In Vivo Assessment of Thermosensitive Liposomes for the Treatment of Port Wine Stains by Antifibrinolytic Site-Specific Pharmaco-Laser Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Tranexamic Acid-Encapsulating Liposome Preparation and Characterization
2.3. Calculation Drug: Lipid Ratio, Encapsulation Efficiency, Trapped Volume, and Endovesicular Tranexamic Acid Concentration
2.4. Heat-Induced Tranexamic Acid Release from Thermosensitive LUVETs in Physiological Buffer
2.5. Calcein-Encapsulating Liposome Preparation and Characterization
2.6. Stability of Thermosensitive Liposomes in Human Plasma
2.7. Animals and Surgical Procedures
2.8. Flow Cytometric Analysis of Hamster and Human Platelet Staining by Molecular Probes, Antibodies, and LUVETs
2.9. Preparation of Fluorophore-, Antibody-, and Liposome-Containing Solutions for Intravenous Administration into Hamsters
2.10. Intravital Fluorescence Microscopy and Laser-Induced Thrombosis Model
2.11. Image Analysis
3. Results
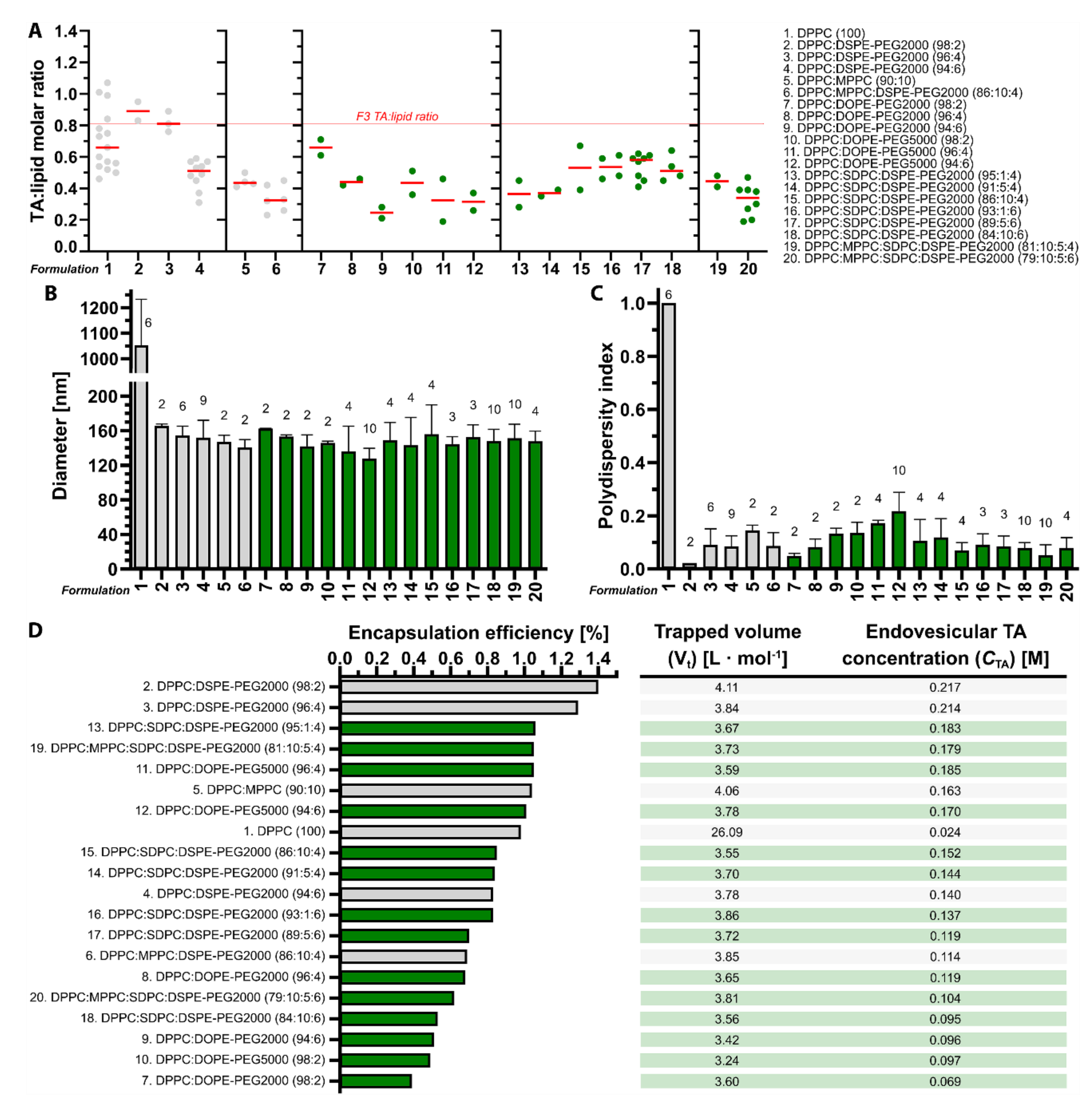
3.1. Tranexamic Acid-Encapsulating DPPC:DSPE-PEG Liposomes Have the Most Favorable Physicochemical Properties for Antifibrinolytic SSPLT
3.2. Tranexamic Acid-Encapsulating PEGylated Thermosensitive Liposomes Release Content at Temperatures Equal to and Above the Phase Transition Temperature in Buffered Solution
3.3. Plasma Stabilizes Thermosensitive Liposomes and Reduces the Extent of Leakage at Body Temperature
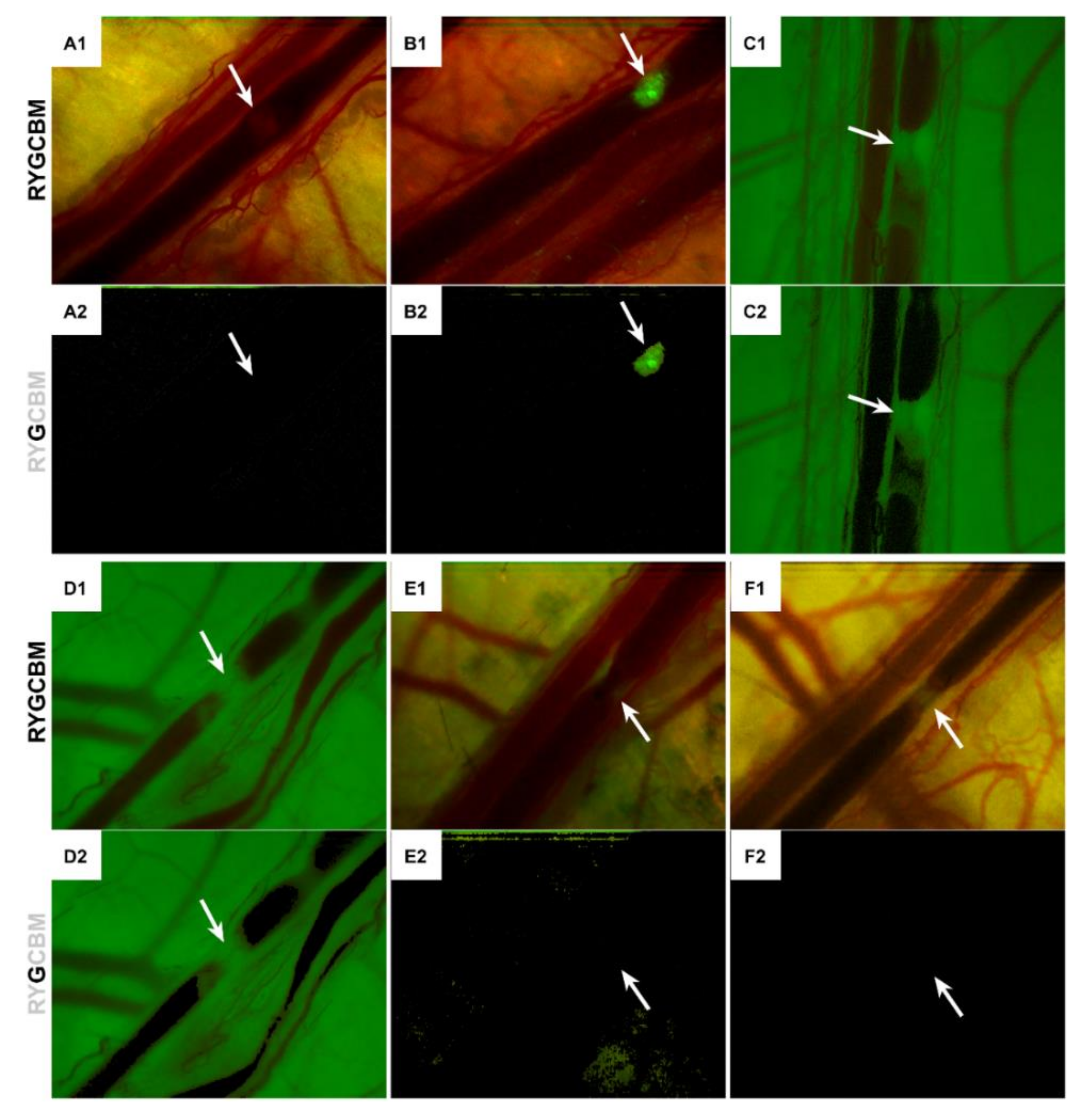
3.4. LUVETs Associate with Hamster and Human Platelets
3.5. LUVETs Do Not Incorporate into Laser-Induced Thrombi In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tan, W.; Wang, J.; Zhou, F.; Gao, L.; Yin, R.; Liu, H.; Sukanthanag, A.; Wang, G.; Mihm, M.C., Jr.; Chen, D.B.; et al. Coexistence of Eph receptor B1 and ephrin B2 in port-wine stain endothelial progenitor cells contributes to clinicopathological vasculature dilatation. Br. J. Dermatol. 2017, 177, 1601–1611. [Google Scholar] [CrossRef]
- Nguyen, V.; Hochman, M.; Mihm, M.C., Jr.; Nelson, J.S.; Tan, W. The Pathogenesis of Port Wine Stain and Sturge Weber Syndrome: Complex Interactions between Genetic Alterations and Aberrant MAPK and PI3K Activation. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Fiskerstrand, E.J.; Svaasand, L.O.; Kopstad, G.; Ryggen, K.; Aase, S. Photothermally induced vessel-wall necrosis after pulsed dye laser treatment: Lack of response in port-wine stains with small sized or deeply located vessels. J. Investig. Dermatol. 1996, 107, 671–675. [Google Scholar] [CrossRef]
- Anderson, R.R.; Parrish, J.A. Selective photothermolysis: Precise microsurgery by selective absorption of pulsed radiation. Science 1983, 220, 524–527. [Google Scholar] [CrossRef]
- Heger, M.; Beek, J.; Stenback, K.; Faber, D.; van Gemert, M.; Ince, C. Darkfield orthogonal polarized spectral imaging for studying endovascular laser-tissue interactions in vivo-a preliminary study. Opt. Express 2005, 13, 702–715. [Google Scholar] [CrossRef]
- Bezemer, R.; Heger, M.; van den Wijngaard, J.P.; Mordon, S.R.; van Gemert, M.J.; Beek, J.F. Laser-induced (endo)vascular photothermal effects studied by combined brightfield and fluorescence microscopy in hamster dorsal skin fold venules. Opt. Express 2007, 15, 8493–8506. [Google Scholar] [CrossRef]
- Heger, M.; Beek, J.F.; Moldovan, N.I.; van der Horst, C.M.; van Gemert, M.J. Towards optimization of selective photothermolysis: Prothrombotic pharmaceutical agents as potential adjuvants in laser treatment of port wine stains. A theoretical study. Thromb. Haemost. 2005, 93, 242–256. [Google Scholar] [CrossRef]
- Hohenleutner, U.; Hilbert, M.; Wlotzke, U.; Landthaler, M. Epidermal damage and limited coagulation depth with the flashlamp-pumped pulsed dye laser: A histochemical study. J. Investig. Dermatol. 1995, 104, 798–802. [Google Scholar] [CrossRef]
- Chen, J.K.; Ghasri, P.; Aguilar, G.; van Drooge, A.M.; Wolkerstorfer, A.; Kelly, K.M.; Heger, M. An overview of clinical and experimental treatment modalities for port wine stains. J. Am. Acad. Dermatol. 2012, 67, 289–304. [Google Scholar] [CrossRef]
- Van Raath, M.I.; Chohan, S.; Wolkerstorfer, A.; van der Horst, C.; Storm, G.; Heger, M. Port wine stain treatment outcomes have not improved over the past three decades. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1369–1377. [Google Scholar] [CrossRef]
- Heger, M.; Bezemer, R.; Huertas-Perez, J.F.; Dekker, H.; Beek, J.F. Endovascular laser-tissue interactions redefined: Shining light on novel windows of therapeutic opportunity beyond selective photothermolysis. Photomed. Laser Surg. 2010, 28, 569–572. [Google Scholar] [CrossRef]
- Van Raath, M.I.; Bambach, C.A.; Dijksman, L.M.; Wolkerstorfer, A.; Heger, M. Prospective analysis of the port-wine stain patient population in the Netherlands in light of novel treatment modalities. J. Cosmet. Laser Ther. 2018, 20, 77–84. [Google Scholar] [CrossRef]
- Aguilar, G.; Choi, B.; Broekgaarden, M.; Yang, O.; Yang, B.; Ghasri, P.; Chen, J.K.; Bezemer, R.; Nelson, J.S.; van Drooge, A.M.; et al. An overview of three promising mechanical, optical, and biochemical engineering approaches to improve selective photothermolysis of refractory port wine stains. Ann. Biomed. Eng. 2012, 40, 486–506. [Google Scholar] [CrossRef]
- Van Raath, M.I.; van Amesfoort, J.E.; Hermann, M.; Ince, Y.; Zwart, M.J.; Echague, A.V.; Chen, Y.; Ding, B.; Huang, X.; Storm, G.; et al. Site-specific pharmaco-laser therapy: A novel treatment modality for refractory port wine stains. J. Clin. Transl. Res. 2019, 5, 1–24. [Google Scholar]
- Heger, M.; Salles, I.I.; Bezemer, R.; Cloos, M.A.; Mordon, S.R.; Begu, S.; Deckmyn, H.; Beek, J.F. Laser-induced primary and secondary hemostasis dynamics and mechanisms in relation to selective photothermolysis of port wine stains. J. Dermatol. Sci. 2011, 63, 139–147. [Google Scholar] [CrossRef]
- Van Raath, M.I.; Weijer, R.; Nguyen, G.H.; Choi, B.; de Kroon, A.I.; Heger, M. Tranexamic Acid-Encapsulating Thermosensitive Liposomes for Site-Specific Pharmaco-Laser Therapy of Port Wine Stains. J. Biomed. Nanotechnol. 2016, 12, 1617–1640. [Google Scholar] [CrossRef]
- Huertas-Perez, J.F.; Heger, M.; Dekker, H.; Krabbe, H.; Lankelma, J.; Ariese, F. Simple, rapid, and sensitive liquid chromatography-fluorescence method for the quantification of tranexamic acid in blood. J. Chromatogr. A 2007, 1157, 142–150. [Google Scholar] [CrossRef]
- Houston, B.L.; Uminski, K.; Mutter, T.; Rimmer, E.; Houston, D.S.; Menard, C.E.; Garland, A.; Ariano, R.; Tinmouth, A.; Abou-Setta, A.M.; et al. Efficacy and Safety of Tranexamic Acid in Major Non-Cardiac Surgeries at High Risk for Transfusion: A Systematic Review and Meta-Analysis. Transfus. Med. Rev. 2020, 34, 51–62. [Google Scholar] [CrossRef]
- Fusca, L.; Perelman, I.; Fergusson, D.; Boutet, M.; Chen, I. The Effectiveness of Tranexamic Acid at Reducing Blood Loss and Transfusion Requirement for Women Undergoing Myomectomy: A Systematic Review and Meta-analysis. J. Obstet. Gynaecol. Can. 2019, 41, 1185–1192.e1. [Google Scholar] [CrossRef]
- Reed, M.R.; Woolley, L.T. Uses of tranexamic acid. Contin. Educ. Anaesth. Crit. Care Pain 2014, 15, 32–37. [Google Scholar] [CrossRef]
- Smithies, D.J.; van Gemert, M.J.; Hansen, M.K.; Milner, T.E.; Nelson, J.S. Three-dimensional reconstruction of port wine stain vascular anatomy from serial histological sections. Phys. Med. Biol. 1997, 42, 1843–1847. [Google Scholar] [CrossRef]
- Selim, M.M.; Kelly, K.M.; Nelson, J.S.; Wendelschafer-Crabb, G.; Kennedy, W.R.; Zelickson, B.D. Confocal microscopy study of nerves and blood vessels in untreated and treated port wine stains: Preliminary observations. Dermatol. Surg. 2004, 30, 892–897. [Google Scholar] [CrossRef]
- Heger, M.; Salles, I.I.; van Vuure, W.; Hamelers, I.H.; de Kroon, A.I.; Deckmyn, H.; Beek, J.F. On the interaction of fluorophore-encapsulating PEGylated lecithin liposomes with hamster and human platelets. Microvasc. Res. 2009, 78, 57–66. [Google Scholar] [CrossRef]
- Heger, M.; Salles, I.I.; de Kroon, A.I.; Deckmyn, H. Platelets and PEGylated lecithin liposomes: When stealth is allegedly picked up on the radar (and eaten). Microvasc. Res. 2009, 78, 1–3. [Google Scholar] [CrossRef]
- Rouser, G.; Fkeischer, S.; Yamamoto, A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 1970, 5, 494–496. [Google Scholar] [CrossRef]
- Udenfriend, S.; Stein, S.; Bohlen, P.; Dairman, W.; Leimgruber, W.; Weigele, M. Fluorescamine: A reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science 1972, 178, 871–872. [Google Scholar] [CrossRef]
- Broekgaarden, M.; de Kroon, A.I.; Gulik, T.M.; Heger, M. Development and in vitro proof-of-concept of interstitially targeted zinc- phthalocyanine liposomes for photodynamic therapy. Curr. Med. Chem. 2014, 21, 377–391. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10. [Google Scholar] [CrossRef]
- Zuidam, N.J.d.V.R.; Crommelin, D.J. Characterization of liposomes. In Liposomes; Torchilin, V.P., Weissig, V., Eds.; Oxford University Press: Oxford, UK, 2003; p. 64. [Google Scholar]
- Nagle, J.F.; Tristram-Nagle, S. Lipid bilayer structure. Curr. Opin. Struct. Biol. 2000, 10, 474–480. [Google Scholar] [CrossRef]
- Majewski, J.; Kuhl, T.L.; Kjaer, K.; Gerstenberg, M.C.; Als-Nielsen, J.; Israelachvili, J.N.; Smith, G.S. X-ray Synchrotron Study of Packing and Protrusions of Polymer−Lipid Monolayers at the Air−Water Interface. J. Am. Chem. Soc. 1998, 120, 1469–1473. [Google Scholar] [CrossRef]
- Chi, L.M.; Wu, W.G. Effective bilayer expansion and erythrocyte shape change induced by monopalmitoyl phosphatidylcholine. Quantitative light microscopy and nuclear magnetic resonance spectroscopy measurements. Biophys J. 1990, 57, 1225–1232. [Google Scholar] [CrossRef]
- Faller, R.; Marrink, S.J. Simulation of domain formation in DLPC-DSPC mixed bilayers. Langmuir 2004, 20, 7686–7693. [Google Scholar] [CrossRef]
- Wheeler, J.J.; Palmer, L.; Ossanlou, M.; MacLachlan, I.; Graham, R.W.; Zhang, Y.P.; Hope, M.J.; Scherrer, P.; Cullis, P.R. Stabilized plasmid-lipid particles: Construction and characterization. Gene Ther. 1999, 6, 271–281. [Google Scholar] [CrossRef]
- De Graaf, W.; Heger, M.; Spruijt, O.; Maas, A.; de Bruin, K.; Hoekstra, R.; Bennink, R.J.; van Gulik, T.M. Quantitative assessment of liver function after ischemia-reperfusion injury and partial hepatectomy in rats. J. Surg. Res. 2012, 172, 85–94. [Google Scholar] [CrossRef]
- Rowan, A.N.; Salem, D.J. The State of the Animals; Humane Society of the United States: Washington, DC, USA, 2001; Volume 8. [Google Scholar]
- Stenberg, P.E.; McEver, R.P.; Shuman, M.A.; Jacques, Y.V.; Bainton, D.F. A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. J. Cell Biol. 1985, 101, 880–886. [Google Scholar] [CrossRef]
- Heger, M.; Salles, I.I.; van Vuure, W.; Deckmyn, H.; Beek, J.F. Fluorescent labeling of platelets with polyanionic fluorescein derivatives. Anal. Quant. Cytol. Histol. 2009, 31, 227–232. [Google Scholar]
- Reiniers, M.J.; van Golen, R.F.; Bonnet, S.; Broekgaarden, M.; van Gulik, T.M.; Egmond, M.R.; Heger, M. Preparation and Practical Applications of 2′,7′-Dichlorodihydrofluorescein in Redox Assays. Anal. Chem. 2017, 89, 3853–3857. [Google Scholar] [CrossRef]
- Guerci, P.; Ergin, B.; Uz, Z.; Ince, Y.; Westphal, M.; Heger, M.; Ince, C. Glycocalyx Degradation Is Independent of Vascular Barrier Permeability Increase in Nontraumatic Hemorrhagic Shock in Rats. Anesth. Analg. 2019, 129, 598–607. [Google Scholar] [CrossRef]
- Dos Santos, N.; Allen, C.; Doppen, A.M.; Anantha, M.; Cox, K.A.; Gallagher, R.C.; Karlsson, G.; Edwards, K.; Kenner, G.; Samuels, L.; et al. Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: Relating plasma circulation lifetimes to protein binding. Biochim. Biophys. Acta 2007, 1768, 1367–1377. [Google Scholar] [CrossRef]
- Mastrotto, F.; Brazzale, C.; Bellato, F.; De Martin, S.; Grange, G.; Mahmoudzadeh, M.; Magarkar, A.; Bunker, A.; Salmaso, S.; Caliceti, P. In Vitro and in Vivo Behavior of Liposomes Decorated with PEGs with Different Chemical Features. Mol. Pharm. 2020, 17, 472–487. [Google Scholar] [CrossRef]
- Maruyama, K.; Yuda, T.; Okamoto, A.; Kojima, S.; Suginaka, A.; Iwatsuru, M. Prolonged circulation time in vivo of large unilamellar liposomes composed of distearoyl phosphatidylcholine and cholesterol containing amphipathic poly(ethylene glycol). Biochim. Biophys. Acta 1992, 1128, 44–49. [Google Scholar] [CrossRef]
- Watabe, N.; Ishida, Y.; Ochiai, A.; Tokuoka, Y.; Kawashima, N. Oxidation decomposition of unsaturated fatty acids by singlet oxygen in phospholipid bilayer membranes. J. Oleo Sci. 2007, 56, 73–80. [Google Scholar] [CrossRef]
- Mori, A.; Klibanov, A.L.; Torchilin, V.P.; Huang, L. Influence of the steric barrier activity of amphipathic poly(ethyleneglycol) and ganglioside GM1 on the circulation time of liposomes and on the target binding of immunoliposomes in vivo. FEBS Lett. 1991, 284, 263–266. [Google Scholar] [CrossRef]
- Manni, M.M.; Tiberti, M.L.; Pagnotta, S.; Barelli, H.; Gautier, R.; Antonny, B. Acyl chain asymmetry and polyunsaturation of brain phospholipids facilitate membrane vesiculation without leakage. Elife 2018, 7. [Google Scholar] [CrossRef]
- Gaber, M.H.; Wu, N.Z.; Hong, K.; Huang, S.K.; Dewhirst, M.W.; Papahadjopoulos, D. Thermosensitive liposomes: Extravasation and release of contents in tumor microvascular networks. Int. J. Radiat Oncol. Biol. Phys. 1996, 36, 1177–1187. [Google Scholar] [CrossRef]
- Han, S.-M.; Na, Y.-G.; Lee, H.-S.; Son, G.-H.; Jeon, S.-H.; Bang, K.-H.; Kim, S.-J.; Lee, H.-J.; Cho, C.-W. Improvement of cellular uptake of hydrophilic molecule, calcein, formulated by liposome. J. Pharm. Investig. 2018, 48, 595–601. [Google Scholar] [CrossRef]
- Mulik, R.; Kulkarni, V.; Murthy, R.S. Chitosan-based thermosensitive hydrogel containing liposomes for sustained delivery of cytarabine. Drug Dev. Ind. Pharm. 2009, 35, 49–56. [Google Scholar] [CrossRef]
- Dicko, A.; Kwak, S.; Frazier, A.A.; Mayer, L.D.; Liboiron, B.D. Biophysical characterization of a liposomal formulation of cytarabine and daunorubicin. Int. J. Pharm. 2010, 391, 248–259. [Google Scholar] [CrossRef]
- Lim, S.K.; Shin, D.H.; Choi, M.H.; Kim, J.S. Enhanced antitumor efficacy of gemcitabine-loaded temperature-sensitive liposome by hyperthermia in tumor-bearing mice. Drug Dev. Ind. Pharm. 2014, 40, 470–476. [Google Scholar] [CrossRef]
- Tong, Q.; Li, H.; Li, W.; Chen, H.; Shu, X.; Lu, X.; Wang, G. In vitro and in vivo anti-tumor effects of gemcitabine loaded with a new drug delivery system. J. Nanosci. Nanotechnol. 2011, 11, 3651–3658. [Google Scholar] [CrossRef]
- Liu, J.J.; Hong, R.L.; Cheng, W.F.; Hong, K.; Chang, F.H.; Tseng, Y.L. Simple and efficient liposomal encapsulation of topotecan by ammonium sulfate gradient: Stability, pharmacokinetic and therapeutic evaluation. Anticancer Drugs 2002, 13, 709–717. [Google Scholar] [CrossRef]
- Dadashzadeh, S.; Vali, A.M.; Rezaie, M. The effect of PEG coating on in vitro cytotoxicity and in vivo disposition of topotecan loaded liposomes in rats. Int. J. Pharm. 2008, 353, 251–259. [Google Scholar] [CrossRef]
- Lim, C.B.; Abuzar, S.M.; Karn, P.R.; Cho, W.; Park, H.J.; Cho, C.W.; Hwang, S.J. Preparation, Characterization, and In Vivo Pharmacokinetic Study of the Supercritical Fluid-Processed Liposomal Amphotericin B. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef]
- Moribe, K.; Tanaka, E.; Maruyama, K.; Iwatsuru, M. Enhanced encapsulation of amphotericin B into liposomes by complex formation with polyethylene glycol derivatives. Pharm. Res. 1998, 15, 1737–1742. [Google Scholar] [CrossRef]
- Javed, I.; Hussain, S.Z.; Ullah, I.; Khan, I.; Ateeq, M.; Shahnaz, G.; Rehman, H.U.; Razi, M.T.; Shah, M.R.; Hussain, I. Synthesis, characterization and evaluation of lecithin-based nanocarriers for the enhanced pharmacological and oral pharmacokinetic profile of amphotericin B. J. Mater. Chem. B 2015, 3, 8359–8365. [Google Scholar] [CrossRef]
- Li, L.; ten Hagen, T.L.; Hossann, M.; Suss, R.; van Rhoon, G.C.; Eggermont, A.M.; Haemmerich, D.; Koning, G.A. Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy. J. Control. Release 2013, 168, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Unezaki, S.; Takahashi, N.; Iwatsuru, M. Enhanced delivery of doxorubicin to tumor by long-circulating thermosensitive liposomes and local hyperthermia. Biochim. Biophys. Acta 1993, 1149, 209–216. [Google Scholar] [CrossRef]
- Tagami, T.; Ernsting, M.J.; Li, S.D. Efficient tumor regression by a single and low dose treatment with a novel and enhanced formulation of thermosensitive liposomal doxorubicin. J. Control. Release 2011, 152, 303–309. [Google Scholar] [CrossRef]
- Shen, S.; Huang, D.; Cao, J.; Chen, Y.; Zhang, X.; Guo, S.; Ma, W.; Qi, X.; Ge, Y.; Wu, L. Magnetic liposomes for light-sensitive drug delivery and combined photothermal-chemotherapy of tumors. J. Mater. Chem. B 2019, 7, 1096–1106. [Google Scholar] [CrossRef]
- Han, B.; Yang, Y.; Chen, J.; Tang, H.; Sun, Y.; Zhang, Z.; Wang, Z.; Li, Y.; Li, Y.; Luan, X.; et al. Preparation, Characterization, and Pharmacokinetic Study of a Novel Long-Acting Targeted Paclitaxel Liposome with Antitumor Activity. Int. J. Nanomed. 2020, 15, 553–571. [Google Scholar] [CrossRef]
- Yang, T.; Cui, F.D.; Choi, M.K.; Cho, J.W.; Chung, S.J.; Shim, C.K.; Kim, D.D. Enhanced solubility and stability of PEGylated liposomal paclitaxel: In vitro and in vivo evaluation. Int. J. Pharm. 2007, 338, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.D.; Bally, M.B.; Loughrey, H.; Masin, D.; Cullis, P.R. Liposomal vincristine preparations which exhibit decreased drug toxicity and increased activity against murine L1210 and P388 tumors. Cancer Res. 1990, 50, 575–579. [Google Scholar] [PubMed]
- Tartau, L.; Cazacu, A.; Melnig, V. Ketoprofen-liposomes formulation for clinical therapy. J. Mater. Sci. Mater. Med. 2012, 23, 2499–2507. [Google Scholar] [CrossRef]
- Maestrelli, F.; Gonzalez-Rodriguez, M.L.; Rabasco, A.M.; Mura, P. Effect of preparation technique on the properties of liposomes encapsulating ketoprofen-cyclodextrin complexes aimed for transdermal delivery. Int. J. Pharm. 2006, 312, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Setua, S.; Kumari, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Gemcitabine Combination Nano Therapies for Pancreatic Cancer. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Hossann, M.; Syunyaeva, Z.; Schmidt, R.; Zengerle, A.; Eibl, H.; Issels, R.D.; Lindner, L.H. Proteins and cholesterol lipid vesicles are mediators of drug release from thermosensitive liposomes. J. Control. Release 2012, 162, 400–406. [Google Scholar] [CrossRef]
- Marsh, D.; Watts, A.; Knowles, P.F. Cooperativity of the phase transition in single- and multibilayer lipid vesicles. Biochim. Biophys. Acta 1977, 465, 500–514. [Google Scholar] [CrossRef]
- Nagle, J.F. Theory of the main lipid bilayer phase transition. Ann. Rev. Phys. Chem. 1980, 31, 157–195. [Google Scholar] [CrossRef]
- Marsh, D. General features of phospholipid phase transitions. Chem. Phys. Lipids 1991, 57, 109–120. [Google Scholar] [CrossRef]
- Nagle, J.F.; Scott, H.L., Jr. Lateral compressibility of lipid mono- and bilayers. Theory of membrane permeability. Biochim. Biophys. Acta 1978, 513, 236–243. [Google Scholar] [CrossRef]
- Ruppel, D.S.E. On defects in different phases of two-dimensional lipid bilayers. J. Phys. 1983, 44, 1025–1034. [Google Scholar] [CrossRef]
- Hosokawa, T.; Sami, M.; Kato, Y.; Hayakawa, E. Alteration in the temperature-dependent content release property of thermosensitive liposomes in plasma. Chem. Pharm. Bull. (Tokyo) 2003, 51, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Panagi, Z.A.K.; Evangelatos, G.; Ithakissios, D.S. Protein-induced CF release from liposomes in vitro and its correlation with the BLOOD/RES biodistribution of liposomes. Int. J. Pharm. 1998, 163, 103–114. [Google Scholar] [CrossRef]
- Burke, C.; Dreher, M.R.; Negussie, A.H.; Mikhail, A.S.; Yarmolenko, P.; Patel, A.; Skilskyj, B.; Wood, B.J.; Haemmerich, D. Drug release kinetics of temperature sensitive liposomes measured at high-temporal resolution with a millifluidic device. Int. J. Hyperth. 2018, 34, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Kneidl, B.; Peller, M.; Winter, G.; Lindner, L.H.; Hossann, M. Thermosensitive liposomal drug delivery systems: State of the art review. Int. J. Nanomed. 2014, 9, 4387–4398. [Google Scholar] [CrossRef]
- Picetti, R.; Shakur-Still, H.; Medcalf, R.L.; Standing, J.F.; Roberts, I. What concentration of tranexamic acid is needed to inhibit fibrinolysis? A systematic review of pharmacodynamics studies. Blood Coagul. Fibrinolysis 2019, 30, 1–10. [Google Scholar] [CrossRef]
- Weijer, R.; Broekgaarden, M.; Kos, M.; van Vught, R.; Rauws, E.A.; Breukink, E.J.; van Gulik, T.M.; Storm, G.; Heger, M. Enhancing photodynamic therapy of refractory solid cancers: Combining second-generation photosensitizers with multi-targeted liposomal delivery. J. Photochem. Photobiol. C 2015, 23, 103–131. [Google Scholar] [CrossRef]
- Weijer, R.; Broekgaarden, M.; van Golen, R.F.; Bulle, E.; Nieuwenhuis, E.; Jongejan, A.; Moerland, P.D.; van Kampen, A.H.; van Gulik, T.M.; Heger, M. Low-power photodynamic therapy induces survival signaling in perihilar cholangiocarcinoma cells. BMC Cancer 2015, 15, 1014. [Google Scholar] [CrossRef]
- Weijer, R.; Clavier, S.; Zaal, E.A.; Pijls, M.M.; van Kooten, R.T.; Vermaas, K.; Leen, R.; Jongejan, A.; Moerland, P.D.; van Kampen, A.H.; et al. Multi-OMIC profiling of survival and metabolic signaling networks in cells subjected to photodynamic therapy. Cell Mol. Life Sci. 2017, 74, 1133–1151. [Google Scholar] [CrossRef]
- Broekgaarden, M.; van Vught, R.; Oliveira, S.; Roovers, R.C.; van Bergen en Henegouwen, P.M.; Pieters, R.J.; Van Gulik, T.M.; Breukink, E.; Heger, M. Site-specific conjugation of single domain antibodies to liposomes enhances photosensitizer uptake and photodynamic therapy efficacy. Nanoscale 2016, 8, 6490–6494. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Weijer, R.; Krekorian, M.; van den Ijssel, B.; Kos, M.; Alles, L.K.; van Wijk, A.C.; Bikadi, Z.; Hazai, E.; van Gulik, T.M.; et al. Inhibition of hypoxia-inducible factor 1 with acriflavine sensitizes hypoxic tumor cells to photodynamic therapy with zinc phthalocyanine-encapsulating cationic liposomes. Nano Res. 2016, 9, 1639–1662. [Google Scholar] [CrossRef]
- Frimer, A.A. The reaction of singlet oxygen with olefins: The question of mechanism. Chem. Rev. 1979, 79, 359–387. [Google Scholar] [CrossRef]
- Ryter, S.W.; Tyrrell, R.M. Singlet molecular oxygen ((1)O2): A possible effector of eukaryotic gene expression. Free Radic. Biol. Med. 1998, 24, 1520–1534. [Google Scholar] [CrossRef]
- Davies, M.J. Reactive species formed on proteins exposed to singlet oxygen. Photochem. Photobiol. Sci. 2004, 3, 17–25. [Google Scholar] [CrossRef]
- Campbell, P.I. Toxicity of some charged lipids used in liposome preparations. Cytobios 1983, 37, 21–26. [Google Scholar] [PubMed]
- Mayhew, E.; Ito, M.; Lazo, R. Toxicity of non-drug-containing liposomes for cultured human cells. Exp. Cell Res. 1987, 171, 195–202. [Google Scholar] [CrossRef]
- Sevanian, A.H.P. Mechanisms and consequences of lipid peroxidation in biological systems. Ann. Rev. Nutr. 1985, 5, 365–390. [Google Scholar] [CrossRef]
- Lakmaker, O.; Pickering, J.W.; van Gemert, M.J. Modeling the color perception of port wine stains and its relation to the depth of laser coagulated blood vessels. Lasers Surg. Med. 1993, 13, 219–226. [Google Scholar] [CrossRef]
- Verkruysse, W.; Lucassen, G.W.; van Gemert, M.J. Simulation of color of port wine stain skin and its dependence on skin variables. Lasers Surg. Med. 1999, 25, 131–139. [Google Scholar] [CrossRef]
- Choi, B.; Tan, W.; Jia, W.; White, S.M.; Moy, W.J.; Yang, B.Y.; Zhu, J.; Chen, Z.; Kelly, K.M.; Nelson, J.S. The Role of Laser Speckle Imaging in Port-Wine Stain Research: Recent Advances and Opportunities. IEEE J. Sel. Top. Quantum. Electron. 2016, 2016. [Google Scholar] [CrossRef]
- Yang, B.; Yang, O.; Guzman, J.; Nguyen, P.; Crouzet, C.; Osann, K.E.; Kelly, K.M.; Nelson, J.S.; Choi, B. Intraoperative, real-time monitoring of blood flow dynamics associated with laser surgery of port wine stain birthmarks. Lasers Surg. Med. 2015, 47, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, G.; Brand, A.; Franklin, E.C. Interaction of immunoglobulins with liposomes. J. Clin. Investig. 1974, 53, 536–543. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mittag, J.J.; Kneidl, B.; Preibeta, T.; Hossann, M.; Winter, G.; Wuttke, S.; Engelke, H.; Radler, J.O. Impact of plasma protein binding on cargo release by thermosensitive liposomes probed by fluorescence correlation spectroscopy. Eur. J. Pharm. Biopharm. 2017, 119, 215–223. [Google Scholar] [CrossRef]
- Wohner, N. Role of cellular elements in thrombus formation and dissolution. Cardiovasc. Hematol. Agents Med. Chem. 2008, 6, 224–228. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; van Raath, M.I.; Khakpour, S.; Seçilir, A.; Sliggers, B.C.; Huang, X.; Ding, B.; Storm, G.; van der Hulst, R.R.; de Kroon, A.I.P.M.; et al. In Vivo Assessment of Thermosensitive Liposomes for the Treatment of Port Wine Stains by Antifibrinolytic Site-Specific Pharmaco-Laser Therapy. Pharmaceutics 2020, 12, 591. https://doi.org/10.3390/pharmaceutics12060591
Li M, van Raath MI, Khakpour S, Seçilir A, Sliggers BC, Huang X, Ding B, Storm G, van der Hulst RR, de Kroon AIPM, et al. In Vivo Assessment of Thermosensitive Liposomes for the Treatment of Port Wine Stains by Antifibrinolytic Site-Specific Pharmaco-Laser Therapy. Pharmaceutics. 2020; 12(6):591. https://doi.org/10.3390/pharmaceutics12060591
Chicago/Turabian StyleLi, Mingjuan, M. Ingmar van Raath, Shervin Khakpour, Ahmet Seçilir, Bart C. Sliggers, Xuan Huang, Baoyue Ding, Gert Storm, René R. van der Hulst, Anton I.P.M. de Kroon, and et al. 2020. "In Vivo Assessment of Thermosensitive Liposomes for the Treatment of Port Wine Stains by Antifibrinolytic Site-Specific Pharmaco-Laser Therapy" Pharmaceutics 12, no. 6: 591. https://doi.org/10.3390/pharmaceutics12060591
APA StyleLi, M., van Raath, M. I., Khakpour, S., Seçilir, A., Sliggers, B. C., Huang, X., Ding, B., Storm, G., van der Hulst, R. R., de Kroon, A. I. P. M., & Heger, M. (2020). In Vivo Assessment of Thermosensitive Liposomes for the Treatment of Port Wine Stains by Antifibrinolytic Site-Specific Pharmaco-Laser Therapy. Pharmaceutics, 12(6), 591. https://doi.org/10.3390/pharmaceutics12060591