Development of a Thymoquinone Polymeric Anticancer Nanomedicine through Optimization of Polymer Molecular Weight and Nanoparticle Architecture

 ,
,
Abstract
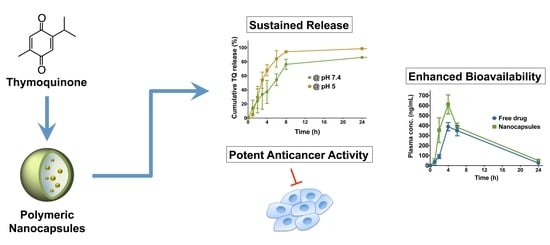
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of mPEG-PCL Copolymers
2.3. Preparation of TQ-Loaded NPs
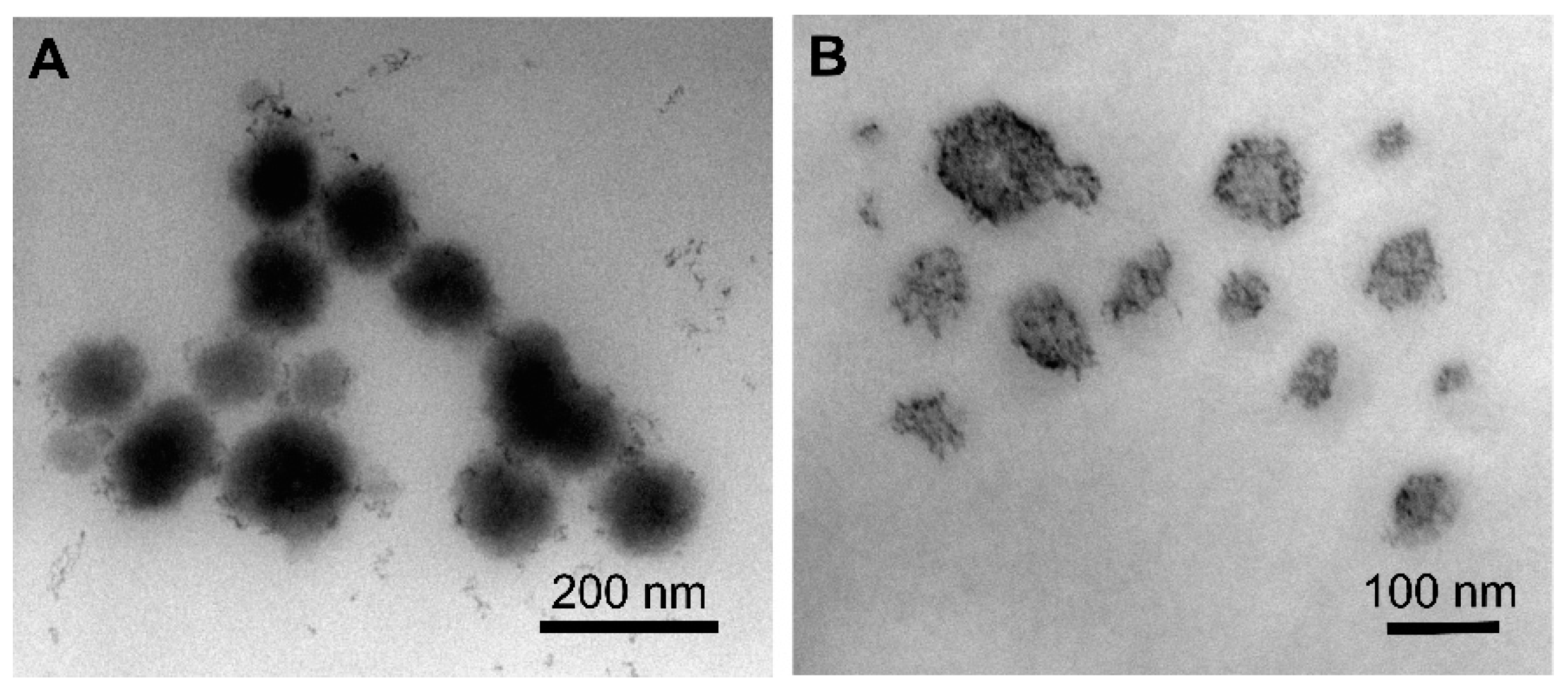
2.4. Characterization of TQ NPs by Dynamic Light Scattering (DLS) and Transmission Electron Microscopy (TEM)
2.5. High-Performance Liquid Chromatography (HPLC) Analysis
2.6. Determination of Drug Loading (DL) and Encapsulation Efficiency (EE)
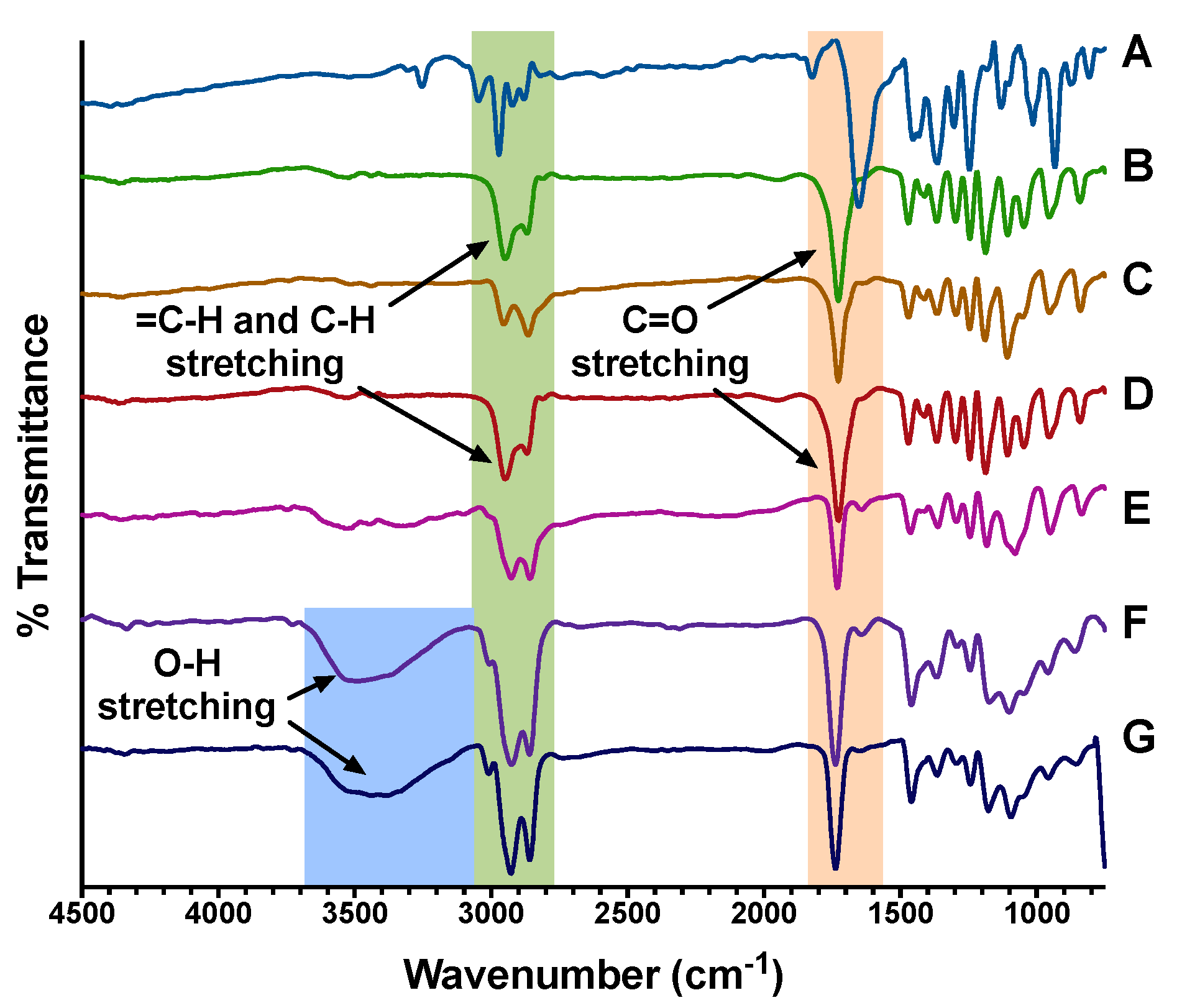
2.7. Characterization by FT-IR
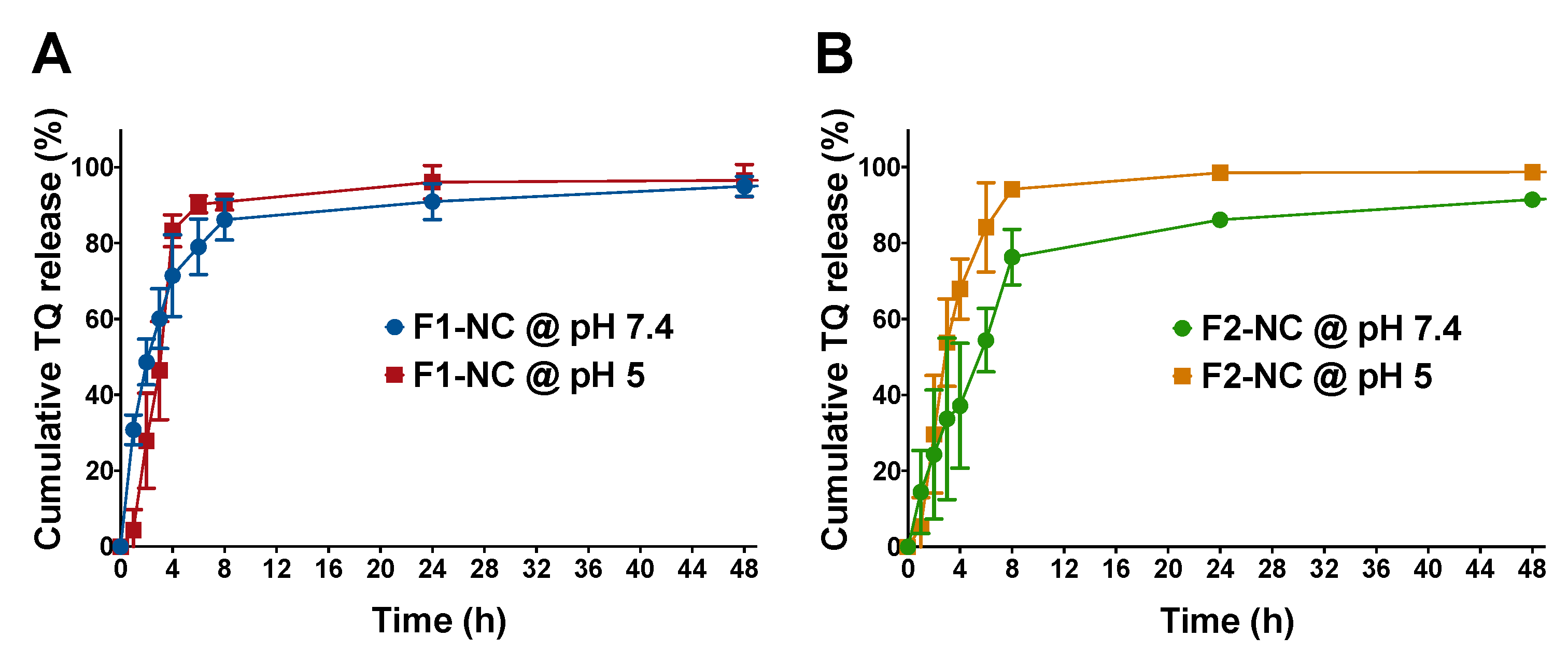
2.8. In Vitro Release of TQ from TQ-NC
2.9. Release Kinetics
2.10. In Vitro Antiproliferative Activity of TQ NPs
2.11. Pilot Pharmacokinetic Study of TQ NPs after Oral Administration
2.12. Analysis of TQ Plasma Levels by Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
2.12.1. Instrumentation
2.12.2. Preparation of Calibration Curve Samples
2.12.3. Preparation of Plasma Samples
2.13. Statistical Analysis
3. Results and Discussion
3.1. Preparation and Characterization of TQ-Loaded NPs with Varying Polymer MW and NP Architectures
3.2. Effect of PCL MW and pH of the Release Medium on TQ Release
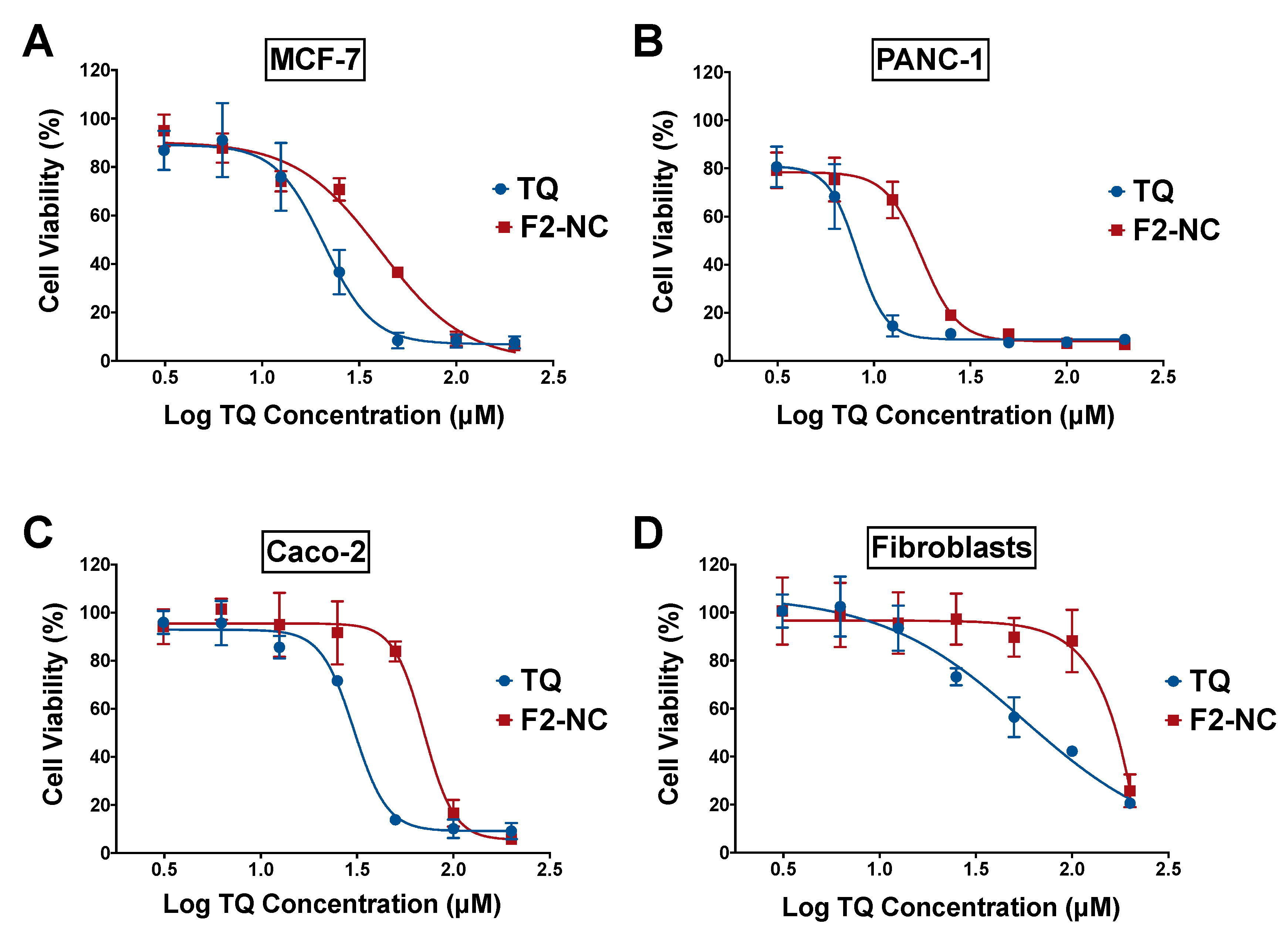
3.3. Antiproliferative Activity of TQ-Loaded NPs in Human Cancer and Normal Cell Lines
3.4. TQ-Loaded NPs Are a Promising Strategy to Increase TQ’s Oral Bioavailability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO Cancer Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 1 April 2020).
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [PubMed]
- Majolo, F.; Delwing LK DO, B.; Marmitt, D.J.; Bustamante-Filho, I.C.; Goettert, M.I. Medicinal plants and bioactive natural compounds for cancer treatment: Important advances for drug discovery. Phytochem. Lett. 2019, 31, 196–207. [Google Scholar] [CrossRef]
- Darakhshan, S.; Bidmeshki Pour, A.; Hosseinzadeh Colagar, A.; Sisakhtnezhad, S. Thymoquinone and its therapeutic potentials. Pharmacol. Res. 2015, 95–96, 138–158. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.C.; Kumar, A.P.; Sethi, G.; Tan, K.H. Thymoquinone: Potential cure for inflammatory disorders and cancer. Biochem. Pharmacol. 2012, 83, 443–451. [Google Scholar] [CrossRef]
- Ballout, F.; Habli, Z.; Rahal, O.N.; Fatfat, M.; Gali-Muhtasib, H. Thymoquinone-based nanotechnology for cancer therapy: Promises and challenges. Drug Discov. Today 2018, 23, 1089–1098. [Google Scholar] [CrossRef]
- Schneider-Stock, R.; Fakhoury, I.H.; Zaki, A.M.; El-Baba, C.O.; Gali-Muhtasib, H.U. Thymoquinone: Fifty years of success in the battle against cancer models. Drug Discov. Today 2014, 19, 18–30. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Sunoqrot, S.; Hamed, R.; Abdel-Halim, H.; Tarawneh, O. Synergistic interplay of medicinal chemistry and formulation strategies in nanotechnology—From drug discovery to nanocarrier design and development. Curr. Top. Med. Chem. 2017, 17, 1451–1468. [Google Scholar] [CrossRef]
- Mohammadabadi, M.R.; Mozafari, M.R. Enhanced efficacy and bioavailability of thymoquinone using nanoliposomal dosage form. J. Drug Deliv. Sci. Technol. 2018, 47, 445–453. [Google Scholar] [CrossRef]
- Odeh, F.; Ismail, S.I.; Abu-Dahab, R.; Mahmoud, I.S.; Al Bawab, A. Thymoquinone in liposomes: A study of loading efficiency and biological activity towards breast cancer. Drug Deliv. 2012, 19, 371–377. [Google Scholar] [CrossRef]
- Singh, A.; Ahmad, I.; Akhter, S.; Jain, G.K.; Iqbal, Z.; Talegaonkar, S.; Ahmad, F.J. Nanocarrier based formulation of Thymoquinone improves oral delivery: Stability assessment, in vitro and in vivo studies. Colloid. Surf. B 2013, 102, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, S.I.; Sheikh, B.Y.; Taha, M.M.E.; How, C.W.; Abdullah, R.; Yagoub, U.; El-Sunousi, R.; Eid, E.E.M. Thymoquinone-loaded nanostructured lipid carriers: Preparation, gastroprotection, in vitro toxicity, and pharmacokinetic properties after extravascular administration. Int. J. Nanomed. 2013, 8, 2163. [Google Scholar] [CrossRef] [PubMed]
- Elmowafy, M.; Samy, A.; Raslan, M.A.; Salama, A.; Said, R.A.; Abdelaziz, A.E.; El-Eraky, W.; El Awdan, S.; Viitala, T. Enhancement of bioavailability and pharmacodynamic effects of thymoquinone via nanostructured lipid carrier (NLC) formulation. AAPS Pharmscitech 2016, 17, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.K.; Saiful Yazan, L.; Yap, L.H.; Wan Nor Hafiza, W.A.G.; How, C.W.; Abdullah, R. Thymoquinone-loaded nanostructured lipid carrier exhibited cytotoxicity towards breast cancer cell lines (MDA-MB-231 and MCF-7) and cervical cancer cell lines (HeLa and SiHa). BioMed Res. Int. 2015. [Google Scholar] [CrossRef]
- Rajput, S.; Puvvada, N.; Kumar, B.N.P.; Sarkar, S.; Konar, S.; Bharti, R.; Dey, G.; Mazumdar, A.; Pathak, A.; Fisher, P.B.; et al. Overcoming Akt Induced Therapeutic Resistance in Breast Cancer through siRNA and Thymoquinone Encapsulated Multilamellar Gold Niosomes. Mol. Pharm. 2015, 12, 4214–4225. [Google Scholar] [CrossRef]
- Goel, S.; Mishra, P. Thymoquinone loaded mesoporous silica nanoparticles retard cell invasion and enhance in vitro cytotoxicity due to ROS mediated apoptosis in HeLa and MCF-7 cell lines. Mater. Sci. Eng. C 2019, 104, 109881. [Google Scholar] [CrossRef]
- Fakhoury, I.; Saad, W.; Bouhadir, K.; Nygren, P.; Schneider-Stock, R.; Gali-Muhtasib, H. Uptake, delivery, and anticancer activity of thymoquinone nanoparticles in breast cancer cells. J. Nanopart. Res. 2016, 18, 210. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Ahir, M.; Patra, P.; Mukherjee, S.; Ghosh, S.; Mazumdar, M.; Chattopadhyay, S.; Das, T.; Chattopadhyay, D.; Adhikary, A. PEGylated-thymoquinone-nanoparticle mediated retardation of breast cancer cell migration by deregulation of cytoskeletal actin polymerization through miR-34a. Biomaterials 2015, 51, 91–107. [Google Scholar] [CrossRef]
- Soni, P.; Kaur, J.; Tikoo, K. Dual drug-loaded paclitaxel–thymoquinone nanoparticles for effective breast cancer therapy. J. Nanopart. Res. 2015, 17, 18. [Google Scholar] [CrossRef]
- Grossen, P.; Witzigmann, D.; Sieber, S.; Huwyler, J. PEG-PCL-based nanomedicines: A biodegradable drug delivery system and its application. J. Control. Release 2017, 260, 46–60. [Google Scholar] [CrossRef]
- Dash, T.K.; Konkimalla, V.B. Polymeric modification and its implication in drug delivery: Poly-epsilon-caprolactone (PCL) as a model polymer. Mol. Pharm. 2012, 9, 2365–2379. [Google Scholar] [CrossRef] [PubMed]
- Gou, M.; Wei, X.; Men, K.; Wang, B.; Luo, F.; Zhao, X.; Wei, Y.; Qian, Z. PCL/PEG copolymeric nanoparticles: Potential nanoplatforms for anticancer agent delivery. Curr. Drug Targets 2011, 12, 1131–1150. [Google Scholar] [CrossRef]
- Sunoqrot, S.; Hasan, L.; Alsadi, A.; Hamed, R.; Tarawneh, O. Interactions of mussel-inspired polymeric nanoparticles with gastric mucin: Implications for gastro-retentive drug delivery. Colloids Surfaces B 2017, 156, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Al-Shalabi, E.; Alkhaldi, M.; Sunoqrot, S. Development and evaluation of polymeric nanocapsules for cirsiliol isolated from Jordanian Teucrium polium L. as a potential anticancer nanomedicine. J. Drug Deliv. Sci. Technol. 2020, 56, 101544. [Google Scholar]
- Sunoqrot, S.; Alsadi, A.; Tarawneh, O.; Hamed, R. Polymer type and molecular weight dictate the encapsulation efficiency and release of Quercetin from polymeric micelles. Colloid Polym. Sci. 2017, 295, 2051–2059. [Google Scholar] [CrossRef]
- Sunoqrot, S.; Abujamous, L. pH-sensitive polymeric nanoparticles of quercetin as a potential colon cancer-targeted nanomedicine. J. Drug Deliv. Sci. Technol. 2019, 52, 670–676. [Google Scholar] [CrossRef]
- Sunoqrot, S.; Al-Debsi, T.; Al-Shalabi, E.; Hasan Ibrahim, L.; Faruqu, F.N.; Walters, A.; Palgrave, R.; Al-Jamal, K.T. Bioinspired polymerization of quercetin to produce a curcumin-loaded nanomedicine with potent cytotoxicity and cancer-targeting potential in vivo. ACS Biomater. Sci. Eng. 2019, 5, 6036–6045. [Google Scholar] [CrossRef]
- Ashcroft, R.E. The Declaration of Helsinki. In The Oxford Textbook of Clinical Research Ethics; Emanuel, E.J., Ed.; Oxford University Press: New York, NY, USA, 2008; pp. 141–148. [Google Scholar]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Mora-Huertas, C.; Fessi, H.; Elaissari, A. Polymer-based nanocapsules for drug delivery. Int. J. Pharm. 2010, 385, 113–142. [Google Scholar] [CrossRef]
- Pohlmann, A.R.; Fonseca, F.N.; Paese, K.; Detoni, C.B.; Coradini, K.; Beck, R.C.; Guterres, S.S. Poly (ϵ-caprolactone) microcapsules and nanocapsules in drug delivery. Expert Opin. Drug Deliv. 2013, 10, 623–638. [Google Scholar] [CrossRef]
- Klippstein, R.; Wang, J.T.W.; El-Gogary, R.I.; Bai, J.; Mustafa, F.; Rubio, N.; Bansal, S.; Al-Jamal, W.T.; Al-Jamal, K.T. Passively targeted curcumin-loaded PEGylated PLGA nanocapsules for colon cancer therapy in vivo. Small 2015, 11, 4704–4722. [Google Scholar] [CrossRef] [PubMed]
- El-Gogary, R.I.; Rubio, N.; Wang, J.T.-W.; Al-Jamal, W.T.; Bourgognon, M.; Kafa, H.; Naeem, M.; Klippstein, R.; Abbate, V.; Leroux, F. Polyethylene glycol conjugated polymeric nanocapsules for targeted delivery of quercetin to folate-expressing cancer cells in vitro and in vivo. ACS Nano 2014, 8, 1384–1401. [Google Scholar] [CrossRef] [PubMed]
- D’souza, A.A.; Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef] [PubMed]
- Khayata, N.; Abdelwahed, W.; Chehna, M.F.; Charcosset, C.; Fessi, H. Preparation of vitamin E loaded nanocapsules by the nanoprecipitation method: From laboratory scale to large scale using a membrane contactor. Int. J. Pharm. 2012, 423, 419–427. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2001, 48, 139–157. [Google Scholar] [CrossRef]
- Majdalawieh, A.F.; Fayyad, M.W.; Nasrallah, G.K. Anti-cancer properties and mechanisms of action of thymoquinone, the major active ingredient of Nigella sativa. Crit. Rev. Food Sci. Nutr. 2017, 57, 3911–3928. [Google Scholar] [CrossRef]
- Ramzy, L.; Metwally, A.A.; Nasr, M.; Awad, G.A.S. Novel thymoquinone lipidic core nanocapsules with anisamide-polymethacrylate shell for colon cancer cells overexpressing sigma receptors. Sci. Rep. 2020, 10, 10987. [Google Scholar] [CrossRef]
- Shahein, S.A.; Aboul-Enein, A.M.; Higazy, I.M.; Abou-Elella, F.; Lojkowski, W.; Ahmed, E.R.; Mousa, S.A.; AbouAitah, K. Targeted anticancer potential against glioma cells of thymoquinone delivered by mesoporous silica core-shell nanoformulations with pH-dependent release. Int. J. Nanomed. 2019, 14, 5503–5526. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Organic Phase a | Aqueous Phase b | ||
|---|---|---|---|---|
| Copolymer Code | Castor Oil (µL) | Span 80 (mg) | Tween 80 (% w/v) | |
| F1-NS c | mPEG5K-PCL10.3K | - | 25 | 0.2 |
| F1-NC d | mPEG5K-PCL10.3K | 150 | 25 | 0.2 |
| F2-NS | mPEG5K-PCL18.5K | - | 25 | 0.2 |
| F2-NC | mPEG5K-PCL18.5K | 150 | 25 | 0.2 |
| Cell Line | TQ | F2-NC | ||
|---|---|---|---|---|
| IC50 (µM) | SI a | IC50 (µM) | SI | |
| MCF-7 | 20.5 ± 3.7 | 2.9 | 40.6 ± 2.1 | 27.9 |
| PANC-1 | 8.8 ± 2.2 | 6.7 | 17.6 ± 1.2 | 64.0 |
| Caco-2 | 30.4 ± 0.5 | 1.9 | 70.4 ± 0.2 | 16.0 |
| Fibroblasts | 58.9 ± 10.3 | - | 1129.5 ± 47.4 | - |
| Parameter | TQ Suspension | F2-NC |
|---|---|---|
| Cmax (ng mL−1) | 388.5 ± 41.5 | 611.4 ± 94.9 |
| tmax (h) | 4.0 | 4.0 |
| t1/2 (h) | 5.0 | 5.8 |
| AUC0→∞ (ng h mL−1) | 4669 ± 508 | 6069 ± 492 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sunoqrot, S.; Alfaraj, M.; Hammad, A.M.; Kasabri, V.; Shalabi, D.; Deeb, A.A.; Hasan Ibrahim, L.; Shnewer, K.; Yousef, I. Development of a Thymoquinone Polymeric Anticancer Nanomedicine through Optimization of Polymer Molecular Weight and Nanoparticle Architecture. Pharmaceutics 2020, 12, 811. https://doi.org/10.3390/pharmaceutics12090811
Sunoqrot S, Alfaraj M, Hammad AM, Kasabri V, Shalabi D, Deeb AA, Hasan Ibrahim L, Shnewer K, Yousef I. Development of a Thymoquinone Polymeric Anticancer Nanomedicine through Optimization of Polymer Molecular Weight and Nanoparticle Architecture. Pharmaceutics. 2020; 12(9):811. https://doi.org/10.3390/pharmaceutics12090811
Chicago/Turabian StyleSunoqrot, Suhair, Malek Alfaraj, Ala’a M. Hammad, Violet Kasabri, Dana Shalabi, Ahmad A. Deeb, Lina Hasan Ibrahim, Khaldoun Shnewer, and Ismail Yousef. 2020. "Development of a Thymoquinone Polymeric Anticancer Nanomedicine through Optimization of Polymer Molecular Weight and Nanoparticle Architecture" Pharmaceutics 12, no. 9: 811. https://doi.org/10.3390/pharmaceutics12090811
APA StyleSunoqrot, S., Alfaraj, M., Hammad, A. M., Kasabri, V., Shalabi, D., Deeb, A. A., Hasan Ibrahim, L., Shnewer, K., & Yousef, I. (2020). Development of a Thymoquinone Polymeric Anticancer Nanomedicine through Optimization of Polymer Molecular Weight and Nanoparticle Architecture. Pharmaceutics, 12(9), 811. https://doi.org/10.3390/pharmaceutics12090811