Improving Tumor Retention of Effector Cells in Adoptive Cell Transfer Therapies by Magnetic Targeting


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Adoptive Cell Therapy (ACT) and Its Limitations
3. Nanobiomedicine
3.1. Nanoparticles as the Base of Localized Delivery System
3.2. Magnetic Nanoparticles
3.3. MNP for ACT
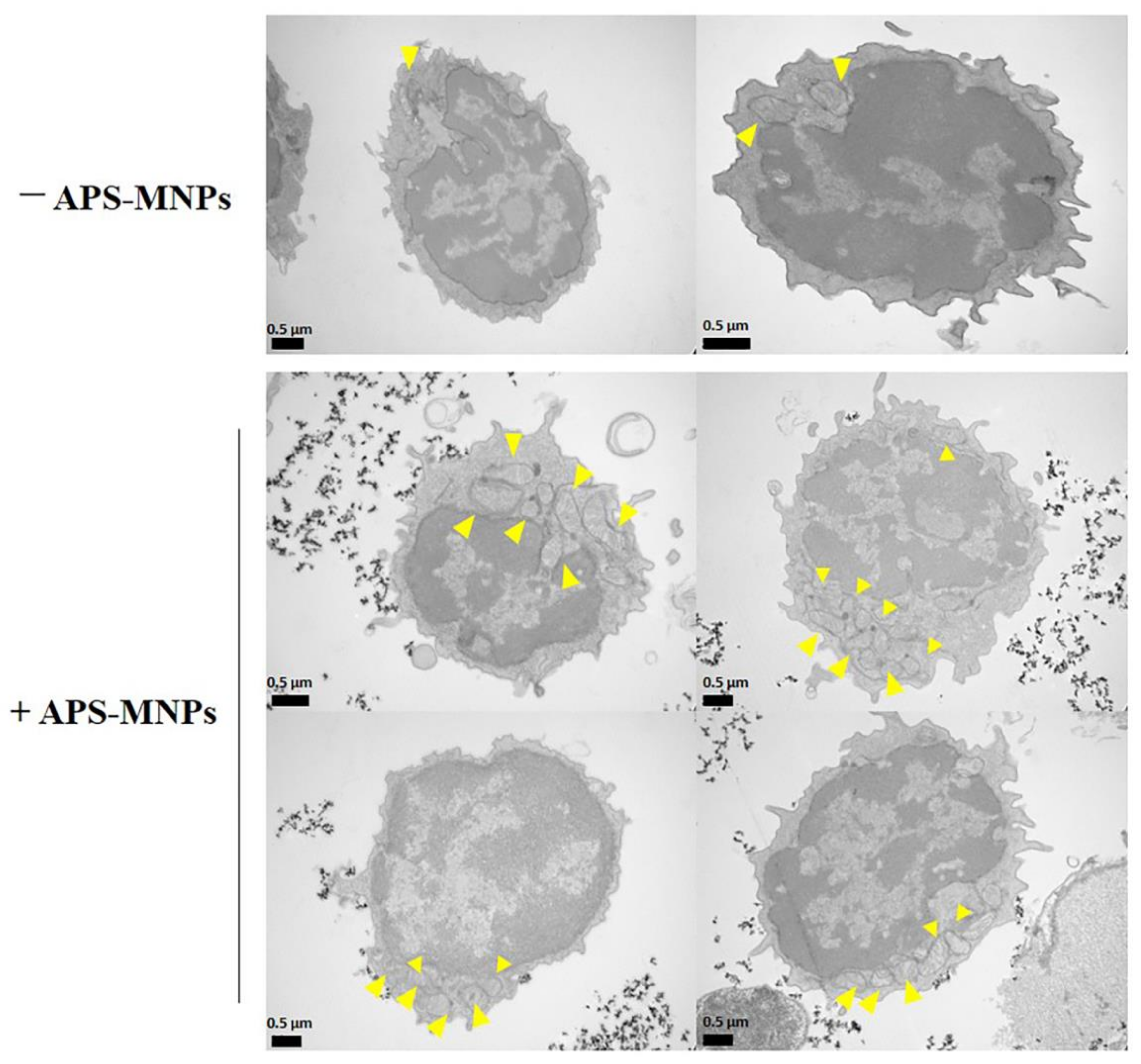
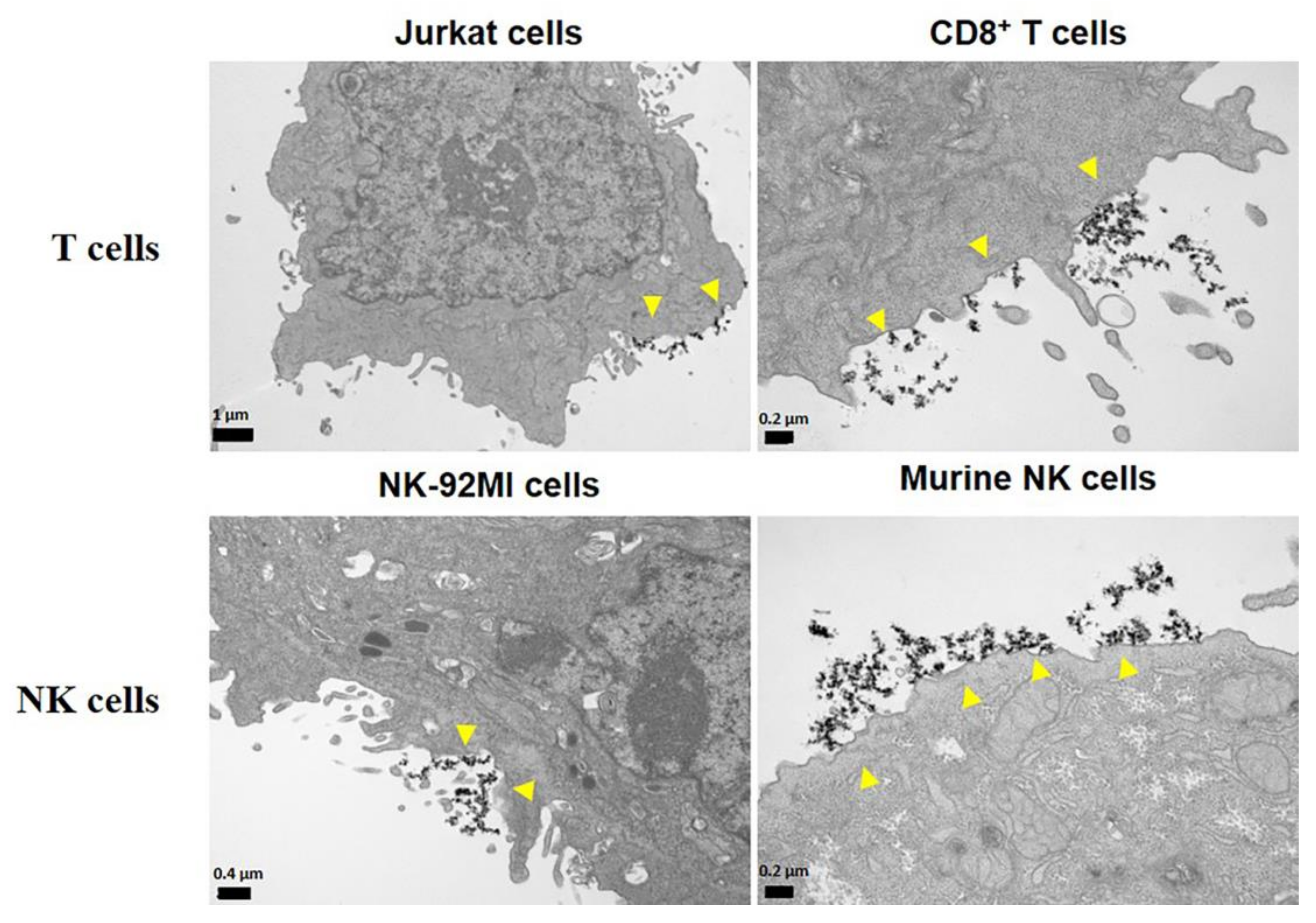
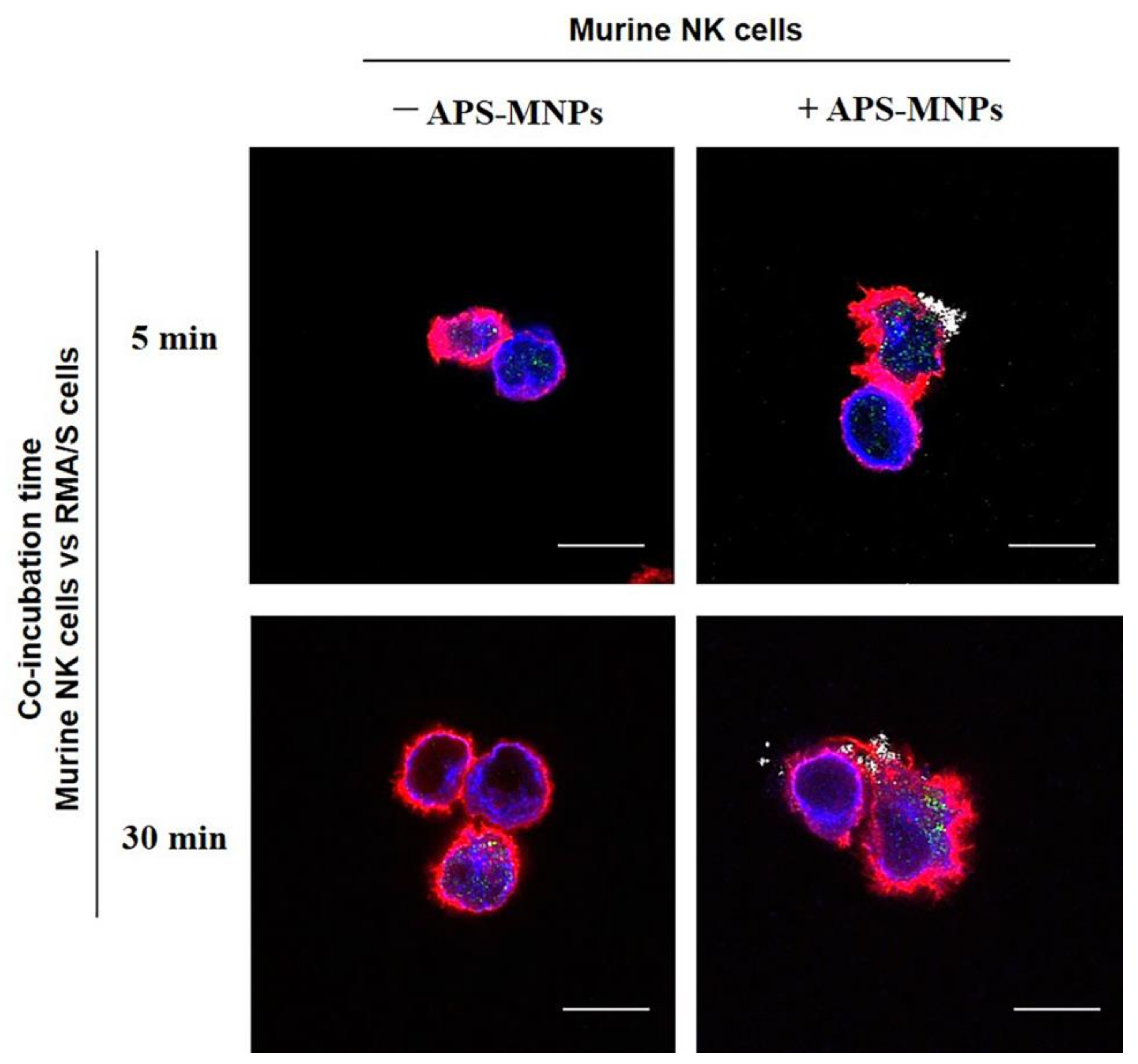
4. The Interaction between MNPs and Lymphoid Cells
5. In Vitro Functionality in MNP-Loaded Lymphoid Cells
6. In Vitro and In Vivo Magnetic Retention of MNP-Loaded Immune Cells
6.1. In Vitro Magnetic Retention
6.2. In Vivo Retention of MNP-Loaded Cells in Lymphoid Tissues
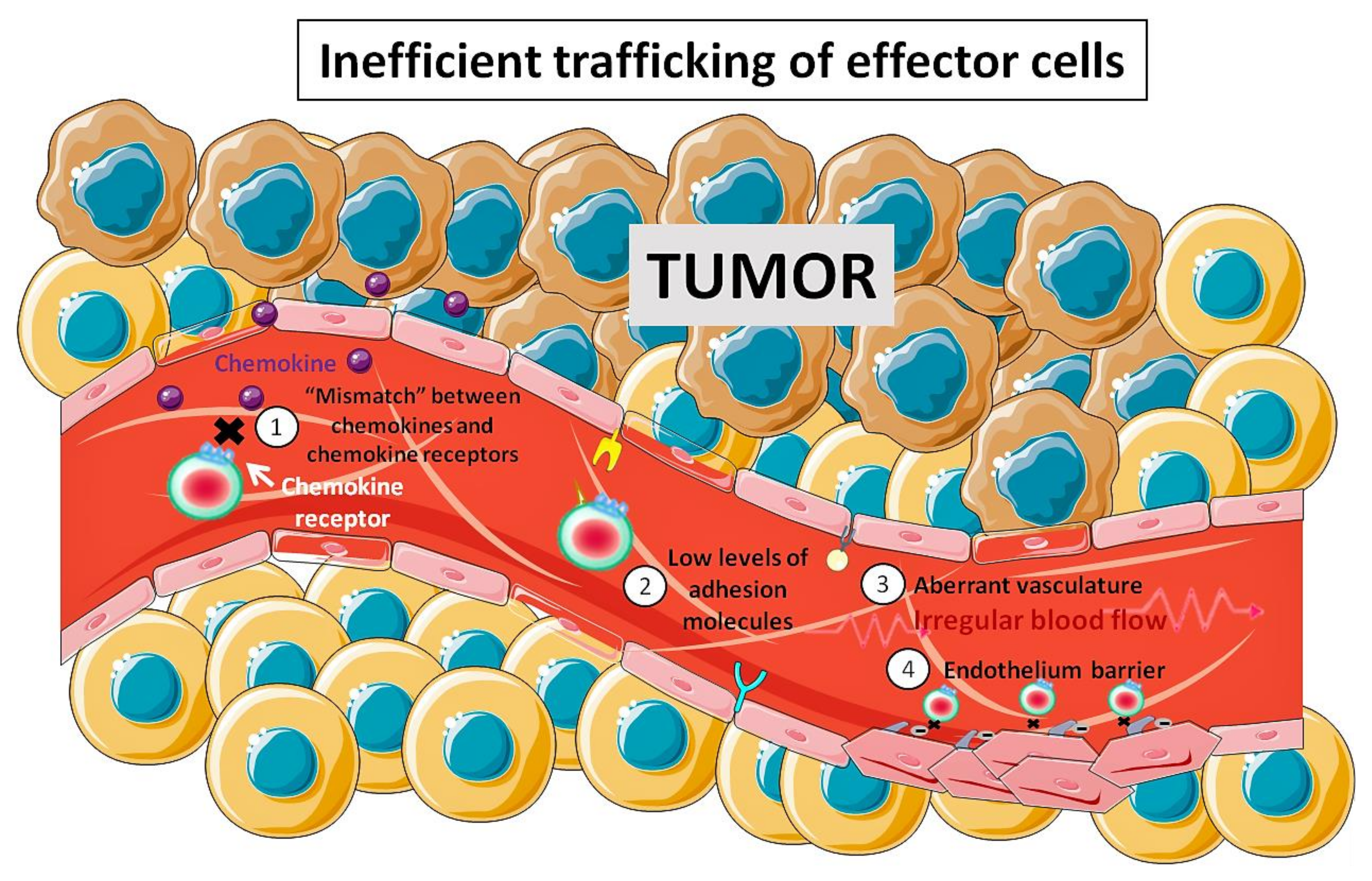
7. In Vivo Magnetic Tumor Targeting of Lymphoid Cells: Not So Straightforward
8. Conclusions and Future Perspectives in the Use of CAR-T Cells
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Caspi, R.R. Immunotherapy of autoimmunity and cancer: The penalty for success. Nat. Rev. Immunol. 2008, 8, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Coukos, G. Deciphering and reversing tumor immune suppression. Immunity 2013, 39, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Blattman, J.N.; Greenberg, P.D. Cancer immunotherapy: A treatment for the masses. Science 2004, 305, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, R.R.; Postow, M.A. Recent advances in understanding antitumor immunity. F1000Research 2016, 5, 2545. [Google Scholar] [CrossRef]
- Dustin, M.L. Cancer immunotherapy: Killers on sterols. Nature 2016, 531, 583–584. [Google Scholar] [CrossRef]
- Kakimi, K.; Karasaki, T.; Matsushita, H.; Sugie, T. Advances in personalized cancer immunotherapy. Breast Cancer 2016, 24, 16–24. [Google Scholar] [CrossRef]
- Mackensen, A.; Meidenbauer, N.; Vogl, S.; Laumer, M.; Berger, J.; Andreesen, R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J. Clin. Oncol. 2006, 24, 5060–5069. [Google Scholar] [CrossRef]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar] [CrossRef]
- Yee, C. Adoptive T cell therapy: Addressing challenges in cancer immunotherapy. J. Transl. Med. 2005, 3, 17. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Mulé, J.J. Immunotherapy of cancer with lymphokine-activated killer cells and recombinant interleukin-2. Surgery 1985, 98, 437–444. [Google Scholar] [PubMed]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T.; et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.Q.; Zhou, J.; Durflinger, K.H.; Langhan, M.M.; Shelton, T.E.; Wunderlich, J.R.; Robbins, P.F.; Rosenberg, S.A.; Dudley, M.E. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J. Immunother. 2008, 31, 742–751. [Google Scholar] [CrossRef]
- Zhou, J.; Dudley, M.E.; Rosenberg, S.A.; Robbins, P.F. Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J. Immunother. 2005, 28, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Dudley, M.E.; Wunderlich, J.; El-Gamil, M.; Li, Y.F.; Zhou, J.; Huang, J.; Powell, D.J.; Rosenberg, S.A. Cutting edge: Persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J. Immunol. 2004, 173, 7125–7130. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef]
- Yee, C.; Thompson, J.A.; Byrd, D.; Riddell, S.R.; Roche, P.; Celis, E.; Greenberg, P.D. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16168–16173. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Hwu, P.; Schwartzentruber, U.J.; Topalian, S.L.; Sherry, R.M.; Marincola, F.M.; Leitman, S.F.; Seipp, C.A.; et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J. Immunother. 2002, 25, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Alatrash, G.; Jakher, H.; Stafford, P.D.; Mittendorf, E.A. Cancer immunotherapies, their safety and toxicity. Expert Opin. Drug Saf. 2013, 12, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.; Øyvind; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef]
- Kim, S.T.; Jeong, H.; Woo, O.H.; Seo, J.H.; Kim, A.; Lee, E.S.; Shin, S.W.; Kim, Y.H.; Kim, J.S.; Park, K. Tumor-infiltrating Lymphocytes, Tumor characteristics, and recurrence in patients with early breast cancer. Am. J. Clin. Oncol. 2013, 36, 224–231. [Google Scholar] [CrossRef]
- Piersma, S.J.; Jordanova, E.S.; Van Poelgeest, M.; Kwappenberg, K.M.; Van Der Hulst, J.M.; Drijfhout, J.W.; Melief, C.J.; Kenter, G.G.; Fleuren, G.J.; Offringa, R.; et al. High number of intraepithelial CD8+Tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007, 67, 354–361. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Zinzindohoué, F.; Bruneval, P.; Cugnenc, P.-H.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Bernhard, H.; Neudorfer, J.; Gebhard, K.; Conrad, H.; Hermann, C.; Nährig, J.; Fend, F.; Weber, W.; Busch, D.H.; Peschel, C. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol. Immunother. 2007, 57, 271–280. [Google Scholar] [CrossRef]
- Pockaj, B.A.; Sherry, R.M.; Wei, J.P.; Yannelli, J.R.; Carter, C.S.; Leitman, S.F.; Carasquillo, J.A.; Steinberg, S.M.; Rosenberg, S.A.; Yang, J.C. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer 1994, 73, 1731–1737. [Google Scholar] [CrossRef]
- John, L.B.; Kershaw, M.H.; Darcy, P. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. OncoImmunology 2013, 2, e26286. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Huang, C.; Loumagne, L.; Tow, C.; Mackay, C.R.; Kato, M.; Nardin, A.; Puaux, A.-L.; Prévost-Blondel, A.; Avril, M.-F.; et al. Chemotherapy Induces intratumoral Expression of Chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. 2011, 71, 6997–7009. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.C.; Balasubramaniam, S.; Hanada, K.-I.; Wrzesinski, C.; Yu, Z.; Farid, S.; Theoret, M.R.; Hwang, L.N.; Klebanoff, C.A.; Gattinoni, L.; et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction1. J. Immunol. 2004, 173, 7209–7216. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Schenkel, J.M. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol. 2013, 13, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Nolz, J.C.; Starbeck-Miller, G.; Harty, J.T. Naive, effector and memory CD8 T-cell trafficking: Parallels and distinctions. Immunotherapy 2011, 3, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, A.M.; Raitman, I.; Feeley, L.; Pinnaduwage, D.; Nguyen, L.T.; O’Malley, F.P.; Ohashi, P.S.; Andrulis, I.L. Tumoral lymphocytic infiltration and expression of the chemokine CXCL10 in breast cancers from the ontario familial breast cancer registry. Clin. Cancer Res. 2012, 19, 336–346. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- 4Musha, H.; Ohtani, H.; Mizoi, T.; Kinouchi, M.; Nakayama, T.; Shiiba, K.; Miyagawa, K.; Nagura, H.; Yoshie, O.; Sasaki, I. Selective infiltration of CCR5+CXCR3+ T lymphocytes in human colorectal carcinoma. Int. J. Cancer 2005, 116, 949–956. [Google Scholar] [CrossRef]
- Bellone, M.; Calcinotto, A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front. Oncol. 2013, 3, 231. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Facciabene, A.; Kim, S.; Benencia, F.; Sasaroli, D.; Balint, K.; Katsaros, D.; O’Brien-Jenkins, A.; Gimotty, P.A.; Coukos, G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med. 2008, 14, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to Chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Ganss, R.; Ryschich, E.; Klar, E.; Arnold, B.; Hämmerling, G.J. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002, 62, 1462–1470. [Google Scholar]
- Johansson, A.; Hamzah, J.; Payne, C.J.; Ganss, R. Tumor-targeted TNF stabilizes tumor vessels and enhances active immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 7841–7846. [Google Scholar] [CrossRef]
- Fu, X.; Rivera, A.; Tao, L.; Zhang, X. Genetically modified T cells targeting neovasculature efficiently destroy tumor blood vessels, shrink established solid tumors and increase nanoparticle delivery. Int. J. Cancer 2013, 133, 2483–2492. [Google Scholar] [CrossRef]
- Chinnasamy, D.; Yu, Z.; Kerkar, S.P.; Zhang, L.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Local delivery of lnterleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin. Cancer Res. 2012, 18, 1672–1683. [Google Scholar] [CrossRef]
- Emerich, D.F.; Thanos, C.G. Nanotechnology and medicine. Expert Opin. Biol. Ther. 2003, 3, 655–663. [Google Scholar] [CrossRef]
- Arruebo, M.; Pacheco, R.; Ibarra, M.R.; Santamaría, J. Magnetic nanoparticles for drug delivery applications. Nano Today 2007, 2, 22–32. [Google Scholar] [CrossRef]
- Shenoy, D.; Little, S.R.; Langer, R.; Amiji, M. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs: Part 2. In vivo distribution and tumor localization studies. Pharm. Res. 2005, 22, 2107–2114. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Gu, Y.; Ma, H.; Bai, J.; Liu, L.; Zhao, P.; He, H. Self-assembled nanoparticle drug delivery systems from galactosylated polysaccharide–doxorubicin conjugate loaded doxorubicin. Int. J. Biol. Macromol. 2010, 46, 245–249. [Google Scholar] [CrossRef]
- McCarthy, D.P.; Hunter, Z.N.; Chackerian, B.; Shea, L.D.; Miller, S.D. Targeted immunomodulation using antigen-conjugated nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 298–315. [Google Scholar] [CrossRef] [PubMed]
- Mejías, R.; Pérez-Yagüe, S.; Gutierrez, L.; Cabrera, L.I.; Spada, R.; Acedo, P.; Serna, C.; Lázaro, F.J.; Villanueva, A.; Morales, M.P.; et al. Dimercaptosuccinic acid-coated magnetite nanoparticles for magnetically guided in vivo delivery of interferon gamma for cancer immunotherapy. Biomaterials 2011, 32, 2938–2952. [Google Scholar] [CrossRef] [PubMed]
- Tartaj, P. Probing Nanomagnets’ Interactions inside colloidal superparamagnetic composites: Aerosol versus surface template methods. ChemPhysChem 2003, 4, 1371–1375. [Google Scholar] [CrossRef]
- Freeman, M.W.; Arrott, A.; Watson, J.H.L. Magnetism in Medicine. J. Appl. Phys. 1960, 31, 404–405. [Google Scholar] [CrossRef]
- Okon, E.; Pouliquen, D.; Okon, P.; Kovaleva, Z.V.; Stepanova, T.P.; Lavit, S.G.; Kudryavtsev, B.N.; Jallet, P. Biodegradation of magnetite dextran nanoparticles in the rat. A histologic and biophysical study. Lab. Investig. 1994, 71, 895–903. [Google Scholar]
- Gutierrez, L.; Lázaro, F.J.; Abadía, A.R.; Romero, M.S.; Quintana, C.; Morales, M.P.; Patiño, C.; Arranz, R. Bioinorganic transformations of liver iron deposits observed by tissue magnetic characterisation in a rat model. J. Inorg. Biochem. 2006, 100, 1790–1799. [Google Scholar] [CrossRef]
- Rojas, J.M.; Gavilán, H.; Del Dedo, V.; Lorente-Sorolla, E.; Sanz-Ortega, L.; Da Silva, G.B.; Costo, R.; Perez-Yagüe, S.; Talelli, M.; Marciello, M.; et al. Time-course assessment of the aggregation and metabolization of magnetic nanoparticles. Acta Biomater. 2017, 58, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Ji, H.; Yu, P.; Niu, J.; Farooq, M.; Akram, M.; Udego, I.O.; Li, H.; Niu, X. Surface modification of magnetic iron oxide nanoparticles. Nanomaterials 2018, 8, 810. [Google Scholar] [CrossRef] [PubMed]
- Biehl, P.; von der Lühe, M.; Dutz, S.; Schacher, F.H. Synthesis, characterization, and applications of magnetic nanoparticles featuring polyzwitterionic coatings. Polymers 2018, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Gul, S.; Khan, S.B.; Rehman, I.U.; Khan, M.A.; Khan, M.I. A Comprehensive review of magnetic nanomaterials modern day theranostics. Front. Mater. 2019, 6. [Google Scholar] [CrossRef]
- Stephan, M.T.; Moon, J.J.; Um, S.H.; Bershteyn, A.; Irvine, D.J. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 2010, 16, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Stephan, M.T.; Stephan, S.B.; Bak, P.; Chen, J.; Irvine, D.J. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials 2012, 33, 5776–5787. [Google Scholar] [CrossRef]
- Huang, B.; Abraham, W.D.; Zheng, Y.; López, S.C.B.; Luo, S.S.; Irvine, D.J. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Transl. Med. 2015, 7, 291ra94. [Google Scholar] [CrossRef]
- Wendel, M.; Galani, I.E.; Suri-Payer, E.; Cerwenka, A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008, 68, 8437–8445. [Google Scholar] [CrossRef]
- Jin, H.; Qian, Y.; Dai, Y.; Qiao, S.; Huang, C.; Lu, L.; Luo, Q.; Chen, J.; Zhang, Z. magnetic enrichment of dendritic cell vaccine in lymph node with fluorescent-magnetic nanoparticles enhanced cancer immunotherapy. Theranostics 2016, 6, 2000–2014. [Google Scholar] [CrossRef]
- Tukmachev, D.; Lunov, O.; Zablotskii, V.; Dejneka, A.; Babič, M.; Syková, E.; Kubinová, Š. An effective strategy of magnetic stem cell delivery for spinal cord injury therapy. Nanoscale 2015, 7, 3954–3958. [Google Scholar] [CrossRef]
- Mou, Y.; Su, H.; An, Y.; Han, W.; Huang, X.; Xia, G.; Ni, Y.; Zhang, Y.; Ma, J.; Hu, Q. The migration of synthetic magnetic nanoparticle labeled dendritic cells into lymph nodes with optical imaging. Int. J. Nanomed. 2013, 8, 3737–3744. [Google Scholar] [CrossRef][Green Version]
- De Chickera, S.N.; Snir, J.; Willert, C.; Rohani, R.; Foley, R.; Foster, P.J.; Dekaban, G.A. Labelling dendritic cells with SPIO has implications for their subsequent in vivo migration as assessed with cellular MRI. Contrast Media Mol. Imaging 2011, 6, 314–327. [Google Scholar] [CrossRef]
- Polyak, B.; Fishbein, I.; Chorny, M.; Alferiev, I.; Williams, D.; Yellen, B.; Friedman, G.; Levy, R.J. High field gradient targeting of magnetic nanoparticle-loaded endothelial cells to the surfaces of steel stents. Proc. Natl. Acad. Sci. USA 2008, 105, 698–703. [Google Scholar] [CrossRef]
- Chertok, B.; David, A.E.; Huang, Y.; Yang, V.C. Glioma selectivity of magnetically targeted nanoparticles: A role of abnormal tumor hydrodynamics. J. Control. Release 2007, 122, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.-S.; Shin, J.-H.; Ren, G.; Park, M.-J.; Cheng, K.; Chen, X.; Wu, J.C.; Sunwoo, J.B.; Cheng, Z. The manipulation of natural killer cells to target tumor sites using magnetic nanoparticles. Biomaterials 2012, 33, 5584–9552. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Casares, X.; Santamaria, P. Nanomedicine in autoimmunity. Immunol. Lett. 2014, 158, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ortega, L.; Portilla, Y.; Pérez-Yagüe, S.; Barber, D.F. Magnetic targeting of adoptively transferred tumour-specific nanoparticle-loaded CD8+ T cells does not improve their tumour infiltration in a mouse model of cancer but promotes the retention of these cells in tumour-draining lymph nodes. J. Nanobiotechnol. 2019, 17, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ortega, L.; Rojas, J.M.; Marcos, A.; Portilla, Y.; Stein, J.V.; Barber, D.F. T cells loaded with magnetic nanoparticles are retained in peripheral lymph nodes by the application of a magnetic field. J. Nanobiotechnol. 2019, 17, 14. [Google Scholar] [CrossRef]
- Sanz-Ortega, L.; Rojas, J.M.; Portilla, Y.; Pérez-Yagüe, S.; Barber, D.F. Magnetic nanoparticles attached to the NK cell surface for tumor targeting in adoptive transfer therapies does not affect cellular effector functions. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Mühlberger, M.; Janko, C.; Unterweger, H.; Friedrich, R.P.; Friedrich, B.; Band, J.; Cebulla, N.; Alexiou, C.; Dudziak, D.; Lee, G.; et al. Functionalization of T Lymphocytes with citrate-coated superparamagnetic iron oxide nanoparticles for magnetically controlled immune therapy. Int. J. Nanomed. 2019, 14, 8421–8432. [Google Scholar] [CrossRef]
- Mühlberger, M.; Unterweger, H.; Band, J.; Lehmann, C.H.K.; Heger, L.; Dudziak, D.; Alexiou, C.; Lee, G.; Janko, C. Loading of primary human T Lymphocytes with citrate-coated superparamagnetic iron oxide nanoparticles does not impair their activation after polyclonal stimulation. Cells 2020, 9, 342. [Google Scholar] [CrossRef]
- Safi, R.; Shokrollahi, H. Physics, chemistry and synthesis methods of nanostructured bismuth ferrite (BiFeO3) as a ferroelectro-magnetic material. Prog. Solid State Chem. 2012, 40, 6–15. [Google Scholar] [CrossRef]
- Patil, R.M.; Thorat, N.D.; Shete, P.B.; Bedge, P.A.; Gavde, S.; Joshi, M.G.; Tofail, S.A.; Bohara, R.A. Comprehensive cytotoxicity studies of superparamagnetic iron oxide nanoparticles. Biochem. Biophys. Rep. 2018, 13, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Jenkins, G.J.S.; Asadi, R.; Doak, S.H. Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano Rev. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Pisanic, T.R.; Blackwell, J.D.; Shubayev, V.I.; Finõnes, R.R.; Jin, S. Nanotoxicity of iron oxide nanoparticle internalization in growing neurons. Biomaterials 2007, 28, 2572–2581. [Google Scholar] [CrossRef]
- Xu, Y.; Sherwood, J.A.; Lackey, K.H.; Qin, Y.; Bao, Y. The responses of immune cells to iron oxide nanoparticles. J. Appl. Toxicol. 2016, 36, 543–553. [Google Scholar] [CrossRef]
- Gaharwar, U.S.; Meena, R.; Rajamani, P. Iron oxide nanoparticles induced cytotoxicity, oxidative stress and DNA damage in lymphocytes. J. Appl. Toxicol. 2017, 37, 1232–1244. [Google Scholar] [CrossRef]
- Zupke, O.; Distler, E.; Jürchott, A.; Paiphansiri, U.; Dass, M.; Thomas, S.; Hartwig, U.F.; Theobald, M.; Landfester, K.; Mailänder, V.; et al. Nanoparticles and antigen-specific T-cell therapeutics: A comprehensive study on uptake and release. Nanomedicine 2015, 10, 1063–1076. [Google Scholar] [CrossRef]
- Shah, A.; Dobrovolskaia, M.A. Immunological effects of iron oxide nanoparticles and iron-based complex drug formulations: Therapeutic benefits, toxicity, mechanistic insights, and translational considerations. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 977–990. [Google Scholar] [CrossRef]
- Strehl, C.; Schellmann, S.; Maurizi, L.; Hofmann-Amtenbrink, M.; Häupl, T.; Hofmann, H.; Buttgereit, F.; Gaber, T. Effects of PVA-coated nanoparticles on human T helper cell activity. Toxicol. Lett. 2016, 245, 52–58. [Google Scholar] [CrossRef]
- Easo, S.L.; Mohanan, P.V. Toxicological evaluation of dextran stabilized iron oxide nanoparticles in human peripheral blood lymphocytes. Biointerphases 2016, 11, 04B302. [Google Scholar] [CrossRef]
- Unterweger, H.; Dezsi, L.; Matuszak, J.; Janko, C.; Pöttler, M.; Jordan, J.; Bäuerle, T.; Szebeni, J.; Fey, T.; Boccaccini, A.R.; et al. Dextran-coated superparamagnetic iron oxide nanoparticles for magnetic resonance imaging: Evaluation of size-dependent imaging properties, storage stability and safety. Int. J. Nanomed. 2018, 13, 1899–1915. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.; Tsourkas, A. Imaging circulating cells and lymphoid tissues with iron oxide nanoparticles. Hematol. Am. Soc. Hematol. Educ. Program 2009, 2009, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, P.; Lavergne, E.; Gazeau, F.; Lewin, M.; Boissonnas, A.; Doan, B.-T.; Gillet, B.; Combadière, C.; Combadière, B.; Clément, O. In vivo cellular imaging of lymphocyte trafficking by MRI: A tumor model approach to cell-based anticancer therapy. Magn. Reson. Med. 2006, 56, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.A.; Shukaliak-Quandt, J.; Jordan, E.K.; Arbab, A.S.; Martin, R.; McFarland, H.; Frank, J.A. Magnetic resonance imaging of labeled T-cells in a mouse model of multiple sclerosis. Ann. Neurol. 2004, 55, 654–659. [Google Scholar] [CrossRef]
- Dodd, C.H.; Hsu, H.-C.; Chu, W.-J.; Yang, P.; Zhang, H.-G.; Zinn, K.R.; Forder, J.; Josephson, L.; Weissleder, R.; Mountz, J.M.; et al. Normal T-cell response and in vivo magnetic resonance imaging of T cells loaded with HIV transactivator-peptide-derived superparamagnetic nanoparticles. J. Immunol. Methods 2001, 256, 89–105. [Google Scholar] [CrossRef]
- Mallett, C.L.; McFadden, C.; Chen, Y.; Foster, P.J. Migration of iron-labeled KHYG-1 natural killer cells to subcutaneous tumors in nude mice, as detected by magnetic resonance imaging. Cytotherapy 2012, 14, 743–751. [Google Scholar] [CrossRef]
- Meier, R.; Golovko, D.; Tavri, S.; Henning, T.D.; Knopp, C.; Piontek, G.; Rudelius, M.; Heinrich, P.; Wels, W.S.; Daldrup-Link, H.E. Depicting adoptive immunotherapy for prostate cancer in an animal model with magnetic resonance imaging. Magn. Reson. Med. 2010, 65, 756–763. [Google Scholar] [CrossRef]
- Daldrup-Link, H.E.; Meier, R.; Rudelius, M.; Piontek, G.; Piert, M.; Metz, S.; Settles, M.; Uherek, C.; Wels, W.; Schlegel, J.; et al. In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur. Radiol. 2005, 15, 4–13. [Google Scholar] [CrossRef]
- Chao, T.; Wang, H.; Ho, P.-C. Mitochondrial control and guidance of cellular activities of T cells. Front. Immunol. 2017, 8, 473. [Google Scholar] [CrossRef]
- Gardiner, C.M.; Finlay, D.K. What Fuels Natural Killers? Metabolism and NK cell responses. Front. Immunol. 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Martín-Cófreces, N.B.; Morlino, G.; Carrasco, Y.R.; Calabia-Linares, C.; Veiga, E.; Serrador, J.M.; Sánchez-Madrid, F. The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. EMBO J. 2011, 30, 1238–1250. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.-R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Chen, H.; Dong, Y.; Luo, X.; Yu, H.; Moore, Z.; Bey, E.A.; Boothman, D.A.; Gao, J. superparamagnetic iron oxide nanoparticles: Amplifying ROS stress to improve anticancer drug efficacy. Theranostics 2013, 3, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Voinov, M.A.; Pagán, J.O.S.; Morrison, E.; Smirnova, T.I.; Smirnov, A.I. Surface-mediated production of Hydroxyl radicals as a mechanism of iron oxide nanoparticle biotoxicity. J. Am. Chem. Soc. 2011, 133, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Cell Biol. 2015, 210, 2104. [Google Scholar] [CrossRef]
- Imanishi, T.; Saito, T. T Cell Co-stimulation and Functional Modulation by Innate Signals. Trends Immunol. 2020, 41, 200–212. [Google Scholar] [CrossRef]
- Mulén, B.; Rojas, J.M.; Pérez-Yagüe, S.; Morales, M.P.; Barber, D.F. Polyethylenimine-coated SPION exhibits potential intrinsic anti-metastatic properties inhibiting migration and invasion of pancreatic tumor cells. J. Control. Release 2015, 216, 78–92. [Google Scholar] [CrossRef]
- Ottersbach, A.; Mykhaylyk, O.; Heidsieck, A.; Eberbeck, D.; Rieck, S.; Zimmermann, K.; Breitbach, M.; Engelbrecht, B.; Brügmann, T.; Hesse, M.; et al. Improved heart repair upon myocardial infarction: Combination of magnetic nanoparticles and tailored magnets strongly increases engraftment of myocytes. Biomaterials 2018, 155, 176–190. [Google Scholar] [CrossRef]
- Zupke, O.; Distler, E.; Baumann, D.; Strand, D.; Meyer, R.; Landfester, K.; Herr, W.; Mailänder, V. Preservation of dendritic cell function upon labeling with amino functionalized polymeric nanoparticles. Biomaterials 2010, 31, 7086–7095. [Google Scholar] [CrossRef]
- Pai, A.; Cao, P.; White, E.E.; Hong, B.; Pailevanian, T.; Wang, M.; Badie, B.; Hajimiri, A.; Berlin, J.M. Dynamically programmable magnetic fields for controlled movement of cells loaded with iron oxide nanoparticles. ACS Appl. Bio Mater. 2020, 3, 4139–4147. [Google Scholar] [CrossRef]
- Berger, C.; Rausch, M.; Schmidt, P.; Rudin, M. Feasibility and limits of magnetically labeling primary cultured rat T cells with ferumoxides coupled with commonly used transfection agents. Mol. Imaging 2006, 5, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Lunov, O.; Syrovets, T.; Loos, C.; Beil, J.; Delacher, M.; Tron, K.; Nienhaus, G.U.; Musyanovych, A.; Mailänder, V.; Landfester, K.; et al. Differential uptake of functionalized polystyrene nanoparticles by human macrophages and a monocytic cell line. ACS Nano 2011, 5, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Hu, Y.; Yin, L.; Tang, C.; Yin, C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 2010, 31, 3657–3666. [Google Scholar] [CrossRef] [PubMed]
- Waiczies, S.; Niendorf, T.; Lombardi, G. Labeling of cell therapies: How can we get it right? OncoImmunology 2017, 6, e1345403. [Google Scholar] [CrossRef] [PubMed]
- Lankoff, A.; Arabski, M.; Węgierek-Ciuk, A.; Kruszewski, M.; Lisowska, H.; Banasik-Nowak, A.; Rózga-Wijas, K.; Wojewódzka, M.; Slomkowski, S. Effect of surface modification of silica nanoparticles on toxicity and cellular uptake by human peripheral blood lymphocytes in vitro. Nanotoxicology 2012, 7, 235–250. [Google Scholar] [CrossRef]
- Smirnov, P. Cellular magnetic resonance imaging using superparamagnetic anionic iron oxide nanoparticles: Applications to in vivo trafficking of lymphocytes and cell-based anticancer therapy. Methods Mol. Biol. 2009, 512, 333–353. [Google Scholar] [CrossRef]
- Garden, O.; Reynolds, P.; Yates, J.; Larkman, D.; Marelli-Berg, F.; Haskard, D.; Edwards, A.; George, A.J. A rapid method for labelling CD4+ T cells with ultrasmall paramagnetic iron oxide nanoparticles for magnetic resonance imaging that preserves proliferative, regulatory and migratory behaviour in vitro. J. Immunol. Methods 2006, 314, 123–133. [Google Scholar] [CrossRef]
- Chen, C.-L.; Siow, T.Y.; Chou, C.-H.; Lin, C.-H.; Lin, M.-H.; Chen, Y.-C.; Hsieh, W.-Y.; Wang, S.-J.; Chang, C. Targeted superparamagnetic iron oxide nanoparticles for in vivo magnetic resonance imaging of t-cells in rheumatoid arthritis. Mol. Imaging Biol. 2016, 19, 233–244. [Google Scholar] [CrossRef]
- Rivolta, I.; Panariti, A.; Miserocchi, G. The effect of nanoparticle uptake on cellular behavior: Disrupting or enabling functions? Nanotechnol. Sci. Appl. 2012, 5, 87–100. [Google Scholar] [CrossRef]
- Kralj, S.; Rojnik, M.; Romih, R.; Jagodič, M.; Kos, J.; Makovec, D. Effect of surface charge on the cellular uptake of fluorescent magnetic nanoparticles. J. Nanoparticle Res. 2012, 14, 1–14. [Google Scholar] [CrossRef]
- Yue, Z.-G.; Wei, W.; Lv, P.-P.; Yue, H.; Wang, L.-Y.; Su, Z.-G.; Ma, G.-H. Surface charge affects cellular uptake and intracellular trafficking of chitosan-based nanoparticles. Biomacromolecules 2011, 12, 2440–2446. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.-L.; Chen, C.; Zhao, Y. Cellular uptake, intracellular trafficking, and Cytotoxicity of nanomaterials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Stellacci, F. Effect of surface properties on nanoparticle-cell interactions. Small 2010, 6, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Orr, G.; Panther, D.J.; Cassens, K.J.; Phillips, J.L.; Tarasevich, B.J.; Pounds, J.G. Syndecan-1 mediates the coupling of positively charged submicrometer amorphous silica particles with actin filaments across the alveolar epithelial cell membrane. Toxicol. Appl. Pharmacol. 2009, 236, 210–220. [Google Scholar] [CrossRef]
- Patil, S.; Sandberg, A.; Heckert, E.; Self, W.T.; Seal, S. Protein adsorption and cellular uptake of cerium oxide nanoparticles as a function of zeta potential. Biomaterials 2007, 28, 4600–4607. [Google Scholar] [CrossRef]
- Meng, Y.; Shi, C.; Hu, B.; Gong, J.; Zhong, X.; Lin, X.; Zhang, X.; Liu, J.; Liu, C.; Xu, H. External magnetic field promotes homing of magnetized stem cells following subcutaneous injection. BMC Cell Biol. 2017, 18, 24. [Google Scholar] [CrossRef]
- Liao, N.; Wu, M.; Pan, F.; Lin, J.; Li, Z.; Zhang, D.; Wang, Y.; Zheng, Y.; Peng, J.; Liu, X.; et al. Poly (dopamine) coated superparamagnetic iron oxide nanocluster for noninvasive labeling, tracking and targeted delivery of adipose tissue-derived stem cells. Sci. Rep. 2016, 6, 18746. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, C.; Zhu, W.; Xia, C.; Wang, D.; Zhang, H.; Wu, J.; Lin, G.; Wu, B.; Gong, Q.; et al. Superparamagnetic MRI probes for in vivo tracking of dendritic cell migration with a clinical 3 T scanner. Biomaterials 2015, 58, 63–71. [Google Scholar] [CrossRef]
- Janic, B.; Rad, A.M.; Jordan, E.K.; Iskander, A.S.M.; Ali, M.; Varma, N.R.S.; Frank, J.A.; Arbab, A.S. Optimization and validation of FePro cell labeling method. PLoS ONE 2009, 4, e5873. [Google Scholar] [CrossRef]
- Iida, H.; Takayanagi, K.; Nakanishi, T.; Kume, A.; Muramatsu, K.; Kiyohara, Y.; Akiyama, Y.; Osaka, T. Preparation of human immune effector T cells containing iron-oxide nanoparticles. Biotechnol. Bioeng. 2008, 101, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.F.; Allport, J.R.; Graves, E.E.; Love, V.; Josephson, L.; Lichtman, A.H.; Weissleder, R. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Res. 2003, 63, 6838–6846. [Google Scholar] [PubMed]
- Yan, L.; Liu, X.; Liu, W.-X.; Tan, X.; Xiong, F.; Gu, N.; Hao, W.; Gao, X.; Cao, J. Fe2O3 nanoparticles suppress Kv1.3 channels via affecting the redox activity of Kvβ2 subunit in Jurkat T cells. Nanotechnology 2015, 26, 505103. [Google Scholar] [CrossRef] [PubMed]
- Alkins, R.; Burgess, A.; Ganguly, M.; Francia, G.; Kerbel, R.; Wels, W.S.; Hynynen, K. Focused ultrasound delivers targeted immune cells to metastatic brain tumors. Cancer Res. 2013, 73, 1892–1899. [Google Scholar] [CrossRef]
- Galli, F.; Histed, S.; Aras, O. NK cell imaging by in vitro and in vivo labelling approaches. Q. J. Nucl. Med. Mol. Imaging 2014, 58, 276–283. [Google Scholar]
- Long, E.O.; Kim, H.S.; Liu, N.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef]
- Tay, C.Y.; Cai, P.; Setyawati, M.I.; Fang, W.; Tan, L.P.; Hong, C.H.L.; Chen, X.; Leong, D.T. Nanoparticles strengthen intracellular tension and retard cellular migration. Nano Lett. 2013, 14, 83–88. [Google Scholar] [CrossRef]
- Diana, V.; Bossolasco, P.; Moscatelli, D.; Silani, V.; Covaa, L. Dose dependent side effect of superparamagnetic iron oxide nanoparticle labeling on cell motility in two fetal stem cell populations. PLoS ONE 2013, 8, e78435. [Google Scholar] [CrossRef]
- Khaleghian, A.; Riazi, G.H.; Ghafari, M.; Rezaie, M.; Takahashi, A.; Nakaya, Y.; Nazari, H. Effect of inganen anticancer properties on microtobule organization. Pak. J. Pharm. Sci. 2010, 23, 273–278. [Google Scholar]
- Soenen, S.J.H.; Nuytten, N.; De Meyer, S.F.; De Smedt, S.C.; De Cuyper, M. High intracellular iron oxide nanoparticle concentrations affect cellular cytoskeleton and focal adhesion kinase-mediated signaling. Small 2010, 6, 832–842. [Google Scholar] [CrossRef]
- Alter, G.; Malenfant, J.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tian, Z.; Wei, H. Developmental and functional control of natural killer cells by Cytokines. Front. Immunol. 2017, 8, 930. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Warren, H.S.; Kinnear, B.F.; Phillips, J.H.; Lanier, L.L. Production of IL-5 by human NK cells and regulation of IL-5 secretion by IL-4, IL-10, and IL-12. J. Immunol. 1995, 154, 5144–5152. [Google Scholar] [PubMed]
- Roda, J.M. Natural killer cells produce T cell-recruiting Chemokines in response to antibody-coated tumor cells. Cancer Res. 2006, 66, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Shah, M.H.; Turner, M.J.; VanDeusen, J.B.; Whitman, S.P.; Cooper, M.A.; Suzuki, K.; Wechser, M.; Goodsaid, F.; Caligiuri, M.A. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: Implications for the innate immune response. J. Immunol. 1999, 162, 4511–4520. [Google Scholar] [PubMed]
- Fauriat, C.; Long, E.O.; Ljunggren, H.-G.; Bryceson, Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef]
- Albuquerque, W.W.C.; Costa, R.M.P.B.; Fernandes, T.D.S.E.; Porto, A.L.F. Evidences of the static magnetic field influence on cellular systems. Prog. Biophys. Mol. Biol. 2016, 121, 16–28. [Google Scholar] [CrossRef]
- Dini, L.; Abbro, L. Bioeffects of moderate-intensity static magnetic fields on cell cultures. Micron 2005, 36, 195–217. [Google Scholar] [CrossRef]
- Rosen, A.D. Mechanism of action of moderate-intensity static magnetic fields on biological systems. Cell Biophys. 2003, 39, 163–174. [Google Scholar] [CrossRef]
- Joseph, N.; Reicher, B.; Barda-Saad, M. The calcium feedback loop and T cell activation: How cytoskeleton networks control intracellular calcium flux. Biochim. Et Biophys. Acta 2014, 1838, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.; Kim, M.-H.; Rossaint, J.; Yamayoshi, I.; Zarbock, A.; Simon, S.I. Leukocyte Function Antigen-1, Kindlin-3, and Calcium Flux Orchestrate Neutrophil Recruitment during Inflammation. J. Immunol. 2012, 189, 5954–5964. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, C.; Friedman, G.; Alamia, J.; Barbee, K.; Polyak, B. Time-varied magnetic field enhances transport of magnetic nanoparticles in viscous gel. Nanomedicine 2010, 5, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kolosnjaj-Tabi, J.; Wilhelm, C.; Clément, O.; Gazeau, F. Cell labeling with magnetic nanoparticles: Opportunity for magnetic cell imaging and cell manipulation. J. Nanobiotechnol. 2013, 11, S7. [Google Scholar] [CrossRef] [PubMed]
- Asperti-Boursin, F.; Real, E.; Bismuth, G.; Trautmann, A.; Donnadieu, E. CCR7 ligands control basal T cell motility within lymph node slices in a phosphoinositide 3–kinase– independent manner. J. Exp. Med. 2007, 204, 1167–1179. [Google Scholar] [CrossRef]
- Wei, S.H.; Safrina, O.; Yu, Y.; Garrod, K.R.; Cahalan, M.D.; Parker, I. Ca2+ signals in CD4+ T cells during early contacts with antigen-bearing dendritic cells in lymph node. J. Immunol. 2007, 179, 1586–1594. [Google Scholar] [CrossRef]
- Dixit, N.; Simon, S.I. Chemokines, selectins and intracellular calcium flux: Temporal and spatial cues for leukocyte arrest. Front. Immunol. 2012, 3, 188. [Google Scholar] [CrossRef]
- Zablotskii, V.; Polyakova, T.; Lunov, O.; Dejneka, A. How a high-gradient magnetic field could affect cell life. Sci. Rep. 2016, 6, 37407. [Google Scholar] [CrossRef]
- Kobukai, S.; Baheza, R.; Cobb, J.G.; Virostko, J.; Xie, J.; Gillman, A.; Koktysh, D.; Kerns, D.; Does, M.; Gore, J.C.; et al. Magnetic nanoparticles for imaging dendritic cells. Magn. Reson. Med. 2010, 63, 1383–1390. [Google Scholar] [CrossRef]
- Goya, G.; Marcos-Campos, I.; Fernández-Pacheco, R.; Saez, B.; Godino, J.; Asin, L.; Lambea, J.; Tabuenca, P.; Mayordomo, J.; Larrad, L. Dendritic cell uptake of iron-based magnetic nanoparticles. Cell Biol. Int. 2008, 32, 1001–1005. [Google Scholar] [CrossRef]
- Cho, N.-H.; Cheong, T.-C.; Min, J.H.; Wu, J.H.; Lee, S.J.; Kim, D.; Yang, J.-S.; Kim, S.; Kim, Y.K.; Seong, S.-Y. A multifunctional core–shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat. Nanotechnol. 2011, 6, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Bedrosian, I.; Mick, R.; Xu, S.; Nisenbaum, H.; Faries, M.; Zhang, P.; Cohen, P.A.; Koski, G.; Czerniecki, B.J. Intranodal administration of peptide-pulsed mature dendritic cell vaccines results in superior CD8+ T-Cell function in melanoma patients. J. Clin. Oncol. 2003, 21, 3826–3835. [Google Scholar] [CrossRef] [PubMed]
- Draube, A.; Klein-Gonzalez, N.; Mattheus, S.; Brillant, C.; Hellmich, M.; Engert, A.; Von Bergwelt-Baildon, M. Dendritic cell based tumor vaccination in prostate and renal cell cancer: A systematic review and meta-analysis. PLoS ONE 2011, 6, e18801. [Google Scholar] [CrossRef] [PubMed]
- Samy, E.T.; Parker, L.A.; Sharp, C.P.; Tung, K.S. Continuous control of autoimmune disease by antigen-dependent polyclonal CD4+CD25+ regulatory T cells in the regional lymph node. J. Exp. Med. 2005, 202, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Ozga, A.J.; Moalli, F.; Abe, J.; Swoger, J.; Sharpe, J.; Zehn, D.; Kreutzfeldt, M.; Merkler, D.; Ripoll, J.; Stein, J.V. pMHC affinity controls duration of CD8+ T cell–DC interactions and imprints timing of effector differentiation versus expansion. J. Exp. Med. 2016, 213, 2811–2829. [Google Scholar] [CrossRef] [PubMed]
- Schell, A.M.; Granger, E.L.; Koczot, F.; Fischer, M.A.; Norbury, C.C. Dendritic cell migration limits the duration of CD8+ T-cell priming to peripheral viral antigen. J. Virol. 2010, 84, 3586–3594. [Google Scholar] [CrossRef]
- Celli, S.; Lemaitre, F.; Bousso, P. Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity 2007, 27, 625–634. [Google Scholar] [CrossRef]
- Sabado, R.L.; Bhardwaj, N. Directing dendritic cell immunotherapy towards successful cancer treatment. Immunotherapy 2010, 2, 37–56. [Google Scholar] [CrossRef]
- Mandl, J.N.; Liou, R.; Klauschen, F.; Vrisekoop, N.; Monteiro, J.P.; Yates, A.J.; Huang, A.Y.; Germain, R.N. Quantification of lymph node transit times reveals differences in antigen surveillance strategies of naive CD4+ and CD8+ T cells. Proc. Natl. Acad. Sci. USA 2012, 109, 18036–18041. [Google Scholar] [CrossRef]
- Ménager, J.; Gorin, J.-B.; Maurel, C.; Drujont, L.; Gouard, S.; Louvet, C.; Chérel, M.; Faivre-Chauvet, A.; Morgenstern, A.; Bruchertseifer, F.; et al. Combining α-radioimmunotherapy and Adoptive T cell therapy to potentiate tumor destruction. PLoS ONE 2015, 10, e0130249. [Google Scholar] [CrossRef]
- Shrikant, P.; Mescher, M.F. Control of syngeneic tumor growth by activation of CD8+ T cells: Efficacy is limited by migration away from the site and induction of nonresponsiveness. J. Immunol. 1999, 162, 2858–2866. [Google Scholar]
- Rotariu, O.; Strachan, N.J. Modelling magnetic carrier particle targeting in the tumor microvasculature for cancer treatment. J. Magn. Magn. Mater. 2005, 293, 639–646. [Google Scholar] [CrossRef]
- Garris, C.S.; Blaho, V.A.; Hla, T.; Han, M.H. Sphingosine-1-phosphate receptor 1 signalling in T cells: Trafficking and beyond. Immunology 2014, 142, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-Phosphate and Lymphocyte egress from lymphoid organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte Sequestration Through S1P Lyase Inhibition and Disruption of S1P Gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef]
- Brinkmann, V.; Lynch, K.R. FTY720: Targeting G-protein-coupled receptors for sphingosine 1-phosphate in transplantation and autoimmunity. Curr. Opin. Immunol. 2002, 14, 569–575. [Google Scholar] [CrossRef]
- Benechet, A.P.; Menon, M.; Xu, D.; Samji, T.; Maher, L.; Murooka, T.T.; Mempel, T.R.; Sheridan, B.S.; Lemoine, F.M.; Khanna, K.M. T cell-intrinsic S1PR1 regulates endogenous effector T-cell egress dynamics from lymph nodes during infection. Proc. Natl. Acad. Sci. USA 2016, 113, 2182–2187. [Google Scholar] [CrossRef]
- Bankovich, A.J.; Shiow, L.R.; Cyster, J.G. CD69 Suppresses Sphingosine 1-Phosophate Receptor-1 (S1P1) Function through Interaction with Membrane Helix 4. J. Biol. Chem. 2010, 285, 22328–22337. [Google Scholar] [CrossRef]
- Shiow, L.R.; Rosen, D.B.; Brdičková, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef]
- Geng, S.; Zhong, Y.; Zhou, X.; Zhao, G.; Xie, X.; Pei, Y.; Liu, H.; Zhang, H.; Shi, Y.; Wang, B. Induced regulatory T cells superimpose their suppressive capacity with effector T cells in lymph nodes via antigen-specific S1p1-dependent egress blockage. Front. Immunol. 2017, 8, 663. [Google Scholar] [CrossRef]
- Mueller, S.N.; Hosiawa-Meagher, K.A.; Konieczny, B.T.; Sullivan, B.M.; Bachmann, M.F.; Locksley, R.M.; Ahmed, R.; Matloubian, M. Regulation of homeostatic Chemokine expression and cell trafficking during immune responses. Science 2007, 317, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Bromley, S.K.; Kan, Z.; Peterson, D.A.; Unanue, E.R. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 3909–3913. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. T-cell activation through immunological synapses and kinapses. Immunol. Rev. 2008, 221, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Cernuda-Morollón, E.; Millán, J.; Shipman, M.; Marelli-Berg, F.M.; Ridley, A.J. Rac Activation by the T-cell receptor inhibits T cell migration. PLoS ONE 2010, 5, e12393. [Google Scholar] [CrossRef]
- Valitutti, S.; Coombs, D.; Dupré, L. The space and time frames of T cell activation at the immunological synapse. FEBS Lett. 2010, 584, 4851–4857. [Google Scholar] [CrossRef]
- Del Pozo, M.A.; Sánchez-Mateos, P.; Nieto, M.; Sánchez-Madrid, F. Chemokines regulate cellular polarization and adhesion receptor redistribution during lymphocyte interaction with endothelium and extracellular matrix. Involvement of cAMP signaling pathway. J. Cell Biol. 1995, 131, 495–508. [Google Scholar] [CrossRef]
- Audemard-Verger, A.; Rivière, M.; Durand, A.; Peranzoni, E.; Guichard, V.; Hamon, P.; Bonilla, N.; Guilbert, T.; Boissonnas, A.; Auffray, C.; et al. Macrophages induce long-term trapping of gammadelta T cells with innate-like properties within secondary lymphoid organs in the steady state. J. Immunol. 2017, 199, 1998–2007. [Google Scholar] [CrossRef]
- Palecek, S.P.; Loftus, J.C.; Ginsberg, M.; Lauffenburger, D.A.; Horwitz, A.F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 1997, 385, 537–540. [Google Scholar] [CrossRef]
- Baeyens, A.; Fang, V.; Chen, C.; Schwab, S.R. Exit Strategies: S1P Signaling and T Cell Migration. Trends Immunol. 2015, 36, 778–787. [Google Scholar] [CrossRef]
- Haig, D.M.; Hopkins, J.; Miller, H.R.P. Local immune responses in afferent and efferent lymph. Immunology 1999, 96, 155–163. [Google Scholar] [CrossRef]
- Steinman, L. Immunology of relapse and remission in multiple sclerosis. Annu. Rev. Immunol. 2014, 32, 257–281. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Liu, H.; Dong, H.; Zhou, N.; Dong, S.; Chen, L.; Zhu, Y.; Hu, H.-m.; Mou, Y. SPIO enhance the cross-presentation and migration of DCs and anionic SPIO influence the nanoadjuvant effects Related to Interleukin-1beta. Nanoscale Res. Lett. 2018, 13, 409. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Liu, L.; Zhu, W.; Li, D.; Yang, L.; Duan, J.; Cai, Z.; Nie, Y.; Zhang, Y.; Gong, Q.; et al. Iron oxide nanoparticles promote macrophage autophagy and inflammatory response through activation of toll-like Receptor-4 signaling. Biomaterials 2019, 203, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Hickey, J.W.; Isser, A.Y.; Vicente, F.P.; Warner, S.; Mao, H.; Schneck, J.P. Efficient magnetic enrichment of antigen-specific T cells by engineering particle properties. Biomaterials 2018, 187, 105–116. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef]
- Seliger, B. Different regulation of MHC Class I antigen processing components in human tumors. J. Immunotoxicol. 2008, 5, 361–367. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; June, C.H. Engineering T cells for cancer: Our synthetic future. Immunol. Rev. 2014, 257, 7–13. [Google Scholar] [CrossRef]
- Kalos, M. Muscle CARs and TcRs: Turbo-charged technologies for the (T cell) masses. Cancer Immunol. Immunother. 2011, 61, 127–135. [Google Scholar] [CrossRef]
- Lipowska-Bhalla, G.; Gilham, D.E.; Hawkins, R.E.; Rothwell, D.G. Targeted immunotherapy of cancer with CAR T cells: Achievements and challenges. Cancer Immunol. Immunother. 2012, 61, 953–962. [Google Scholar] [CrossRef]
- Ramos, C.A.; Dotti, G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.N.; Raubitschek, A.; Forman, S.J.; Jensen, M.C.; Yin, N.; Wang, D.; Zhang, H.; Yi, X.; et al. Specific recognition and killing of glioblastoma multiforme by Interleukin 13-Zetakine redirected Cytolytic T cells. Cancer Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Shpall, E.J.; Grupp, S.A. Chimeric antigen receptor T-cell therapy for ALL. Hematology 2014, 2014, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric antigen receptor-engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Suck, G. Novel approaches using natural killer cells in cancer therapy. Semin. Cancer Biol. 2006, 16, 412–418. [Google Scholar] [CrossRef]
- Nie, W.; Wei, W.; Zuo, L.; Lv, C.; Zhang, F.; Lu, G.-H.; Li, F.; Wu, G.; Huang, L.-L.; Xi, X.; et al. Magnetic nanoclusters armed with responsive PD-1 antibody synergistically improved adoptive T-cell therapy for solid tumors. ACS Nano 2019, 13, 1469–1478. [Google Scholar] [CrossRef]
- Pellico, J.; Ruíz-Cabello, J.; Fernandez-Barahona, I.; Gutierrez, L.; Lechuga-Vieco, A.V.; Enriquez, J.A.; Morales, M.P.; Herranz, F. One-step fast synthesis of nanoparticles for MRI: Coating chemistry as the key variable determining positive or negative contrast. Langmuir 2017, 33, 10239–10247. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanz-Ortega, L.; Rojas, J.M.; Barber, D.F. Improving Tumor Retention of Effector Cells in Adoptive Cell Transfer Therapies by Magnetic Targeting. Pharmaceutics 2020, 12, 812. https://doi.org/10.3390/pharmaceutics12090812
Sanz-Ortega L, Rojas JM, Barber DF. Improving Tumor Retention of Effector Cells in Adoptive Cell Transfer Therapies by Magnetic Targeting. Pharmaceutics. 2020; 12(9):812. https://doi.org/10.3390/pharmaceutics12090812
Chicago/Turabian StyleSanz-Ortega, Laura, José Manuel Rojas, and Domingo F. Barber. 2020. "Improving Tumor Retention of Effector Cells in Adoptive Cell Transfer Therapies by Magnetic Targeting" Pharmaceutics 12, no. 9: 812. https://doi.org/10.3390/pharmaceutics12090812
APA StyleSanz-Ortega, L., Rojas, J. M., & Barber, D. F. (2020). Improving Tumor Retention of Effector Cells in Adoptive Cell Transfer Therapies by Magnetic Targeting. Pharmaceutics, 12(9), 812. https://doi.org/10.3390/pharmaceutics12090812