukin-1: Relevance to intracerebral antigen presentation. J. Neuroimmunol. 1989, 23, 117–124. [Google Scholar] [CrossRef]
- Gimsa, U.; Mitchison, N.A.; Brunner-Weinzierl, M.C. Immune privilege as an intrinsic CNS property: Astrocytes protect the CNS against T-cell-mediated neuroinflammation. Mediators Inflamm. 2013, 2013, 320519. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Newman, E.A. Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef] [PubMed]
- Sreeramkumar, V.; Fresno, M.; Cuesta, N. Prostaglandin E2 and T cells: Friends or foes? Immunol. Cell Biol. 2012, 90, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Covelo, A.; Araque, A. Neuronal activity determines distinct gliotransmitter release from a single astrocyte. Elife 2018, 7, e32237. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Axtell, R.; Mitra, A.; Miranda, M.; Lock, C.; Tsien, R.W.; Steinman, L. Inhibitory role for GABA in autoimmune inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2580–2585. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S. The short-term stress response—Mother nature’s mechanism for enhancing protection and performance under conditions of threat, challenge, and opportunity. Front. Neuroendocrinol. 2018, 49, 175–192. [Google Scholar] [CrossRef]
- Kubick, N.; Brosamle, D.; Mickael, M.E. Molecular evolution and functional divergence of the IgLON family. Evol. Bioinform. Online 2018, 14. [Google Scholar] [CrossRef]
- Vallee, A.; Lecarpentier, Y. Alzheimer disease: Crosstalk between the canonical Wnt/Beta-Catenin pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Muller, W.A. Mechanisms of leukocyte transendothelial migration. Annu. Rev. Pathol. 2011, 6, 323–344. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Lopes Pinheiro, M.A.; Kamermans, A.; Garcia-Vallejo, J.J.; van Het Hof, B.; Wierts, L.; O’Toole, T.; Boeve, D.; Verstege, M.; van der Pol, S.M.; van Kooyk, Y.; et al. Internalization and presentation of myelin antigens by the brain endothelium guides antigen-specific T cell migration. Elife 2016, 5, e13149. [Google Scholar] [CrossRef] [PubMed]
- Wennstrom, M.; Nielsen, H.M. Cell adhesion molecules in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 65–77. [Google Scholar] [CrossRef] [PubMed]
- McQuillan, K.; Lynch, M.A.; Mills, K.H. Activation of mixed glia by Abeta-specific Th1 and Th17 cells and its regulation by Th2 cells. Brain Behav. Immun. 2010, 24, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Abeta production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef]
- Chun, H.; Marriott, I.; Lee, C.J.; Cho, H. Elucidating the interactive roles of glia in Alzheimer’s disease using established and newly developed experimental models. Front. Neurol. 2018, 9, 797. [Google Scholar] [CrossRef]
- Li, C.; Zhao, R.; Gao, K.; Wei, Z.; Yin, M.Y.; Lau, L.T.; Chui, D.; Yu, A.C. Astrocytes: Implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 67–80. [Google Scholar] [CrossRef]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Hatton, R.D.; Weaver, C.T. The Th17 family: Flexibility follows function. Immunol. Rev. 2013, 252, 89–103. [Google Scholar] [CrossRef]
- Xing, L.; Yang, T.; Cui, S.; Chen, G. Connexin hemichannels in astrocytes: Role in CNS disorders. Front. Mol. Neurosci. 2019, 12, 23. [Google Scholar] [CrossRef]
- Melnikova, I. Therapies for Alzheimer’s disease. Nat. Rev. Drug Discov. 2007, 6, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Slowikowski, S.P.; Westaway, D.; Mount, H.T. Interactions between beta-amyloid and central cholinergic neurons: Implications for Alzheimer’s disease. J. Psychiatry Neurosci. 2004, 29, 427–441. [Google Scholar] [PubMed]
- Seltzer, B. Donepezil: A review. Expert Opin. Drug Metab. Toxicol. 2005, 1, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Drug delivery to the brain in Alzheimer’s disease: Consideration of the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Jozwik, A.; Landowski, J.; Bidzan, L.; Fulop, T.; Bryl, E.; Witkowski, J.M. Beta-amyloid peptides enhance the proliferative response of activated CD4CD28 lymphocytes from Alzheimer disease patients and from healthy elderly. PLoS ONE 2012, 7, e33276. [Google Scholar] [CrossRef]
- Jiang, Y.; Zou, Y.; Chen, S.; Zhu, C.; Wu, A.; Liu, Y.; Ma, L.; Zhu, D.; Ma, X.; Liu, M.; et al. The anti-inflammatory effect of donepezil on experimental autoimmune encephalomyelitis in C57 BL/6 mice. Neuropharmacology 2013, 73, 415–424. [Google Scholar] [CrossRef]
- Onor, M.L.; Trevisiol, M.; Aguglia, E. Rivastigmine in the treatment of Alzheimer’s disease: An update. Clin. Interv. Aging 2007, 2, 17–32. [Google Scholar] [CrossRef]
- Busse, S.; Steiner, J.; Glorius, S.; Dobrowolny, H.; Greiner-Bohl, S.; Mawrin, C.; Bommhardt, U.; Hartig, R.; Bogerts, B.; Busse, M. VGF expression by T lymphocytes in patients with Alzheimer’s disease. Oncotarget 2015, 6, 14843–14851. [Google Scholar] [CrossRef]
- Darreh-Shori, T.; Jelic, V. Safety and tolerability of transdermal and oral rivastigmine in Alzheimer’s disease and Parkinson’s disease dementia. Expert Opin. Drug Saf. 2010, 9, 167–176. [Google Scholar] [CrossRef]
- Nizri, E.; Irony-Tur-Sinai, M.; Faranesh, N.; Lavon, I.; Lavi, E.; Weinstock, M.; Brenner, T. Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J. Neuroimmunol. 2008, 203, 12–22. [Google Scholar] [CrossRef]
- Raskind, M.A.; Peskind, E.R.; Wessel, T.; Yuan, W. Galantamine in AD: A 6-month randomized, placebo-controlled trial with a 6-month extension. The Galantamine USA-1 Study Group. Neurology 2000, 54, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.; Murray, J. Galantamine: A randomized, double-blind, dose comparison in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2001, 16, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Hanes, W.M.; Olofsson, P.S.; Kwan, K.; Hudson, L.K.; Chavan, S.S.; Pavlov, V.A.; Tracey, K.J. Galantamine attenuates type 1 diabetes and inhibits anti-insulin antibodies in nonobese diabetic mice. Mol. Med. 2015, 21, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine-searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef]
- Lowinus, T.; Bose, T.; Busse, S.; Busse, M.; Reinhold, D.; Schraven, B.; Bommhardt, U.H. Immunomodulation by memantine in therapy of Alzheimer’s disease is mediated through inhibition of Kv1.3 channels and T cell responsiveness. Oncotarget 2016, 7, 53797–53807. [Google Scholar] [CrossRef]
- Kahlfuss, S.; Simma, N.; Mankiewicz, J.; Bose, T.; Lowinus, T.; Klein-Hessling, S.; Sprengel, R.; Schraven, B.; Heine, M.; Bommhardt, U. Immunosuppression by N-methyl-D-aspartate receptor antagonists is mediated through inhibition of Kv1.3 and KCa3.1 channels in T cells. Mol. Cell Biol. 2014, 34, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Di, X.; Liu, Y. Protective effects of Donepezil against endothelial permeability. Eur. J. Pharmacol. 2017, 811, 60–65. [Google Scholar] [CrossRef]
- Wazea, S.A.; Wadie, W.; Bahgat, A.K.; El-Abhar, H.S. Galantamine anti-colitic effect: Role of alpha-7 nicotinic acetylcholine receptor in modulating Jak/STAT3, NF-kappaB/HMGB1/RAGE and p-AKT/Bcl-2 pathways. Sci. Rep. 2018, 8, 5110. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, T.-T.; Wu, C.-X.; Lan, X.; Du, G.-H. Targeting the neurovascular unit: Development of a new model and consideration for novel strategy for Alzheimer’s disease. Brain Res. Bull. 2011, 86, 13–21. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, Y.; Xu, Y.; Qu, P.; Wang, M. Memantine protects against ischemia/reperfusion-induced brain endothelial permeability. IUBMB Life 2018, 70, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zou, Z.; Gong, Y.; Yuan, D.; Chen, X.; Sun, T. Regulation of human brain microvascular endothelial cell adhesion and barrier functions by memantine. J. Mol. Neurosci. 2017, 62, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sanchez, K.; Ariza-Salamanca, D.; Mora-Munoz, L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhao, L.; Chen, X.; Cheng, X.; Zhang, Y. Galantamine attenuates amyloid-beta deposition and astrocyte activation in APP/PS1 transgenic mice. Exp. Gerontol. 2015, 72, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, K.; Obara-Michlewska, M.; Zielinska, M.; Albrecht, J. NMDA receptors in astrocytes: In search for roles in neurotransmission and astrocytic homeostasis. Int. J. Mol. Sci. 2019, 20, 309. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, L.; Wu, C.Y. Triptolide inhibits the differentiation of Th17 cells and suppresses collagen-induced arthritis. Scand. J. Immunol. 2008, 68, 383–390. [Google Scholar] [CrossRef]
- Wang, Q.; Xiao, B.; Cui, S.; Song, H.; Qian, Y.; Dong, L.; An, H.; Cui, Y.; Zhang, W.; He, Y.; et al. Triptolide treatment reduces Alzheimer’s disease (AD)-like pathology through inhibition of BACE1 in a transgenic mouse model of AD. Dis. Model. Mech. 2014, 7, 1385–1395. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. S1), S167–S174. [Google Scholar] [CrossRef]
- Stenner, M.P.; Waschbisch, A.; Buck, D.; Doerck, S.; Einsele, H.; Toyka, K.V.; Wiendl, H. Effects of natalizumab treatment on Foxp3+ T regulatory cells. PLoS ONE 2008, 3, e3319. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Ma, D.; Gong, Y.; Yu, T.; Yao, G. Salvianolic acid B ameliorates CNS autoimmunity by suppressing Th1 responses. Neurosci. Lett. 2016, 619, 92–99. [Google Scholar] [CrossRef]
- Perez-Jeldres, T.; Tyler, C.J.; Boyer, J.D.; Karuppuchamy, T.; Yarur, A.; Giles, D.A.; Yeasmin, S.; Lundborg, L.; Sandborn, W.J.; Patel, D.R.; et al. Targeting cytokine signaling and lymphocyte traffic via small molecules in inflammatory bowel disease: JAK inhibitors and S1PR agonists. Front. Pharmacol. 2019, 10, 212. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD4 Type | Event | Effect | Effect |
|---|---|---|---|
| Amyloid reactive CD4+ T cells | Produce proinflammatory cytokines | Prolonged inflammation in Alzheimer’s | Negative |
| CD4+ T cells | Depletion | Decreased impairment | Negative |
| CD4+ T cells | Apoptosis | Reverse AD | Positive |
| CD4+ T cells | Deficient mice | Larger growth of AD | Positive |
| Th2 CD4+ T cells | Induce of Aβ autoantibodies | Protective against AD | Positive |
| Treg CD4+ T cells | Increased microglia specific for plaques | Enhance cognitive abilities | Positive |
| Th1 CD4+ T cells | Produce IFNg, increase Aβ | Worsen cognitive abilities | Negative |
| Th17 CD4+ T cells | Produce proinflammatory cytokine | Increase inflammation in Alzheimer brain | Negative |
| Characteristics | Component | Function |
|---|---|---|
| High structural integrity | Tight junctions (occludin, JAMs, and zonulae occludens) | Restrict paracellular diffusion of CD4+ T cells |
| High structural integrity | Adherens junction (cadherins, catenin, and actin) | Mediate cell-cell adhesion to reduce CD4+ T cells infiltration |
| Low transcytosis rates of macromolecules promoting CD4+ T cells | albumin and leptin | Reduce T cells proliferation |
| High transcytosis rates of macromolecules reducing CD4+ T cells proliferation | GLUT1 | Regulate T cells numbers |
| Low expression of leucocytes adhesion molecules | E-selectin and ICAM1 | Reduce CD4+ T cells adhesion to the endothelial cells |
| Stage | Component | Result of Stage Completion |
|---|---|---|
| Rolling stage | VCAM1 bind to VLA4 on CD4+ T cells | Weak bonds that reduce the speed of CD4+ T cells |
| Activation stage | e.g., CCL19 and CCL21 expressed on endothelial cells activate CD4+ T cells | CD4+ T cells are activated |
| Arrest stage | VCAM1 and ICAM bind to their ligands to mediate T cells arrest | CD4+ T cells attached to the endothelial cells |
| Diapedesis | V-cadherin gets phosphorylated | the catenin is released |
| PECAM upregulation | Activate kinesin molecular motors | |
| LRBC trafficking | Expand the distance between endothelial cells |
| Interaction | Effect |
|---|---|
| Astrocytes express high levels of Cx43 | Reduce CD4+ T cells infiltration |
| Astrocytes express low levels of MHC2 | Reduce CD4+ T cells activation |
| Astrocytes express low levels of ICAM | Reduce CD4+ T cells adhesion |
| Astrocytes activate FASL pathway | Induce apoptosis in CD4+ T cells |
| Astrocytes production of PGE2 | Induce Treg and suppress Th1, Th12, Th17 |
| Astrocytes production of GABA | Reduces Th-cell induced inflammation and hence migration |
| Characteristics | Component | Function |
|---|---|---|
| Low structural integrity | Reduction in the expression of TJ proteins, occludin, claudin5, and ZO1 | Reduce restriction of CD4+ T cells paracellular infiltration |
| Low structural integrity | Reduction in B catenein expression | Reduce adhesion between endothelial cells |
| Low transcytosis rates of macromolecules reducing CD4+ T cells proliferation | GLUT1 | Reducing restriction of CD4+ T cells proliferation |
| Increase adhesion molecules | ICAM1 and VCAM1 | Increase CD4+ T cells adhesion to the endothelial cells layer |
| Interaction | Effect on CD4+ T Cells Migration |
|---|---|
| Astrocytes present Aβ to Th2 | Th2 activation and acquire regulatory phenotype and inhibit Th1 and Th17 |
| Astrocytes downregulate Cx43 (earlier stage) | Increase interaction with CD4+ T cells |
| Astrocytes produce IL-6 | Activation of Th2, Th17 and inhibit Treg and Th1 |
| Uptake TNF-α+IFN-γ produced by proinflammatory CD4+ T cells | Increase astrocytes activation and increase production of plaque |
| Astrocytes upregulate Cx43 (later stage) | decrease interaction with CD4+ T cells |
| Astrocytes slightly upregulate MHC2 | Prevent further interaction with CD4+ T cells |
| Astrocytes upregulate FASL | Cause apoptosis |
| Drug | Effect CD4+ T Cells in AD |
|---|---|
| Donepezil | Inhibit Th1, promote Th2 |
| Rivastigmine | Inhibit Th1, Th17 but not Th2 |
| Galantamine | Decrease CD4+ T cells |
| Memantine | Inhibit Th1, promote Th2 |
| Drug | Effect Endothelial Cells |
|---|---|
| Donepezil | Increased BBB integrity, upregulated VE-cadherin and ZO1 |
| Rivastigmine | Increased transendothelial electrical resistance, decreased ICAM1 and VAM1 and preserved occluding and ZO1 |
| Galantamine | Significantly reduces ICAM1 |
| Memantine | Inhibit ICAM1, increase BBB integrity, upregulate VE-cadherin and occludin |
| Drug | Effect on CD4+ T Cells Interaction with Astrocytes |
|---|---|
| Donepezil | Reduce astrocytes activation |
| Rivastigmine | Reduce astrocytes activation |
| memantine | Reduce astrocytes activation |
| Molecule | Effect on Th17/Treg Axis | Effect on AD |
|---|---|---|
| Triptolide | Inhibit Th17 cells [85] | Potential candidate for drug [86]. |
| Caffeine (1,3,7-Trimethylpurine-2,6-dione) | Favors autoimmunity Treg and decreases cytokines needed for Th17 and Th1 | Improve prognosis [87] |
| Albumin | Albumin functions as an inhibitor of T cell adhesion in vitro. Negatively correlated with Th17, positively correlated with Treg | Improve prognosis |
| Huperzine A | reduce lymphocyte proliferation and the secretion of proinflammatory cytokines | Choline esterase inhibitor |
| Insulin | Increase Treg | Improve prognosis |
| Ladostigil | reduce lymphocyte proliferation and the secretion of proinflammatory cytokines | a cholinesterase and monoamine oxidase inhibitor |
| Quercetin | Immunosuppressive, inhibit Th17 | Improve prognosis |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubick, N.; Flournoy, P.C.H.; Enciu, A.-M.; Manda, G.; Mickael, M.-E. Drugs Modulating CD4+ T Cells Blood–Brain Barrier Interaction in Alzheimer’s Disease. Pharmaceutics 2020, 12, 880. https://doi.org/10.3390/pharmaceutics12090880
Kubick N, Flournoy PCH, Enciu A-M, Manda G, Mickael M-E. Drugs Modulating CD4+ T Cells Blood–Brain Barrier Interaction in Alzheimer’s Disease. Pharmaceutics. 2020; 12(9):880. https://doi.org/10.3390/pharmaceutics12090880
Chicago/Turabian StyleKubick, Norwin, Patrick C. Henckell Flournoy, Ana-Maria Enciu, Gina Manda, and Michel-Edwar Mickael. 2020. "Drugs Modulating CD4+ T Cells Blood–Brain Barrier Interaction in Alzheimer’s Disease" Pharmaceutics 12, no. 9: 880. https://doi.org/10.3390/pharmaceutics12090880
APA StyleKubick, N., Flournoy, P. C. H., Enciu, A.-M., Manda, G., & Mickael, M.-E. (2020). Drugs Modulating CD4+ T Cells Blood–Brain Barrier Interaction in Alzheimer’s Disease. Pharmaceutics, 12(9), 880. https://doi.org/10.3390/pharmaceutics12090880