Injectable Lipid-Based Depot Formulations: Where Do We Stand?



Abstract
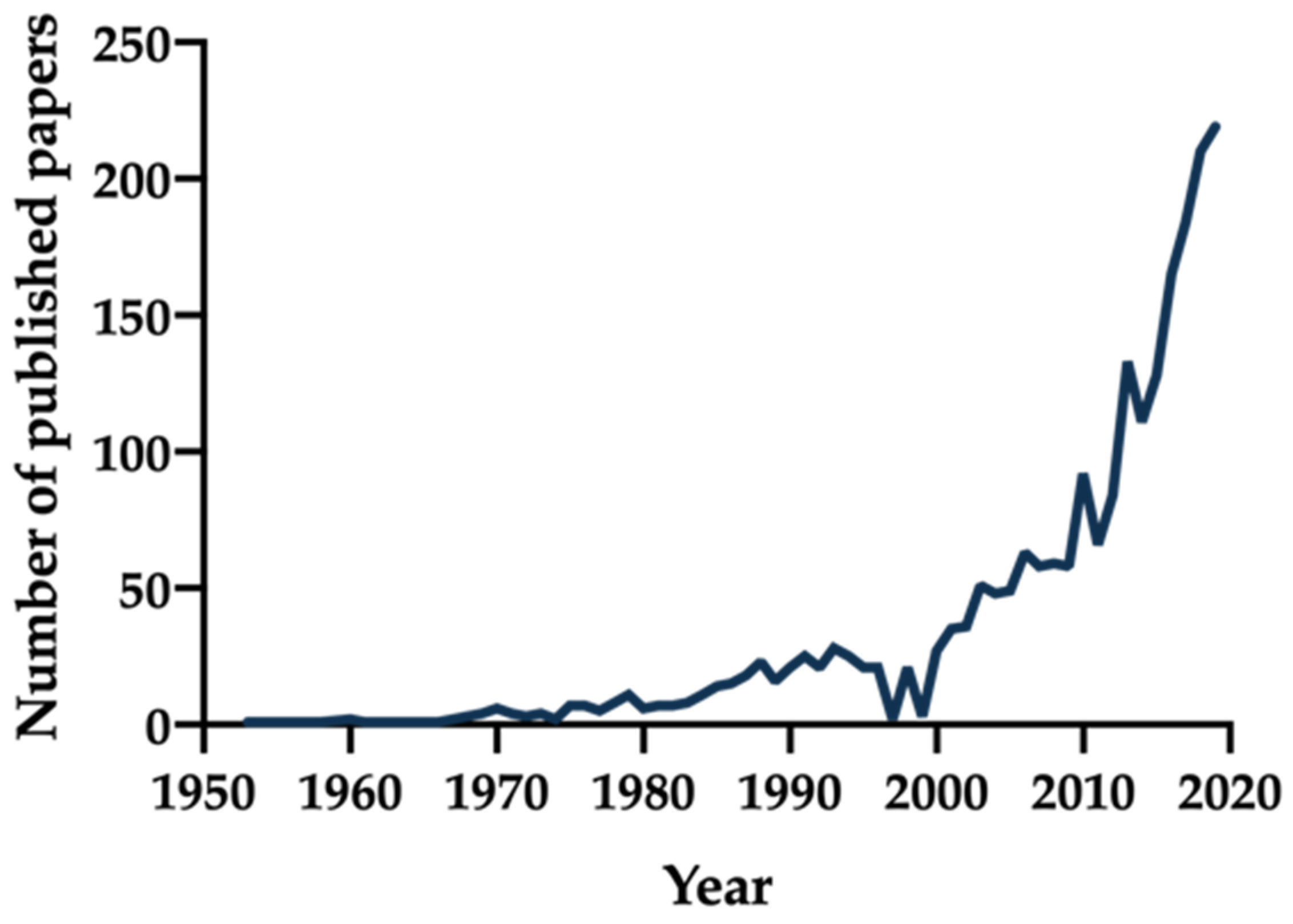
1. Introduction
2. The Ideal Depot Delivery System
2.1. Excipients for Formulating Depots
2.2. Parenteral Administration Routes for Depot Injectables
2.3. Biocompatibility
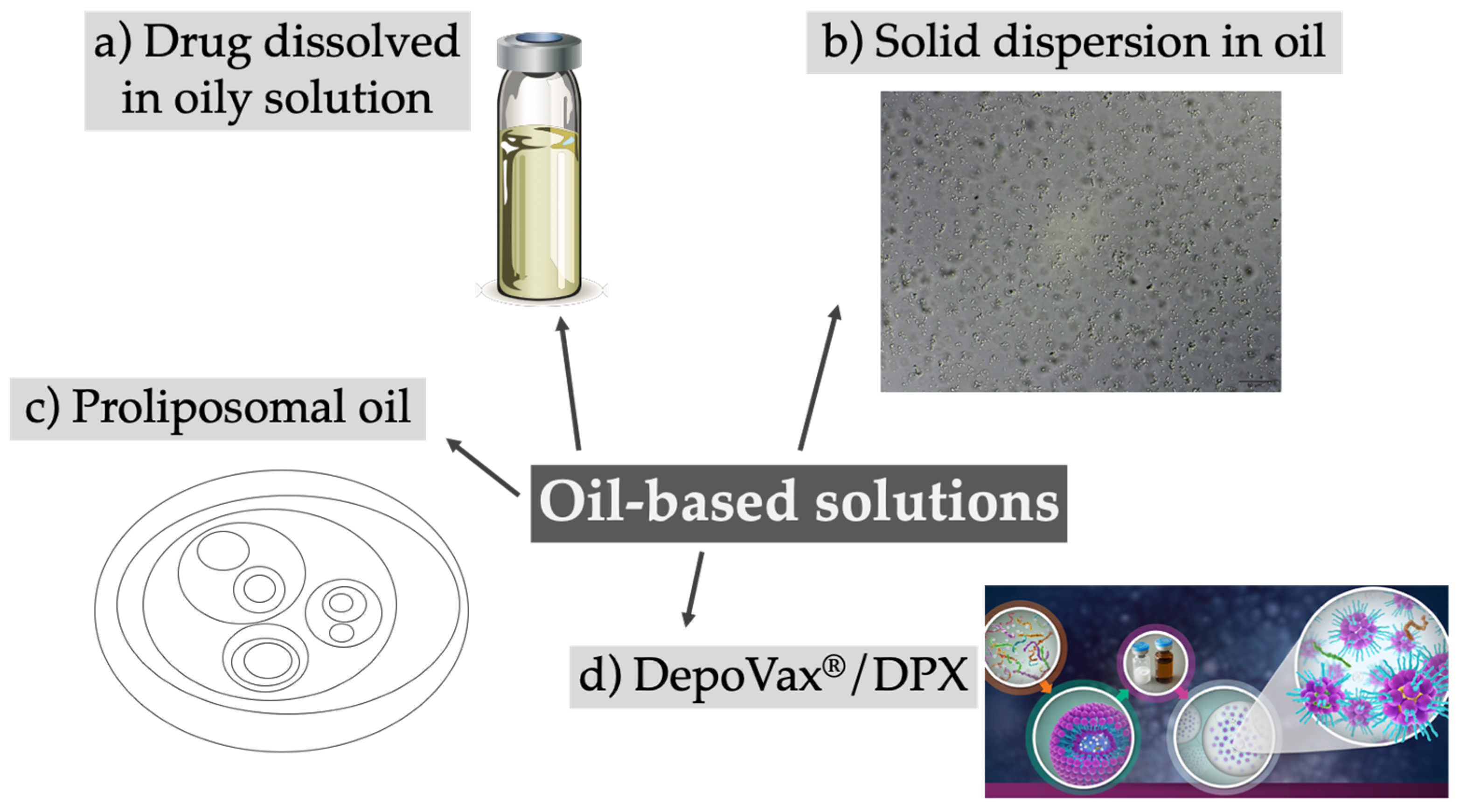
3. Oil-Based Solutions
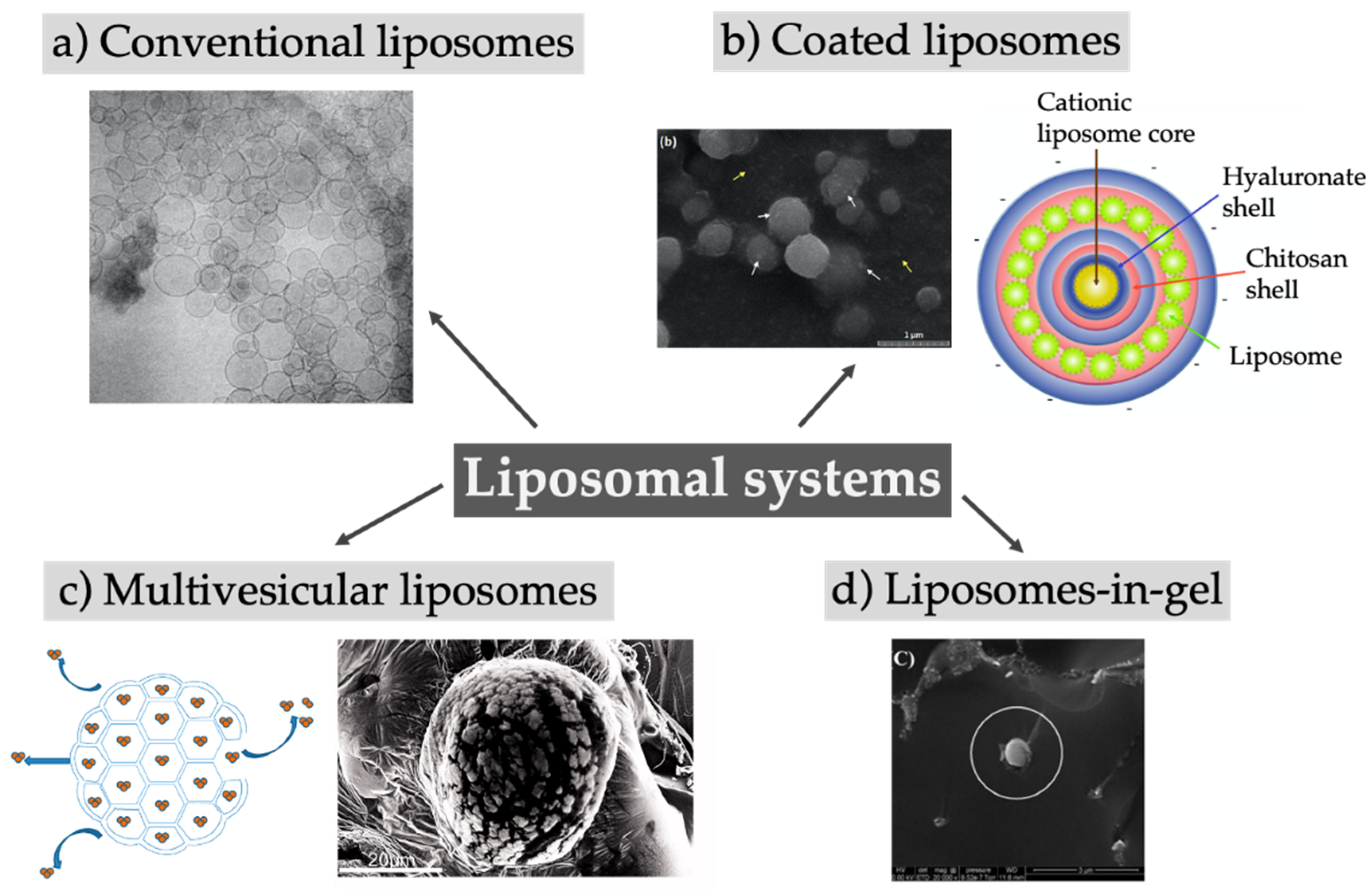
4. Liposomal Systems
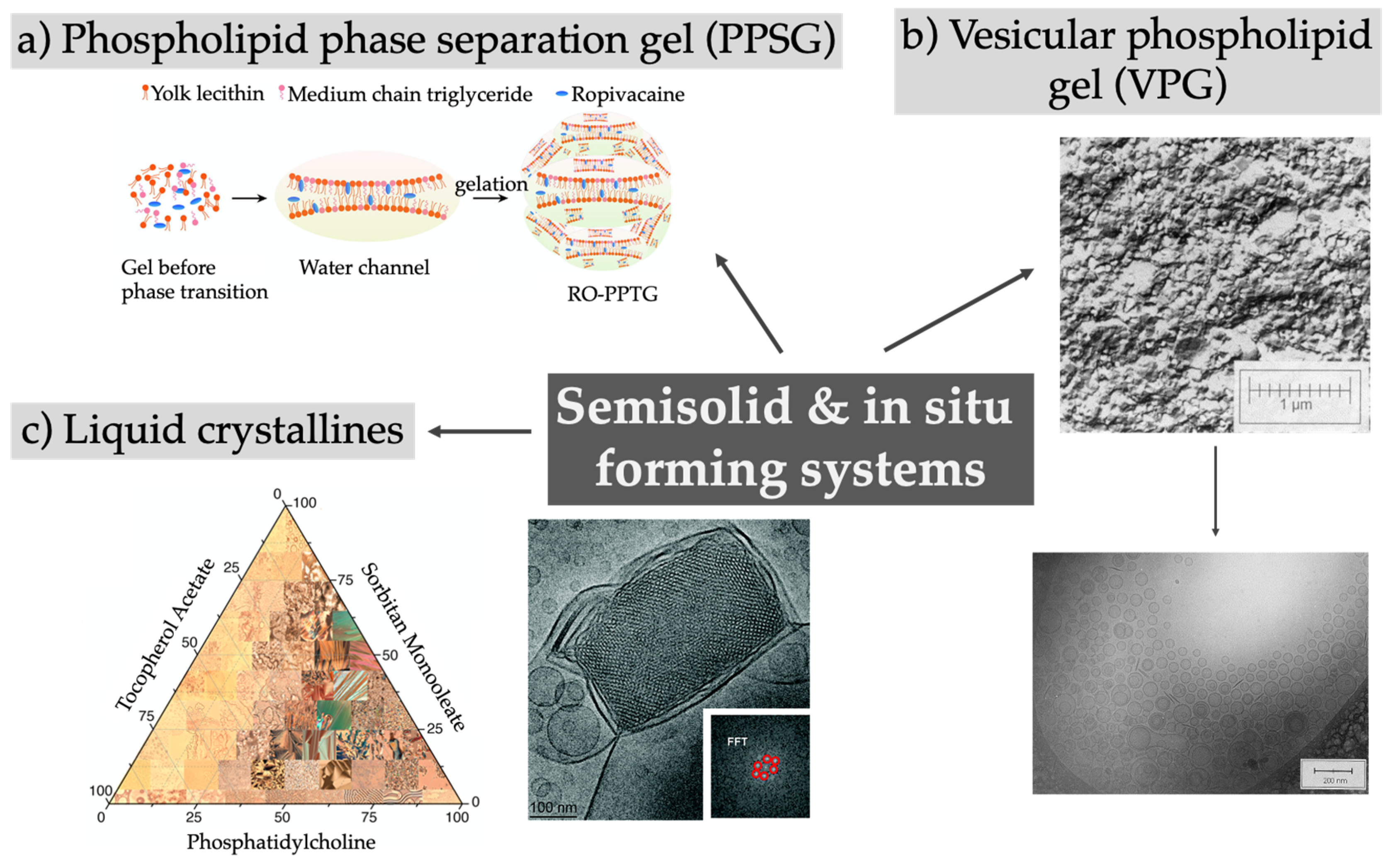
5. Semi-Solid Preparations and In Situ Forming Systems
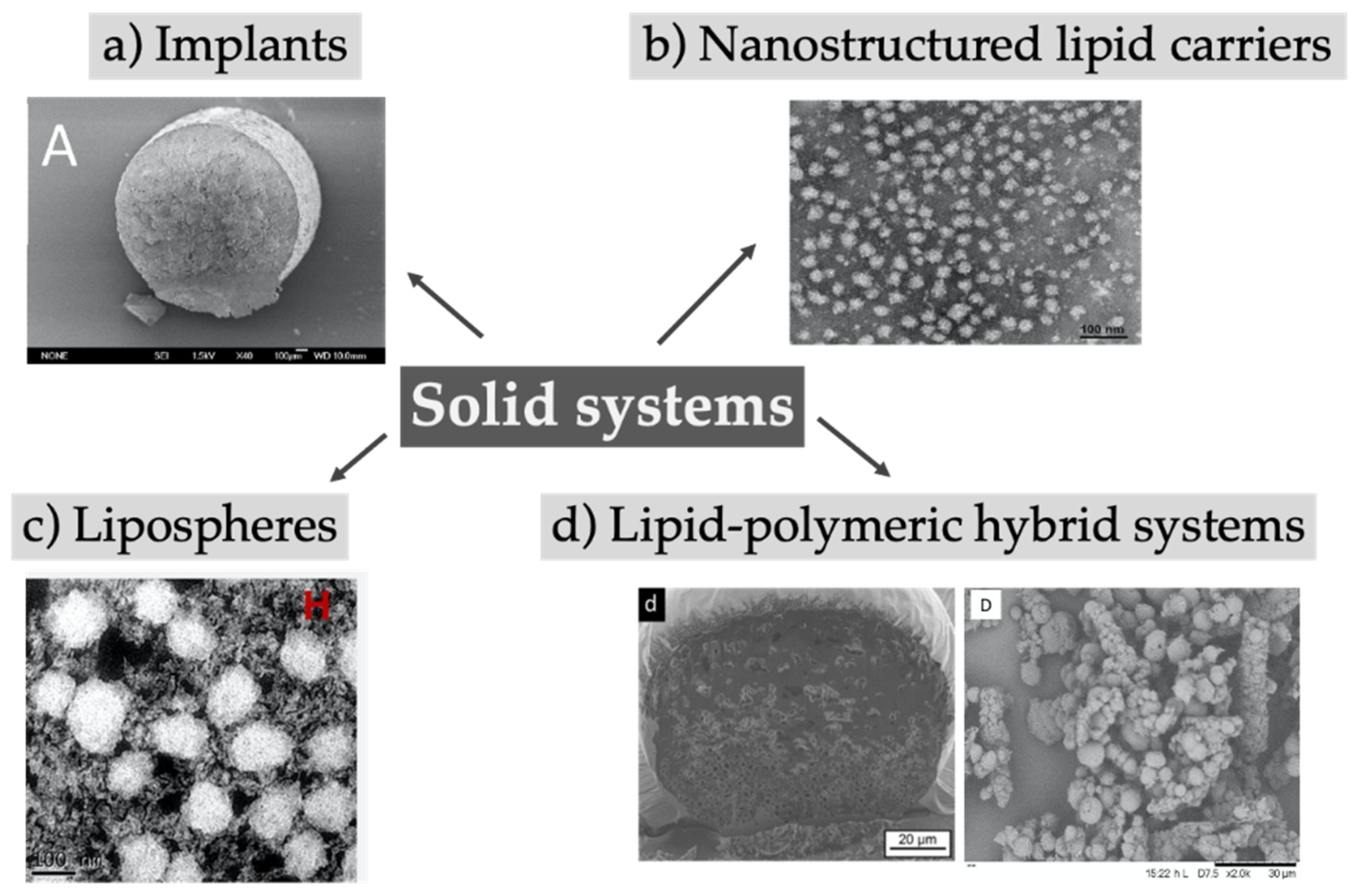
6. Solid Particles and Implants
7. From Design to Application: Considerations about In Vitro Release Testing
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jin, J.; Sklar, G.E.; Min Sen Oh, V.; Li, S.C. Factors affecting therapeutic compliance: A review from the patient’s perspective. Ther. Clin. Risk Manag. 2008, 4, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Sabaté, E.; De Geest, S. Adherence to Long-Term Therapies: Evidence for Action; World Health Organization: Geneva, Switzerland, 2003; Volume 2, ISBN 9241545992. [Google Scholar]
- Lee, W.Y.; Asadujjaman, M.; Jee, J.-P. Long acting injectable formulations: The state of the arts and challenges of poly(lactic-co-glycolic acid) microsphere, hydrogel, organogel and liquid crystal. J. Pharm. Investig. 2019. [Google Scholar] [CrossRef]
- 〈1151〉 Pharmaceutical Dosage Forms; United States Pharmacopeia: Rockville, MD, USA, 2019.
- Glossary. In European Pharmacopoeia 9.2, 1502; European Pharmacopoeia: Strasbourg, France, 2017.
- Pubmed Search. Available online: https://www.ncbi.nlm.nih.gov/pubmed?term=((((((%22longactinginjectable%22)OR%22longactingparental%22)OR%22longactingdepot%22)OR%22depotformulation%22))OR%22sustainedreleaseparental%22)OR%22controlledreleaseparental%22 (accessed on 25 March 2020).
- Bassyouni, F.; ElHalwany, N.; Abdel Rehim, M.; Neyfeh, M. Advances and new technologies applied in controlled drug delivery system. Res. Chem. Intermed. 2015, 41, 2165–2200. [Google Scholar] [CrossRef]
- Vhora, I.; Bardoliwala, D.; Ranamalla, S.R.; Javia, A. Parenteral Controlled and Prolonged Drug Delivery Systems: Therapeutic Needs and Formulation Strategies. In Novel Drug Delivery Technologies; Springer: Singapore, 2019; pp. 183–260. [Google Scholar]
- Kohane, D.S.; Langer, R.; Kinney, R.C.; Lipp, M.; Lotan, N.; Louis, D.N. Biocompatibility of lipid-protein-sugar particles containing bupivacaine in the epineurium. J. Biomed. Mater. Res. 2002, 59, 450–459. [Google Scholar] [CrossRef]
- Capen, R.; Christopher, D.; Forenzo, P.; Ireland, C.; Liu, O.; Lyapustina, S.; O’Neill, J.; Patterson, N.; Quinlan, M.; Sandell, D.; et al. On the shelf life of pharmaceutical products. AAPS PharmSciTech 2012, 13, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Faham, A.; Clas, S.D.; Boyd, B.J.; Jannin, V.; Bernkop-Schnürch, A.; Zhao, H.; Lecommandoux, S.; Evans, J.C.; Allen, C.; et al. Lipids and polymers in pharmaceutical technology: Lifelong companions. Int. J. Pharm. 2019, 558, 128–142. [Google Scholar] [CrossRef]
- Paolini, M.S.; Fenton, O.S.; Bhattacharya, C.; Andresen, J.L.; Langer, R. Polymers for extended-release administration. Biomed. Microdevices 2019, 21. [Google Scholar] [CrossRef]
- George, A.; Shah, P.A.; Shrivastav, P.S. Natural biodegradable polymers based nano-formulations for drug delivery: A review. Int. J. Pharm. 2019, 561, 244–264. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. Eur. J. Lipid Sci. Technol. 2005, 107, 337–364. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; Van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids 1. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef]
- LIPID MAPS Structure Database (LMSD). Available online: http://www.lipidmaps.org/data/classification/LM_classification_exp.php (accessed on 8 July 2019).
- Ruiz, M.E.; Scioli Montoto, S. Routes of Drug Administration. In ADME Processes in Pharmaceutical Sciences: Dosage, Design, and Pharmacotherapy Success; Talevi, A., Quiroga, P.A.M., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 97–133. ISBN 978-3-319-99593-9. [Google Scholar]
- Usach, I.; Martinez, R.; Festini, T.; Peris, J.E. Subcutaneous Injection of Drugs: Literature Review of Factors Influencing Pain Sensation at the Injection Site. Adv. Ther. 2019, 36, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, E.; Jahnke, J.; Fischer, A.; Kapitza, C.; Forst, T. Impact of Injection Speed, Volume, and Site on Pain Sensation. J. Diabetes Sci. Technol. 2018, 12, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Toh, M.-R.; Chiu, G.N.C. Liposomes as sterile preparations and limitations of sterilisation techniques in liposomal manufacturing. Asian J. Pharm. Sci. 2013, 8, 88–95. [Google Scholar] [CrossRef]
- Morais, J.M.; Papadimitrakopoulos, F.; Burgess, D.J. Biomaterials/Tissue Interactions: Possible Solutions to Overcome Foreign Body Response. AAPS J. 2010, 12, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; McNally, A.K. Biocompatibility of implants: Lymphocyte/macrophage interactions. Semin. Immunopathol. 2011, 33, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Onuki, Y.; Bhardwaj, U.; Papadimitrakopoulos, F.; Burgess, D.J. A review of the biocompatibility of implantable devices: Current challenges to overcome foreign body response. J. Diabetes Sci. Technol. 2008, 2, 1003–1015. [Google Scholar] [CrossRef]
- Anderson, J.M. Host Response to Long Acting Injections and Implants. In Long Acting Injections and Implants; Wright, J.C., Burgess, D.J., Eds.; Springer: Boston, MA, USA, 2012; pp. 25–55. ISBN 978-1-4614-0554-2. [Google Scholar]
- Chen, W.; Yung, B.C.; Qian, Z.; Chen, X. Improving long-term subcutaneous drug delivery by regulating material-bioenvironment interaction. Adv. Drug Deliv. Rev. 2018, 127, 20–34. [Google Scholar] [CrossRef]
- Doty, A.C.; Hirota, K.; Olsen, K.F.; Sakamoto, N.; Ackermann, R.; Feng, M.R.; Wang, Y.; Choi, S.; Qu, W.; Schwendeman, A.; et al. Validation of a cage implant system for assessing in vivo performance of long-acting release microspheres. Biomaterials 2016, 109, 88–96. [Google Scholar] [CrossRef]
- Kim, J.; Dadsetan, M.; Ameenuddin, S.; Windebank, A.J.; Yaszemski, M.J.; Lu, L. In vivo biodegradation and biocompatibility of PEG/sebacic acid-based hydrogels using a cage implant system. J. Biomed. Mater. Res. Part A 2010, 95, 191–197. [Google Scholar] [CrossRef]
- Marchant, R.; Hiltner, A.; Hamlin, C.; Rabinovitch, A.; Slobodkin, R.; Anderson, J.M. In vivo biocompatibility studies. I. The cage implant system and a biodegradable hydrogel. J. Biomed. Mater. Res. 1983, 17, 301–325. [Google Scholar] [CrossRef]
- Rawat, M.; Singh, D.; Saraf, S.; Saraf, S. Lipid Carriers: A Versatile Delivery Vehicle for Proteins and Peptides. YAKUGAKU ZASSHI 2008, 128, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Mohamed, M.; Daftardar, S.B.; Patel, M.; Boddu, S.H.S.; Nesamony, J. Solid Lipid Nanoparticles in Drug Delivery: Opportunities and Challenges. In Emerging Nanotechnologies for Diagnostics, Drug Delivery and Medical Devices; Elsevier: Amsterdam, The Netherlands, 2017; pp. 291–330. ISBN 9780323429979. [Google Scholar]
- Katre, N.V.; Asherman, J.; Schaefer, H.; Hora, M. Multivesicular Liposome (DepoFoam) Technology for the Sustained Delivery of Insulin-Like Growth Factor-I (IGF-I). J. Pharm. Sci. 1998, 87, 1341–1346. [Google Scholar] [CrossRef]
- Roehrborn, A.A.; Hansbrough, J.F.; Gualdoni, B.; Kim, S. Lipid-based slow-release formulation of amikacin sulfate reduces foreign body-associated infections in mice. Antimicrob. Agents Chemother. 1995, 39, 1752–1755. [Google Scholar] [CrossRef]
- Rhee, Y.-S.; Park, C.-W.; DeLuca, P.P.; Mansour, H.M. Sustained-Release Injectable Drug Delivery. Pharm. Technol. 2010, 34, 6–13. [Google Scholar]
- Altamura, A.C.; Sassella, F.; Santini, A.; Montresor, C.; Fumagalli, S.; Mundo, E. Intramuscular Preparations of Antipsychotics. Drugs 2003, 63, 493–512. [Google Scholar] [CrossRef]
- Weng Larsen, S.; Larsen, C. Critical Factors Influencing the In Vivo Performance of Long-acting Lipophilic Solutions—Impact on In Vitro Release Method Design. AAPS J. 2009, 11, 762–770. [Google Scholar] [CrossRef]
- Park, E.J.; Amatya, S.; Kim, M.S.; Park, J.H.; Seol, E.; Lee, H.; Shin, Y.H.; Na, D.H. Long-acting injectable formulations of antipsychotic drugs for the treatment of schizophrenia. Arch. Pharm. Res. 2013, 36, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Set of Different Medical Ampoules on White Background-Download Free Vectors, Clipart Graphics & Vector Art. Available online: https://www.vecteezy.com/vector-art/141828-set-of-different-medical-ampoules-on-white-background (accessed on 12 May 2020).
- Zhang, S.; Wan, Q.; Xu, X.; Xing, Y.; Ding, J.; Yang, S.; Sun, W.; Lu, M.; Pan, B. A novel oil-based suspension of a micro-environmental, pH-modifying solid dispersion for parenteral delivery: Formulation and stability evaluation. Colloids Surf. B Biointerfaces 2019, 179, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Platform | IMV. Available online: https://imv-inc.com/platform/ (accessed on 25 March 2020).
- Nguyen, T.T.L.; Duong, V.A.; Maeng, H.J.; Chi, S.C. Preparation of an oil suspension containing ondansetron hydrochloride as a sustained release parenteral formulation. Drug Deliv. Transl. Res. 2020, 10, 282–295. [Google Scholar] [CrossRef]
- Davidson, E.M.; Haroutounian, S.; Kagan, L.; Naveh, M.; Aharon, A.; Ginosar, Y. A Novel Proliposomal Ropivacaine Oil. Anesth. Analg. 2016, 122, 1663–1672. [Google Scholar] [CrossRef]
- Ginosar, Y.; Haroutounian, S.; Kagan, L.; Naveh, M.; Aharon, A.; Davidson, E.M. Proliposomal Ropivacaine Oil. Anesth. Analg. 2016, 122, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xiong, D.; Sun, W.; Yu, T.; Hu, Z.; Ding, J.; Cai, Y.; Yang, S.; Pan, B. Sustained release ivermectin-loaded solid lipid dispersion for subcutaneous delivery: In vitro and in vivo evaluation. Drug Deliv. 2017, 24, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Cai, Y.; Yang, S.; Wan, Q.; Pan, B. A single subcutaneous administration of a sustained-release ivermectin suspension eliminates Psoroptes cuniculi infection in a rabbit farm. Drug Dev. Ind. Pharm. 2018, 44, 2000–2004. [Google Scholar] [CrossRef] [PubMed]
- Hobson, J.J.; Curley, P.; Savage, A.C.; Al-Khouja, A.; Siccardi, M.; Flexner, C.; Meyers, C.F.; Owen, A.; Rannard, S.P. Anhydrous nanoprecipitation for the preparation of nanodispersions of tenofovir disoproxil fumarate in oils as candidate long-acting injectable depot formulations. Nanoscale Adv. 2019, 1, 4301–4307. [Google Scholar] [CrossRef]
- Brewer, K.D.; Weir, G.M.; Dude, I.; Davis, C.; Parsons, C.; Penwell, A.; Rajagopalan, R.; Sammatur, L.; Bowen, C.V.; Stanford, M.M. Unique depot formed by an oil based vaccine facilitates active antigen uptake and provides effective tumour control. J. Biomed. Sci. 2018, 25, 1–12. [Google Scholar] [CrossRef]
- Weir, G.M.; MacDonald, L.D.; Rajagopalan, R.; Sivko, G.S.; Valderas, M.W.; Rayner, J.; Berger, B.J.; Sammatur, L.; Stanford, M.M. Single dose of DPX-rPA, an enhanced-delivery anthrax vaccine formulation, protects against a lethal Bacillus anthracis spore inhalation challenge. NPJ Vaccines 2019, 4, 1–9. [Google Scholar] [CrossRef]
- Karkada, M.; Weir, G.M.; Quinton, T.; Fuentes-Ortega, A.; Mansour, M. A liposome-based platform, VacciMax®, and its modified water-free platform DepoVax™ enhance efficacy of in vivo nucleic acid delivery. Vaccine 2010, 28, 6176–6182. [Google Scholar] [CrossRef]
- Clinical Trials | IMV. Available online: https://imv-inc.com/clinical-trials/ongoing-trials/ (accessed on 18 March 2020).
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z.; Li, F.; Gao, J.; Wang, L.; Huang, G. Liposomes for systematic delivery of vancomycin hydrochloride to decrease nephrotoxicity: Characterization and evaluation. Asian J. Pharm. Sci. 2015, 10, 212–222. [Google Scholar] [CrossRef]
- Couto, V.M.; Prieto, M.J.; Igartúa, D.E.; Feas, D.A.; Ribeiro, L.N.M.; Silva, C.M.G.; Castro, S.R.; Guilherme, V.A.; Dantzger, D.D.; Machado, D.; et al. Dibucaine in Ionic-Gradient Liposomes: Biophysical, Toxicological, and Activity Characterization. J. Pharm. Sci. 2018, 107, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Rwei, A.Y.; Sherburne, R.T.; Zurakowski, D.; Wang, B.; Kohane, D.S. Prolonged duration local anesthesia using liposomal bupivacaine combined with liposomal dexamethasone and dexmedetomidine. Anesth. Analg. 2018, 126, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Rahnfeld, L.; Thamm, J.; Steiniger, F.; Van Hoogevest, P.; Luciani, P. Study on the in situ aggregation of liposomes with negatively charged phospholipids for use as injectable depot formulation. Colloids Surf. B Biointerfaces 2018, 168, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.Y.; Seong, J.S.; Park, S.N. Preparation of novel capsosome with liposomal core by layer-by-Layer self-assembly of sodium hyaluronate and chitosan. Colloids Surf. B Biointerfaces 2016, 144, 99–107. [Google Scholar] [CrossRef]
- Billard, A.; Pourchet, L.; Malaise, S.; Alcouffe, P.; Montembault, A.; Ladavière, C. Liposome-loaded chitosan physical hydrogel: Toward a promising delayed-release biosystem. Carbohydr. Polym. 2015, 115, 651–657. [Google Scholar] [CrossRef]
- Mu, H.; Wang, Y.; Chu, Y.; Jiang, Y.; Hua, H.; Chu, L.; Wang, K.; Wang, A.; Liu, W.; Li, Y.; et al. Multivesicular liposomes for sustained release of bevacizumab in treating laser-induced choroidal neovascularization. Drug Deliv. 2018, 25, 1372–1383. [Google Scholar] [CrossRef]
- Blazaki, S.; Pachis, K.; Tzatzarakis, M.; Tsilimbaris, M.; Antimisiaris, S.G. Novel Liposome Aggregate Platform (LAP) system for sustained retention of drugs in the posterior ocular segment following intravitreal injection. Int. J. Pharm. 2020, 576, 118987. [Google Scholar] [CrossRef]
- Schlich, M.; Sinico, C.; Valenti, D.; Gulati, A.; Joshi, M.D.; Meli, V.; Murgia, S.; Xanthos, T. Towards long-acting adrenaline for cardiopulmonary resuscitation: Production and characterization of a liposomal formulation. Int. J. Pharm. 2019, 557, 105–111. [Google Scholar] [CrossRef]
- Christensen, D.; Bøllehuus Hansen, L.; Leboux, R.; Jiskoot, W.; Christensen, J.P.; Andersen, P.; Dietrich, J. A Liposome-Based Adjuvant Containing Two Delivery Systems with the Ability to Induce Mucosal Immunoglobulin A Following a Parenteral Immunization. ACS Nano 2019, 13, 1116–1126. [Google Scholar] [CrossRef]
- Ji, X.; Yan, Y.; Sun, T.; Zhang, Q.; Wang, Y.; Zhang, M.; Zhang, H.; Zhao, X. Glucosamine sulphate-loaded distearoyl phosphocholine liposomes for osteoarthritis treatment: Combination of sustained drug release and improved lubrication. Biomater. Sci. 2019, 7, 2716–2728. [Google Scholar] [CrossRef]
- Karumanchi, D.K.; Skrypai, Y.; Thomas, A.; Gaillard, E.R. Rational design of liposomes for sustained release drug delivery of bevacizumab to treat ocular angiogenesis. J. Drug Deliv. Sci. Technol. 2018, 47, 275–282. [Google Scholar] [CrossRef]
- Vyas, S.P.; Rawat, M.; Rawat, A.; Mahor, S.; Gupta, P.N. Pegylated Protein Encapsulated Multivesicular Liposomes: A Novel Approach for Sustained Release of Interferon α. Drug Dev. Ind. Pharm. 2006, 32, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.M.G.; Franz-Montan, M.; Limia, C.E.G.; Ribeiro, L.N.d.M.; Braga, M.A.; Guilherme, V.A.; Da Silva, C.B.; Casadei, B.R.; Cereda, C.M.S.; De Paula, E. Encapsulation of ropivacaine in a combined (donor-acceptor, ionic-gradient) liposomal system promotes extended anesthesia time. PLoS ONE 2017, 12, e0185828. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.M.G.; Fraceto, L.F.; Franz-Montan, M.; Couto, V.M.; Casadei, B.R.; Cereda, C.M.S.; De Paula, E. Development of egg PC/cholesterol/α-tocopherol liposomes with ionic gradients to deliver ropivacaine. J. Liposome Res. 2016, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ding, L.; Tang, C.; Li, Y.; Yang, L. Liraglutide-loaded multivesicular liposome as a sustained-delivery reduces blood glucose in SD rats with diabetes. Drug Deliv. 2016, 23, 3358–3363. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, Z.; Zhang, X.; Huang, J.; Yu, X.; Li, J.; Xiong, D.; Sun, X.; Zhong, Z. Effect of a controlled-release drug delivery system made of oleanolic acid formulated into multivesicular liposomes on hepatocellular carcinoma in vitro and in vivo. Int. J. Nanomed. 2016, 11, 3111–3129. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, Y.; Li, C.; Zhang, X.; Pi, C.; Yu, L.; Wang, S.; Zhong, Z. Optimized formulation of multivesicular liposomes loaded with oleanolic acid enhanced anticancer effect in vitro. Drug Des. Dev. Ther. 2017, 11, 955–968. [Google Scholar] [CrossRef]
- Vafaei, S.Y.; Dinarvand, R.; Esmaeili, M.; Mahjub, R.; Toliyat, T. Controlled-release drug delivery system based on fluocinolone acetonide–cyclodextrin inclusion complex incorporated in multivesicular liposomes. Pharm. Dev. Technol. 2015, 20, 775–781. [Google Scholar] [CrossRef]
- Lee, C.Y.; Robinson, D.A.; Johnson, C.A.; Zhang, Y.; Wong, J.; Joshi, D.J.; Wu, T.T.; Knight, P.A. A Randomized Controlled Trial of Liposomal Bupivacaine Parasternal Intercostal Block for Sternotomy. Ann. Thorac. Surg. 2019, 107, 128–134. [Google Scholar] [CrossRef]
- Yalmanchili, H.M.; Buchanan, S.N.; Chambers, L.W.; Thorns, J.D.; McKenzie, N.A.; Reiss, A.D.; Page, M.P.; Dizon, V.V.; Brooks, S.E.; Shaffer, L.E.; et al. Postlaparotomy pain management: Comparison of patient-controlled analgesia pump alone, with subcutaneous bupivacaine infusion, or with injection of liposomal bupivacaine suspension. J. Opioid Manag. 2019, 15, 169. [Google Scholar] [CrossRef]
- Fu, H.; Huang, L.; Xu, C.; Zhang, J.; Li, D.; Ding, L.; Liu, L.; Dong, Y.; Wang, W.; Duan, Y. Highly biocompatible thermosensitive nanocomposite gel for combined therapy of hepatocellular carcinoma via the enhancement of mitochondria related apoptosis. Nanomed. Nanotechnol. Biol. Med. 2019, 21, 102062. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wong, B.C.K.; Chen, H.; Zhang, S.; Bian, Z.; Zhang, G.; Lin, C.; Riaz, M.K.; Tyagi, D.; Lu, A.; et al. Long-lasting Insulin Treatment Via a Single Subcutaneous Administration of Liposomes in Thermoreversible Pluronic® F127 Based Hydrogel. Curr. Pharm. Des. 2017, 23, 6079–6085. [Google Scholar] [CrossRef] [PubMed]
- Pachis, K.; Blazaki, S.; Tzatzarakis, M.; Klepetsanis, P.; Naoumidi, E.; Tsilimbaris, M.; Antimisiaris, S.G. Sustained release of intravitreal flurbiprofen from a novel drug-in-liposome-in-hydrogel formulation. Eur. J. Pharm. Sci. 2017, 109, 324–333. [Google Scholar] [CrossRef]
- Cao, D.; Zhang, X.; Akabar, M.D.; Luo, Y.; Wu, H.; Ke, X.; Ci, T. Liposomal doxorubicin loaded PLGA-PEG-PLGA based thermogel for sustained local drug delivery for the treatment of breast cancer. Artif. Cells Nanomed. Biotechnol. 2019, 47, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Guo, B.; Wang, S.; Ding, J.; Zhou, W. A thermo-responsive and self-healing liposome-in-hydrogel system as an antitubercular drug carrier for localized bone tuberculosis therapy. Int. J. Pharm. 2019, 558, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Dai, Y.; Li, C.; Qiu, Z.; Wang, X.; Tian, F.; Zhou, S.; Liu, Q.; Xing, H.; Lu, Y.; et al. Pharmacokinetics and pharmacodynamics evaluation of a thermosensitive chitosan based hydrogel containing liposomal doxorubicin. Eur. J. Pharm. Sci. 2016, 92, 137–145. [Google Scholar] [CrossRef]
- Peng, S.; Zou, L.; Liu, W.; Li, Z.; Liu, W.; Hu, X.; Chen, X.; Liu, C. Hybrid liposomes composed of amphiphilic chitosan and phospholipid: Preparation, stability and bioavailability as a carrier for curcumin. Carbohydr. Polym. 2017, 156, 322–332. [Google Scholar] [CrossRef]
- Ravar, F.; Saadat, E.; Gholami, M.; Dehghankelishadi, P.; Mahdavi, M.; Azami, S.; Dorkoosh, F.A. Hyaluronic acid-coated liposomes for targeted delivery of paclitaxel, in-vitro characterization and in-vivo evaluation. J. Control. Release 2016, 229, 10–22. [Google Scholar] [CrossRef]
- Nascimento Vieira, A.L.; Franz-Montan, M.; Cabeça, L.F.; De Paula, E. Anaesthetic benefits of a ternary drug delivery system (Ropivacaine-in-Cyclodextrin-in-Liposomes): In-Vitro and In-Vivo evaluation. J. Pharm. Pharmacol. 2020, 72, 396–408. [Google Scholar] [CrossRef]
- Nagarsekar, K.; Ashtikar, M.; Steiniger, F.; Thamm, J.; Schacher, F.; Fahr, A. Understanding cochleate formation: Insights into structural development. Soft Matter 2016, 12, 3797–3809. [Google Scholar] [CrossRef]
- Nagarsekar, K.; Ashtikar, M.; Steiniger, F.; Thamm, J.; Schacher, F.H.; Fahr, A. Micro-spherical cochleate composites: Method development for monodispersed cochleate system. J. Liposome Res. 2017, 27, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Nagarsekar, K.; Ashtikar, M.; Thamm, J.; Steiniger, F.; Schacher, F.; Fahr, A.; May, S. Electron Microscopy and Theoretical Modeling of Cochleates. Langmuir 2014, 30, 13143–13151. [Google Scholar] [CrossRef]
- Duan, Y.; Liu, Y.; Zhang, C.; Chen, Z.; Wen, S. Insight into the Tribological Behavior of Liposomes in Artificial Joints. Langmuir ACS J. Surf. Colloids 2016, 32, 10957–10966. [Google Scholar] [CrossRef] [PubMed]
- Sivan, S.; Schroeder, A.; Verberne, G.; Merkher, Y.; Diminsky, D.; Priev, A.; Maroudas, A.; Halperin, G.; Nitzan, D.; Etsion, I.; et al. Liposomes act as effective biolubricants for friction reduction in human synovial joints. Langmuir 2010, 26, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Angst, M.S.; Drover, D.R. Pharmacology of Drugs Formulated with DepoFoam™: A Sustained Release Drug Delivery System for Parenteral Administration Using Multivesicular Liposome Technology. Clin. Pharmacokinet. 2006, 45, 1153–1176. [Google Scholar] [CrossRef]
- Crommelin, D.J.A.; Van Hoogevest, P.; Storm, G. The role of liposomes in clinical nanomedicine development. What now? Now what? J. Control. Release 2020, 318, 256–263. [Google Scholar] [CrossRef]
- Mantripragada, S. A lipid based depot (DepoFoam® technology) for sustained release drug delivery. Prog. Lipid Res. 2002, 41, 392–406. [Google Scholar] [CrossRef]
- Kim, S.; Turker, M.S.; Chi, E.Y.; Sela, S.; Martin, G.M. Preparation of multivesicular liposomes. Biochim. Biophys. Acta 1983, 728, 339–348. [Google Scholar] [CrossRef]
- Katre, N.V. Liposome-based depot injection technologies: How versatile are they? Am. J. Drug Deliv. 2004, 2, 213–227. [Google Scholar] [CrossRef]
- Shen, Y.; Ji, Y.; Xu, S.; Chen, D.Q.; Tu, J. Multivesicular liposome formulations for the sustained delivery of ropivacaine hydrochloride: Preparation, characterization, and pharmacokinetics. Drug Deliv. 2011, 18, 361–366. [Google Scholar] [CrossRef]
- Public Statement EMA. Available online: https://www.ema.europa.eu/en/documents/public-statement/public-statement-depocyte-withdrawal-marketing-authorisation-european-union_en.pdf (accessed on 12 May 2020).
- Burnett, A.; Faley, B.; Nyirenda, T.; Bamboat, Z.M. Liposomal bupivacaine reduces narcotic use and time to flatus in a retrospective cohort of patients who underwent laparotomy. Int. J. Surg. 2018, 59, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, T.; Zhu, Y.; Fu, Q.; Wu, W.; Deng, J.; Lan, L.; Shi, S. An in situ-forming phospholipid-based phase transition gel prolongs the duration of local anesthesia for ropivacaine with minimal toxicity. Acta Biomater. 2017, 58, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Tardi, C.; Drechsler, M.; Bauer, K.H.; Brandl, M. Steam sterilisation of vesicular phospholipid gels. Int. J. Pharm. 2001, 217, 161–172. [Google Scholar] [CrossRef]
- Brandl, M.; Drechsler, M.; Bachmann, D.; Tardi, C.; Schmidtgen, M.; Bauer, K.-H. Preparation and characterization of semi-solid phospholipid dispersions and dilutions thereof. Int. J. Pharm. 1998, 170, 187–199. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; Otte, A.; Mun, E.A.; Soh, B.K.; Song, C.G.; Lee, Y.-N.; Park, K. Formulation and characterization of a liquid crystalline hexagonal mesophase region of phosphatidylcholine, sorbitan monooleate, and tocopherol acetate for sustained delivery of leuprolide acetate. Int. J. Pharm. 2016, 514, 314–321. [Google Scholar] [CrossRef]
- Angelov, B.; Angelova, A.; Drechsler, M.; Garamus, V.M.; Mutafchieva, R.; Lesieur, S. Identification of large channels in cationic PEGylated cubosome nanoparticles by synchrotron radiation SAXS and Cryo-TEM imaging. Soft Matter 2015, 11, 3686–3692. [Google Scholar] [CrossRef]
- Zhang, T.; Luo, J.; Peng, Q.; Dong, J.; Wang, Y.; Gong, T.; Zhang, Z. Injectable and biodegradable phospholipid-based phase separation gel for sustained delivery of insulin. Colloids Surf. B Biointerfaces 2019, 176, 194–201. [Google Scholar] [CrossRef]
- Chen, T.; Gong, T.; Zhao, T.; Liu, X.; Fu, Y.; Zhang, Z.; Gong, T. Paclitaxel loaded phospholipid-based gel as a drug delivery system for local treatment of glioma. Int. J. Pharm. 2017, 528, 127–132. [Google Scholar] [CrossRef]
- Luo, J.W.; Zhang, T.; Zhang, Q.; Cao, X.; Zeng, X.; Fu, Y.; Zhang, Z.R.; Gong, T. A novel injectable phospholipid gel co-loaded with doxorubicin and bromotetrandrine for resistant breast cancer treatment by intratumoral injection. Colloids Surf. B Biointerfaces 2016, 140, 538–547. [Google Scholar] [CrossRef]
- Xiang, N.; Zhou, X.; He, X.; Zhang, Y.; Zhang, J.; Zhang, Z.R.; Sun, X.; Gong, T.; Fu, Y. An Injectable Gel Platform for the Prolonged Therapeutic Effect of Pitavastatin in the Management of Hyperlipidemia. J. Pharm. Sci. 2016, 105, 1148–1155. [Google Scholar] [CrossRef]
- Zhang, T.; Peng, Q.; San, F.Y.; Luo, J.W.; Wang, M.X.; Wu, W.Q.; Gong, T.; Zhang, Z.R. A high-efficiency, low-toxicity, phospholipids-based phase separation gel for long-term delivery of peptides. Biomaterials 2015, 45, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Xue, J.; Wang, L.; Peng, K.; Zhang, Z.; Gong, T.; Sun, X. An injectable, low-toxicity phospholipid-based phase separation gel that induces strong and persistent immune responses in mice. Biomaterials 2016, 105, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhang, Y.; Xiang, N.; Zhong, Y.; Gong, T.; Zhang, Z.R.; Fu, Y. Long-Acting Phospholipid Gel of Exenatide for Long-Term Therapy of Type II Diabetes. Pharm. Res. 2016, 33, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Gong, T.; Zhang, Z.; Ding, R.; Fu, Y. Preparation and evaluation of a phospholipid-based injectable gel for the long term delivery of leuprolide acetate. Acta Pharm. Sin. B 2016, 6, 329–335. [Google Scholar] [CrossRef]
- Wang, M.; Shan, F.; Zou, Y.; Sun, X.; Zhang, Z.R.; Fu, Y.; Gong, T. Pharmacokinetic and pharmacodynamic study of a phospholipid-based phase separation gel for once a month administration of octreotide. J. Control. Release 2016, 230, 45–56. [Google Scholar] [CrossRef]
- Yang, L.; Song, X.; Gong, T.; Wang, L.; Zhao, T.; Li, L.; Zhou, C.; Sun, X.; Zhang, Z.; Gong, T. Enhanced anti-tumor and anti-metastasis efficacy against breast cancer with an intratumoral injectable phospholipids-based phase separation gel co-loaded with 5-fluotouracil and magnesium oxide by neutralizing acidic microenvironment. Int. J. Pharm. 2018, 547, 181–189. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, M.; Wei, G.; Jia, M.; Gong, T.; Liu, J. An injectable in situ lipid phase transition system for sustained delivery of dabigatran etexilate with low burst release. RSC Adv. 2017, 7, 56594–56601. [Google Scholar] [CrossRef]
- Neuhofer, C.T.K.-H. Development of Lipid Based Depot Formulations Using Interferon-Beta-1b as a Model Protein. Ph.D. Thesis, Ludwig-Maximilians-Universität München, Munich, Germany, 2015. Available online: https://edoc.ub.uni-muenchen.de/18948/1/Neuhofer_Christian.pdf (accessed on 18 May 2020).
- Balsevich, G.; Häusl, A.S.; Meyer, C.W.; Karamihalev, S.; Feng, X.; Pöhlmann, M.L.; Dournes, C.; Uribe-Marino, A.; Santarelli, S.; Labermaier, C.; et al. Stress-responsive FKBP51 regulates AKT2-AS160 signaling and metabolic function. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Pöhlmann, M.L.; Häusl, A.S.; Harbich, D.; Balsevich, G.; Engelhardt, C.; Feng, X.; Breitsamer, M.; Hausch, F.; Winter, G.; Schmidt, M.V. Pharmacological Modulation of the Psychiatric Risk Factor FKBP51 Alters Efficiency of Common Antidepressant Drugs. Front. Behav. Neurosci. 2018, 12, 1–8. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhong, Y.; Hu, M.; Xiang, N.; Fu, Y.; Gong, T.; Zhang, Z. In vitro and in vivo sustained release of exenatide from vesicular phospholipid gels for type II diabetes. Drug Dev. Ind. Pharm. 2016, 42, 1042–1049. [Google Scholar] [CrossRef]
- Breitsamer, M.; Stulz, A.; Heerklotz, H.H.; Winter, G. Do interactions between protein and phospholipids influence the release behavior from lipid-based exenatide depot systems? Eur. J. Pharm. Biopharm. 2019, 142, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.X.; Chen, H. Phospholipid Depot. U.S. Patent 2017/0294648 A1, 6 July 2017. [Google Scholar]
- Chen, A.X.; Chen, H. Phospholipid Depot. U.S. Patent 9.517.202 B2, 13 December 2016. [Google Scholar]
- Xia, M.; Liu, L.; Tian, C.; Li, Q.; Hu, R.; Gui, S.; Chu, X. Pharmacokinetics of sinomenine hydrochloride cubic liquid crystal injection based on microdialysis technology. J. Drug Deliv. Sci. Technol. 2019, 52, 553–558. [Google Scholar] [CrossRef]
- Pavel, M.; Borson-Chazot, F.; Cailleux, A.; Hörsch, D.; Lahner, H.; Pivonello, R.; Tauchmanova, L.; Darstein, C.; Olsson, H.; Tiberg, F.; et al. Octreotide SC depot in patients with acromegaly and functioning neuroendocrine tumors: A phase 2, multicenter study. Cancer Chemother. Pharmacol. 2019, 83, 375–385. [Google Scholar] [CrossRef]
- Schoubben, A.; Ricci, M.; Giovagnoli, S. Meeting the unmet: From traditional to cutting-edge techniques for poly lactide and poly lactide-co-glycolide microparticle manufacturing. J. Pharm. Investig. 2019. [Google Scholar] [CrossRef]
- Wang, X.; Huang, L.; Zhang, Y.; Meng, F.; Donoso, M.; Haskell, R.; Luo, L. Tunable Two-Compartment On-Demand Sustained Drug Release Based on Lipid Gels. J. Pharm. Sci. 2020, 109, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Xie, Y.; Huang, Y.; Wang, B.; Chen, J.; Quan, G.; Pan, X.; Liu, H.; Wang, L.; Liu, X.; et al. Injectable in situ forming gel based on lyotropic liquid crystal for persistent postoperative analgesia. Acta Biomater. 2018, 67, 99–110. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, X.; Ma, P.; Tao, Y.; Wu, X.; Wu, X.; Chu, X.; Gui, S. Phytantriol-Based In Situ Liquid Crystals with Long-Term Release for Intra-articular Administration. AAPS PharmSciTech 2015, 16, 846–854. [Google Scholar] [CrossRef]
- Otte, A.; Báez-Santos, Y.M.; Mun, E.A.; Soh, B.K.; Lee, Y.-n.; Park, K. The in vivo transformation and pharmacokinetic properties of a liquid crystalline drug delivery system. Int. J. Pharm. 2017, 532, 345–351. [Google Scholar] [CrossRef]
- Li, Y.; Angelova, A.; Liu, J.; Garamus, V.M.; Li, N.; Drechsler, M.; Gong, Y.; Zou, A. In situ phase transition of microemulsions for parenteral injection yielding lyotropic liquid crystalline carriers of the antitumor drug bufalin. Colloids Surf. B Biointerfaces 2019, 173, 217–225. [Google Scholar] [CrossRef]
- Jiang, X.; Liang, X.; Wang, S.; Guo, J.; Tao, Y.; Gui, S. An injectable in situ hexagonal mesophase system for local delivery of minocycline hydrochloride: Preparation and pharmacodynamics in rats. Pharmazie 2017, 72, 249–256. [Google Scholar] [CrossRef]
- Wang, B.; Huang, Y.; Huang, Z.; Wang, H.; Chen, J.; Pan, X.; Wu, C. Self-assembling in situ gel based on lyotropic liquid crystals containing VEGF for tissue regeneration. Acta Biomater. 2019, 99, 84–99. [Google Scholar] [CrossRef]
- Rarokar, N.R.; Saoji, S.D.; Raut, N.A.; Taksande, J.B.; Khedekar, P.B.; Dave, V.S. Nanostructured Cubosomes in a Thermoresponsive Depot System: An Alternative Approach for the Controlled Delivery of Docetaxel. AAPS PharmSciTech 2016, 17, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Borgheti-Cardoso, L.N.; Depieri, L.V.; Kooijmans, S.A.A.; Diniz, H.; Calzzani, R.A.J.; De Carvalho Vicentini, F.T.M.; Van Der Meel, R.; De Abreu Fantini, M.C.; Iyomasa, M.M.; Schiffelers, R.M.; et al. An in situ gelling liquid crystalline system based on monoglycerides and polyethylenimine for local delivery of siRNAs. Eur. J. Pharm. Sci. 2015, 74, 103–117. [Google Scholar] [CrossRef]
- Frost, M.; Bailey, G.L.; Lintzeris, N.; Strang, J.; Dunlop, A.; Nunes, E.V.; Jansen, J.B.; Frey, L.C.; Weber, B.; Haber, P.; et al. Long-term safety of a weekly and monthly subcutaneous buprenorphine depot (CAM2038) in the treatment of adult outpatients with opioid use disorder. Addiction 2019, 114, 1416–1426. [Google Scholar] [CrossRef]
- Lofwall, M.R.; Walsh, S.L.; Nunes, E.V.; Bailey, G.L.; Sigmon, S.C.; Kampman, K.M.; Frost, M.; Tiberg, F.; Linden, M.; Sheldon, B.; et al. Weekly and Monthly Subcutaneous Buprenorphine Depot Formulations vs Daily Sublingual Buprenorphine With Naloxone for Treatment of Opioid Use Disorder: A Randomized Clinical Trial. JAMA Intern. Med. 2018, 178, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Tiberg, F.; Roberts, J.; Cervin, C.; Johnsson, M.; Sarp, S.; Tripathi, A.P.; Linden, M. Octreotide s.c. depot provides sustained octreotide bioavailability and similar IGF-1 suppression to octreotide LAR in healthy volunteers. Br. J. Clin. Pharmacol. 2015, 80, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Rachmawati, H.; Arvin, Y.A.; Asyarie, S.; Anggadiredja, K.; Tjandrawinata, R.R.; Storm, G. Local sustained delivery of bupivacaine HCl from a new castor oil-based nanoemulsion system. Drug Deliv. Transl. Res. 2018, 8, 515–524. [Google Scholar] [CrossRef]
- Li, Z.; Cao, J.; Li, H.; Liu, H.; Han, F.; Liu, Z.; Tong, C.; Li, S. Self-assembled drug delivery system based on low-molecular-weight bis-amide organogelator: Synthesis, properties and in vivo evaluation. Drug Deliv. 2016, 23, 3168–3178. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Yan, H.; Sun, Y.; Chen, X.; Sun, Y.; Li, S.; Jing, Y.; Li, H. Organogels based on amino acid derivatives and their optimization for drug release using response surface methodology. Artif. Cells Nanomed. Biotechnol. 2020, 48, 266–275. [Google Scholar] [CrossRef]
- Breitsamer, M.; Winter, G. Vesicular phospholipid gels as drug delivery systems for small molecular weight drugs, peptides and proteins: State of the art review. Int. J. Pharm. 2019, 557, 1–8. [Google Scholar] [CrossRef]
- Breitsamer, M.; Winter, G. Needle-Free Injection of Vesicular Phospholipid Gels—A Novel Approach to Overcome an Administration Hurdle for Semisolid Depot Systems. J. Pharm. Sci. 2017, 106, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.X.; Chen, H. One-Phase Gel Composition Comprising Phospholipids. WO 2011/075623 A1, 23 June 2011. [Google Scholar]
- Singhvi, G.; Banerjee, S.; Khosa, A. Lyotropic liquid crystal nanoparticles. In Organic Materials as Smart Nanocarriers for Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2018; pp. 471–517. ISBN 9780128136638. [Google Scholar]
- Otte, A.; Soh, B.K.; Yoon, G.; Park, K. Liquid crystalline drug delivery vehicles for oral and IV/subcutaneous administration of poorly soluble (and soluble) drugs. Int. J. Pharm. 2018, 539, 175–183. [Google Scholar] [CrossRef]
- Madheswaran, T.; Kandasamy, M.; Bose, R.J.C.; Karuppagounder, V. Current potential and challenges in the advances of liquid crystalline nanoparticles as drug delivery systems. Drug Discov. Today 2019, 24, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Bisset, N.B.; Boyd, B.J.; Dong, Y. Da Tailoring liquid crystalline lipid nanomaterials for controlled release of macromolecules. Int. J. Pharm. 2015, 495, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Lim, S.; Shim, J.; Song, J.E.; Chang, J.S.; Jin, K.S.; Cho, E.C. A simple evaporation method for large-scale production of liquid crystalline lipid nanoparticles with various internal structures. ACS Appl. Mater. Interfaces 2015, 7, 20438–20446. [Google Scholar] [CrossRef]
- Tiberg, F.; Harwigsson, I.; Johnsson, M. High Bioavailability Opioid Formulations. U.S. Patent 2013/0190341 A1, 25 July 2013. [Google Scholar]
- Camurus Technologies | Camurus. Available online: https://www.camurus.com/technologies/#injectiondepot (accessed on 20 June 2017).
- Esposito, C.L.; Kirilov, P.; Roullin, V.G. Organogels, promising drug delivery systems: An update of state-of-the-art and recent applications. J. Control. Release 2018, 271, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Gordillo-Galeano, A.; Mora-Huertas, C.E. Solid lipid nanoparticles and nanostructured lipid carriers: A review emphasizing on particle structure and drug release. Eur. J. Pharm. Biopharm. 2018, 133, 285–308. [Google Scholar] [CrossRef] [PubMed]
- Rosiaux, Y.; Jannin, V.; Hughes, S.; Marchaud, D. Solid lipid excipients-matrix agents for sustained drug delivery. J. Control. Release 2014, 188, 18–30. [Google Scholar] [CrossRef]
- Santamaria, C.M.; Woodruff, A.; Yang, R.; Kohane, D.S. Drug delivery systems for prolonged duration local anesthesia. Mater. Today 2017, 20, 22–31. [Google Scholar] [CrossRef]
- Makoni, P.A.; Kasongo, K.W.; Walker, R.B. Short term stability testing of efavirenz-loaded solid lipid nanoparticle (SLN) and nanostructured lipid carrier (NLC) dispersions. Pharmaceutics 2019, 11, 397. [Google Scholar] [CrossRef]
- Vollrath, M.; Engert, J.; Winter, G. Long-term release and stability of pharmaceutical proteins delivered from solid lipid implants. Eur. J. Pharm. Biopharm. 2017, 117, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tang, D.; Yang, T.; Wang, C. Facile preparation of biocompatible nanostructured lipid carrier with ultra-small size as a tumor-penetration delivery system. Colloids Surf. B Biointerfaces 2018, 170, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Pooladanda, V.; Bulbake, U.; Doppalapudi, S.; Rafeeqi, T.A.; Godugu, C.; Khan, W. Liposphere mediated topical delivery of thymoquinone in the treatment of psoriasis. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2251–2262. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Baldursdottir, S.; Yang, M.; Mu, H. Lipid and PLGA hybrid microparticles as carriers for protein delivery. J. Drug Deliv. Sci. Technol. 2018, 43, 65–72. [Google Scholar] [CrossRef]
- Janich, C.; Friedmann, A.; Martins de Souza e Silva, J.; Santos de Oliveira, C.; Souza, L.E.d.; Rujescu, D.; Hildebrandt, C.; Beck-Broichsitter, M.; Schmelzer, C.E.H.; Mäder, K. Risperidone-Loaded PLGA–Lipid Particles with Improved Release Kinetics: Manufacturing and Detailed Characterization by Electron Microscopy and Nano-CT. Pharmaceutics 2019, 11, 665. [Google Scholar] [CrossRef]
- Vollrath, M.; Engert, J.; Winter, G. New insights into process understanding of solid lipid extrusion (SLE) of extruded lipid implants for sustained protein delivery. Eur. J. Pharm. Biopharm. 2018, 130, 11–21. [Google Scholar] [CrossRef]
- Duque, L.; Körber, M.; Bodmeier, R. Impact of change of matrix crystallinity and polymorphism on ovalbumin release from lipid-based implants. Eur. J. Pharm. Sci. 2018, 117, 128–137. [Google Scholar] [CrossRef]
- Even, M.P.; Bobbala, S.; Gibson, B.; Hook, S.; Winter, G.; Engert, J. Twin-screw extruded lipid implants containing TRP2 peptide for tumour therapy. Eur. J. Pharm. Biopharm. 2017, 114, 79–87. [Google Scholar] [CrossRef]
- Jensen, S.S.; Jensen, H.; Møller, E.H.; Cornett, C.; Siepmann, F.; Siepmann, J.; Østergaard, J. In vitro release studies of insulin from lipid implants in solution and in a hydrogel matrix mimicking the subcutis. Eur. J. Pharm. Sci. 2016, 81, 103–112. [Google Scholar] [CrossRef]
- Even, M.P.; Bobbala, S.; Kooi, K.L.; Hook, S.; Winter, G.; Engert, J. Impact of implant composition of twin-screw extruded lipid implants on the release behavior. Int. J. Pharm. 2015, 493, 102–110. [Google Scholar] [CrossRef]
- Duong, V.A.; Nguyen, T.T.L.; Maeng, H.J.; Chi, S.C. Preparation of Ondansetron Hydrochloride-Loaded Nanostructured Lipid Carriers Using Solvent Injection Method for Enhancement of Pharmacokinetic Properties. Pharm. Res. 2019, 36, 138. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yang, F.; Xin, J.; Bai, X. An efficient and long-acting local anesthetic: Ropivacaine-loaded lipid-polymer hybrid nanoparticles for the control of pain. Int. J. Nanomed. 2019, 14, 913–920. [Google Scholar] [CrossRef]
- Wang, B.; Friess, W. Lipid-coated mannitol core microparticles for sustained release of protein. Eur. J. Pharm. Biopharm. 2018, 128, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Van de Weert, M.; Baldursdottir, S.G.; Yang, M.; Mu, H. Effect of excipients on encapsulation and release of insulin from spray-dried solid lipid microparticles. Int. J. Pharm. 2018, 550, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Li, T.; Xing, H.; Wang, S.; Sun, Y.; Sheng, X.; Wang, K. Local anesthetic effects of bupivacaine loaded lipid-polymer hybrid nanoparticles: In vitro and in vivo evaluation. Biomed. Pharmacother. 2017, 89, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Abo Elela, M.M.; ElKasabgy, N.A.; Basalious, E.B. Bio-shielding In Situ Forming Gels (BSIFG) Loaded with Lipospheres for Depot Injection of Quetiapine Fumarate: In Vitro and In Vivo Evaluation. AAPS PharmSciTech 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Pineda-Hernández, M.T.; Pérez-Urizar, J.T.; Ganem-Rondero, A. Thermo-reversible in situ forming implant with nanostructured lipid carriers (NLC) as a delivery system for the administration of estradiol valerate. Drug Deliv. Transl. Res. 2020, 1–10. [Google Scholar] [CrossRef]
- Estey, T.; Kang, J.; Schwendeman, S.P.; Carpenter, J.F. BSA degradation under acidic conditions: A model for protein instability during release from PLGA delivery systems. J. Pharm. Sci. 2006, 95, 1626–1639. [Google Scholar] [CrossRef]
- Duque, L.; Körber, M.; Bodmeier, R. Improving release completeness from PLGA-based implants for the acid-labile model protein ovalbumin. Int. J. Pharm. 2018, 538, 139–146. [Google Scholar] [CrossRef]
- Kreye, F.; Siepmann, F.; Siepmann, J. Drug release mechanisms of compressed lipid implants. Int. J. Pharm. 2011, 404, 27–35. [Google Scholar] [CrossRef]
- Kreye, F.; Siepmann, F.; Willart, J.F.; Descamps, M.; Siepmann, J. Drug release mechanisms of cast lipid implants. Eur. J. Pharm. Biopharm. 2011, 78, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Kreye, F.; Siepmann, F.; Zimmer, A.; Willart, J.F.; Descamps, M.; Siepmann, J. Cast lipid implants for controlled drug delivery: Importance of the tempering conditions. J. Pharm. Sci. 2011, 100, 3471–3481. [Google Scholar] [CrossRef] [PubMed]
- Manna, S.; Wu, Y.; Wang, Y.; Koo, B.; Chen, L.; Petrochenko, P.; Dong, Y.; Choi, S.; Kozak, D.; Oktem, B.; et al. Probing the mechanism of bupivacaine drug release from multivesicular liposomes. J. Control. Release 2019, 294, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.H.; Kapoor, Y.; Alleyne, C.; Walsh, E.; Leithead, A.; Habulihaz, B.; Salituro, G.M.; Bak, A.; Rhodes, T. Development of a Convenient In Vitro Gel Diffusion Model for Predicting the In Vivo Performance of Subcutaneous Parenteral Formulations of Large and Small Molecules. AAPS PharmSciTech 2017, 18, 2203–2213. [Google Scholar] [CrossRef]
- Ye, F.; Larsen, S.W.; Yaghmur, A.; Jensen, H.; Larsen, C.; Østergaard, J. Real-time UV imaging of piroxicam diffusion and distribution from oil solutions into gels mimicking the subcutaneous matrix. Eur. J. Pharm. Sci. 2012, 46, 72–78. [Google Scholar] [CrossRef]
- Moronkeji, K.; Todd, S.; Dawidowska, I.; Barrett, S.D.; Akhtar, R. The role of subcutaneous tissue stiffness on microneedle performance in a representative in vitro model of skin. J. Control. Release 2017, 265, 102–112. [Google Scholar] [CrossRef]
- Sun, Y.; Jensen, H.; Petersen, N.J.; Larsen, S.W.; Østergaard, J. Concomitant monitoring of implant formation and drug release of in situ forming poly (lactide-co-glycolide acid) implants in a hydrogel matrix mimicking the subcutis using UV–vis imaging. J. Pharm. Biomed. Anal. 2018, 150, 95–106. [Google Scholar] [CrossRef]
- Zou, L.; Stenslik, M.J.; Giles, M.B.; Ormes, J.D.; Marsales, M.; Santos, C.; Kassim, B.; Smith, J.P.; Gonzalez, J.J.; Bu, X. Direct visualization of drug release in injectable implant by laser induced breakdown spectroscopy (LIBS). J. Anal. At. Spectrom. 2019, 1351–1354. [Google Scholar] [CrossRef]
- Kinnunen, H.M.; Sharma, V.; Contreras-Rojas, L.R.; Yu, Y.; Alleman, C.; Sreedhara, A.; Fischer, S.; Khawli, L.; Yohe, S.T.; Bumbaca, D.; et al. A novel in vitro method to model the fate of subcutaneously administered biopharmaceuticals and associated formulation components. J. Control. Release 2015, 214, 94–102. [Google Scholar] [CrossRef]
- Application Note 501/15: A Novel In Vitro Model for Subcutaneous Injection, Pion Scissor. Available online: https://pion-inc.com/resource/226/download (accessed on 17 June 2020).
- HypoSkin® Human Skin Model for Subcutaneous Injections. Available online: https://www.genoskin.com/en/tissue-samples/skin-model-subcutaneous-injections/ (accessed on 9 March 2020).
- Russell, W.; Burch, R. The Principles of Humane Experimental Technique; Methuen: London, UK, 1959; ISBN 9780900767784. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Subgroup Examples |
|---|---|
| Fatty acyls | Fatty esters, fatty amides, fatty acyl glycosides |
| Glycerolipids | Mono-, di-, triradylglycerols |
| Glycerophospholipids | Glycerophosphocholines, -serines, -glycerols, -inositols |
| Sphingolipids | Sphingoid bases, ceramides, phosphosphingolipids |
| Sterol Lipids | Sterols, steroids, secosteroids, bile acids, and derivatives |
| Prenol Lipids | Isoprenoids, quinones and hydroquinones, polyprenols |
| Saccharolipids | Acylaminosugars, acylaminosugar glycans, acyltrehaloses |
| Polyketides | Flavonoids, macrolides, linear or aromatic polyketides |
| Principle | Encapsulated Drug | Release Time Range | Advantages (+) and Disadvantages (–) | Ref. |
|---|---|---|---|---|
| Classical oil-based solutions with dissolved drug | ||||
| Oily solution | Paliperidone palmitate, medroxy-progesterone, haloperidol decanoate, testosterone (in general antipsychotics and hormones | Up to several months, in vivo |
| [33,34] |
| Drug combined with second carrier in oil-based solutions | ||||
| Microparticles in oily solution | Ondansetron | Hours, in vitro and in vivo |
| [40] |
| Proliposomal oil | Ropivacaine | Days, in vivo | [41,42] | |
| Solid dispersion in oil | Ivermectin, toltrazuril (veterinary) | Days, in vivo | [38,43,44] | |
| Nanoprecipitates in oil | Tenofovir disoproxil fumarate | Hours, in vitro | [45] | |
| Lyophilized liposomes dispersed in oil (DPX/DepoVax™) | Vaccines, antigens | Weeks, in vivo | [46,47] | |
| Principle (Used Phospholipids *) | Encapsulated Drug/Marker | Release Time Range | Advantages (+) and Disadvantages (–) | Ref. |
|---|---|---|---|---|
| Conventional liposomes | ||||
| Aggregation of negatively charged liposomes (DPPC, EPC, HPC, DSPG, DPPG, DOPA, DPPA) | Flurbiprofen, Calcein, FITC-dextran-4000 | Days, in vitro and in vivo |
| [55,59] |
| PEGylated liposomes (DPPC, DSPE-PEG2000) | Adrenaline | Hours, in vitro | [60] | |
| Combination of fast and slow release liposomes (DSPC, DLPC, DMPC, DSPC) | Vaccine | Not specified | [61] | |
| Small unilamellar liposomes (DSPC) | D-glucosamine sulphate, lubrication effect of phospholipids | Weeks, in vitro, days in vivo | [62] | |
| Conventional and stealth liposomes (DPPG, DPPE-PEG2000) | Bevacizumab | Months in vitro, weeks in vivo | [63] | |
| Multivesicular liposomes | ||||
| MVLs, DepoFoam® (EPC, SPC, HSPC, DEPC, DOPC, DPPG) | Bupivacaine (Exparel®), ropivacaine, oleanolic acid, fluocinolone acetonide in cyclodextrin liraglutide, bevacizumab | Days, in vitro and in vivo |
| [58,64,65,66,67,68,69,70,71,72] |
| Liposomes in gel, coated liposomes | ||||
| Liposomes in Pluronic® F127 hydrogel (HPC, DPPG, DSPE-PEG2000) | Insulin, flurbiprofen, doxorubicin, paclitaxel | Days to weeks, in vivo |
| [73,74,75] |
| Liposomes in PLGA-PEG-PLGA hydrogel (SPC) | Isoniazid, doxorubicin | Days, in vitro and in vivo | [76,77] | |
| Liposomes in chitosan hydrogel (SPC, DPPC) | Doxorubicin, Carboxy-fluorescein | Days, in vitro and in vivo or not specified | [57,78] | |
| Hybrid liposomes with chitosan (S75) | Curcumin | Days, in vitro | [79] | |
| Hyaluronic acid-coated liposomes (DPPC, DOTAP) | Paclitaxel | Hours, in vitro | [80] | |
| Ternary drug-cyclodextrin-liposomes complex (EPC) | Ropivacaine | Hours in vitro and in vivo | [81] | |
| Capsosomes (EPC, DOTAP) | None | Not specified | [56] | |
| Principle | Encapsulated Drug | Release Time Range | Advantages (+) and Disadvantages (–) | Ref. |
|---|---|---|---|---|
| Phospholipid-based phase separation gel (PPSG) | Ropivacaine, pitavastatin, 5-fluorouracil, paclitaxel, doxorubicin, bromo-tetrandrine, dabigatran etexilate, exenatide, leuprolide, octreotide, insulin, incomplete Freund’s adjuvant | Days to weeks, in vitro and in vivo |
| [95,100,101,102,103,104,105,106,107,108,109,110] |
| Vesicular phospholipid gels | Interferon-beta-1b, granulocyte-colony stimulating factor, exenatide, FKBP51 antagonist | Days to weeks in vitro and in vivo |
| [111,112,113,114,115] |
| Phospholipid gel depo (PG Depot™) | Small molecules, proteins, peptides | Days |
| [116,117] |
| Liquid crystalline phases (cubosomes, hexosomes), FluidCrystal®, hybrid systems | Bufalin, bupivacaine, minocycline, indomethacin, finasteride, doxorubicin, buprenorphine (Buvidal®, Brixadi®), sinomenine, docetaxel, leuprolide, octreotide, setmelanotide, Huperzine A, VEGF siRNA (combined with polyethylene-imine), | Days to weeks in vitro and in vivo |
| [98,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132] |
| Nanoemulsion | Bupivacaine | Hours, in vitro and in vivo |
| [133] |
| Organogel | Candesartan cilexetil, risperidone | Days in vitro and in vivo |
| [134,135] |
| Principle | Encapsulated Drug | Release Time Range | Advantages (+) and Disadvantages (–) | Ref. |
|---|---|---|---|---|
| Implants | Ovalbumin, monoclonal antibody, ranibizumab, insulin, tumor peptide TRP2 | Weeks in vitro |
| [151,156,157,158,159,160] |
| Nanostructured lipid carriers | Ondansetron | Days in vitro and in vivo |
| [161] |
| Core-shell systems of lipid nano-/microparticles, combinations with polymers | Ropivacaine, bupivacaine, risperidone, monoclonal antibody, lysozyme, insulin | Days to weeks in vitro and in vivo |
| [154,155,162,163,164,165] |
| Lipospheres or nanostructured lipid carriers in hydrogel | Quetiapine, estradiol valerate | Hours to days in vitro and in vivo | [166,167] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahnfeld, L.; Luciani, P. Injectable Lipid-Based Depot Formulations: Where Do We Stand? Pharmaceutics 2020, 12, 567. https://doi.org/10.3390/pharmaceutics12060567
Rahnfeld L, Luciani P. Injectable Lipid-Based Depot Formulations: Where Do We Stand? Pharmaceutics. 2020; 12(6):567. https://doi.org/10.3390/pharmaceutics12060567
Chicago/Turabian StyleRahnfeld, Lisa, and Paola Luciani. 2020. "Injectable Lipid-Based Depot Formulations: Where Do We Stand?" Pharmaceutics 12, no. 6: 567. https://doi.org/10.3390/pharmaceutics12060567
APA StyleRahnfeld, L., & Luciani, P. (2020). Injectable Lipid-Based Depot Formulations: Where Do We Stand? Pharmaceutics, 12(6), 567. https://doi.org/10.3390/pharmaceutics12060567