Recent Developments in Microfluidic Technologies for Central Nervous System Targeted Studies

 , , ,
, , ,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
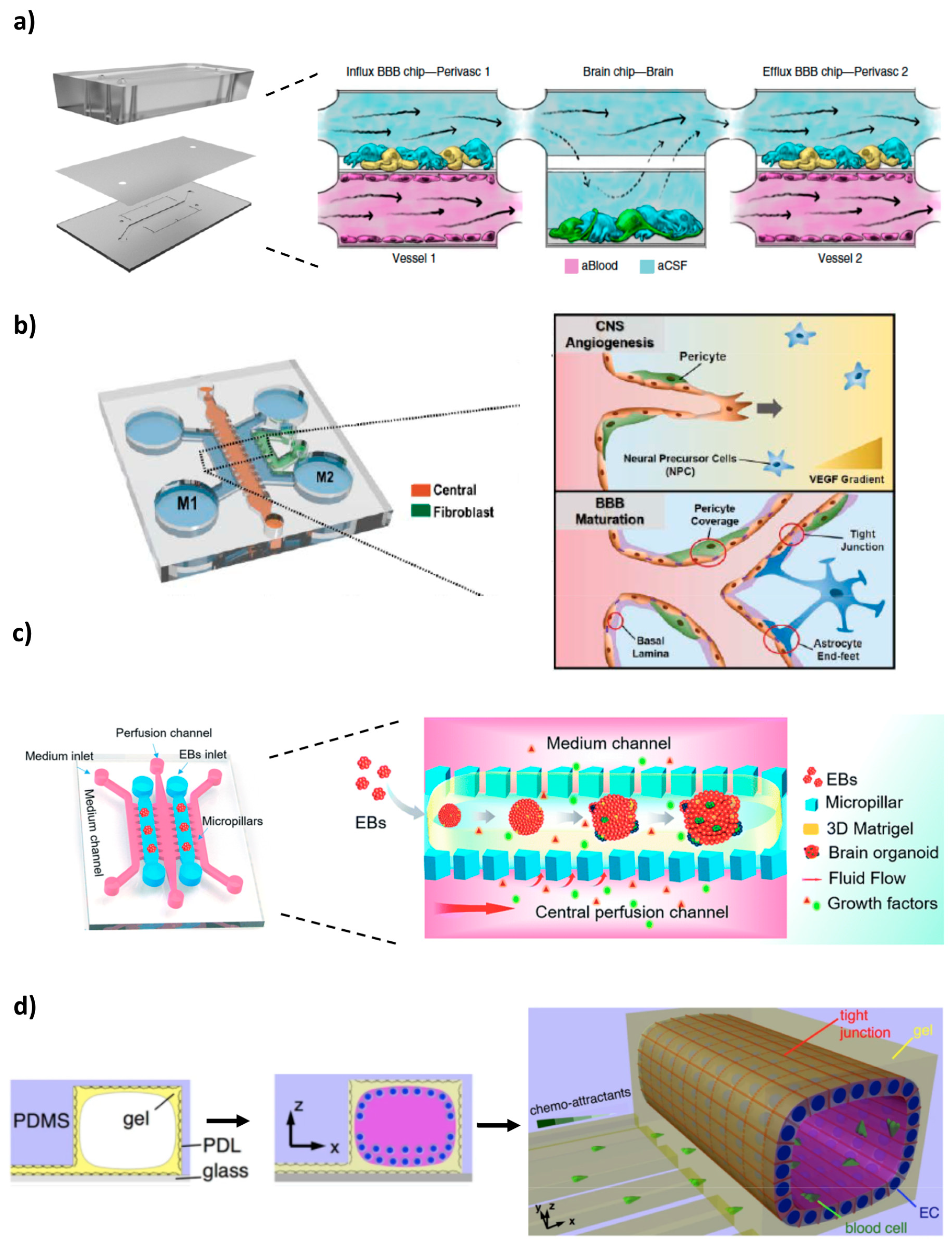{kind=link}
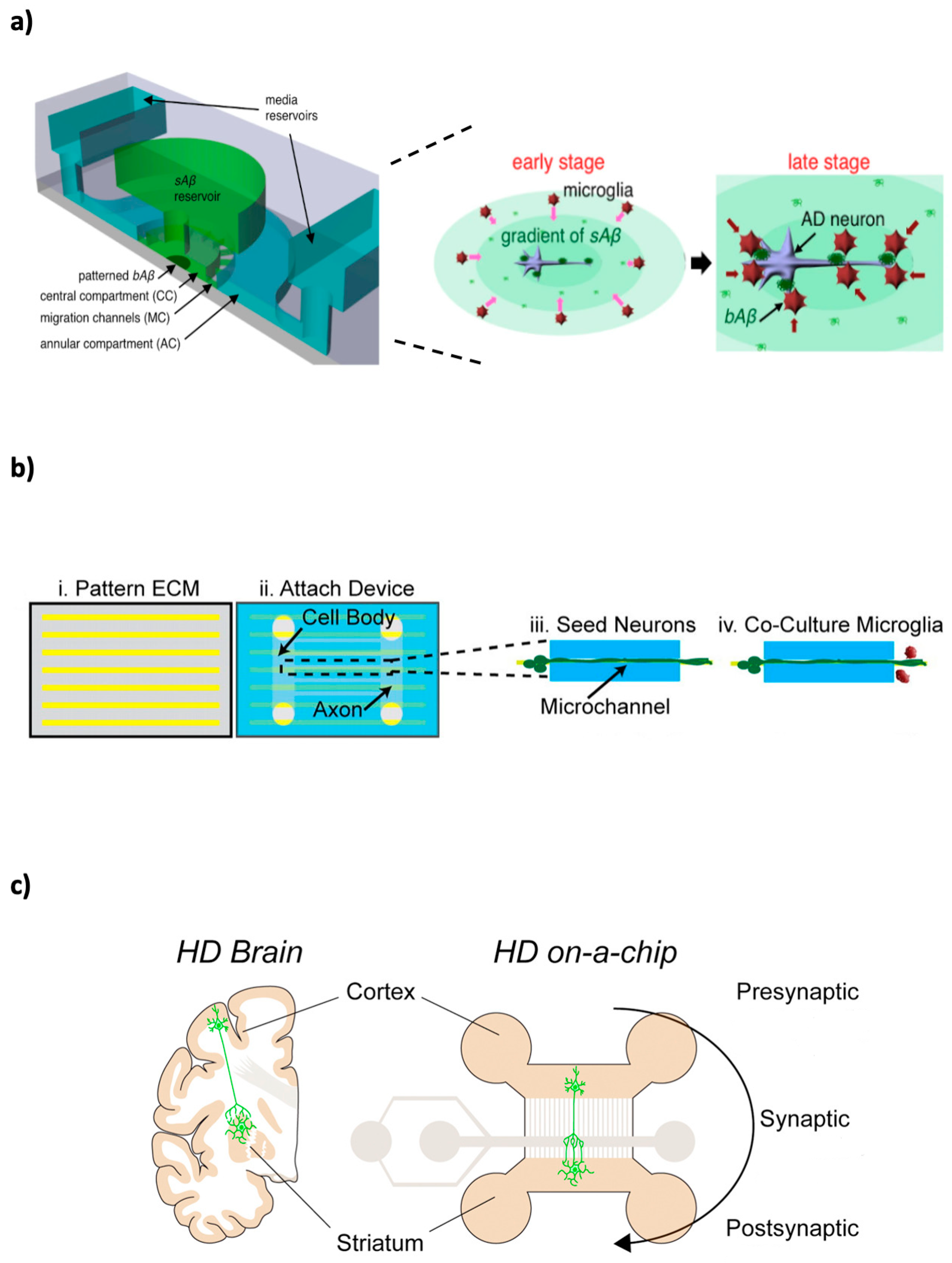{kind=link}
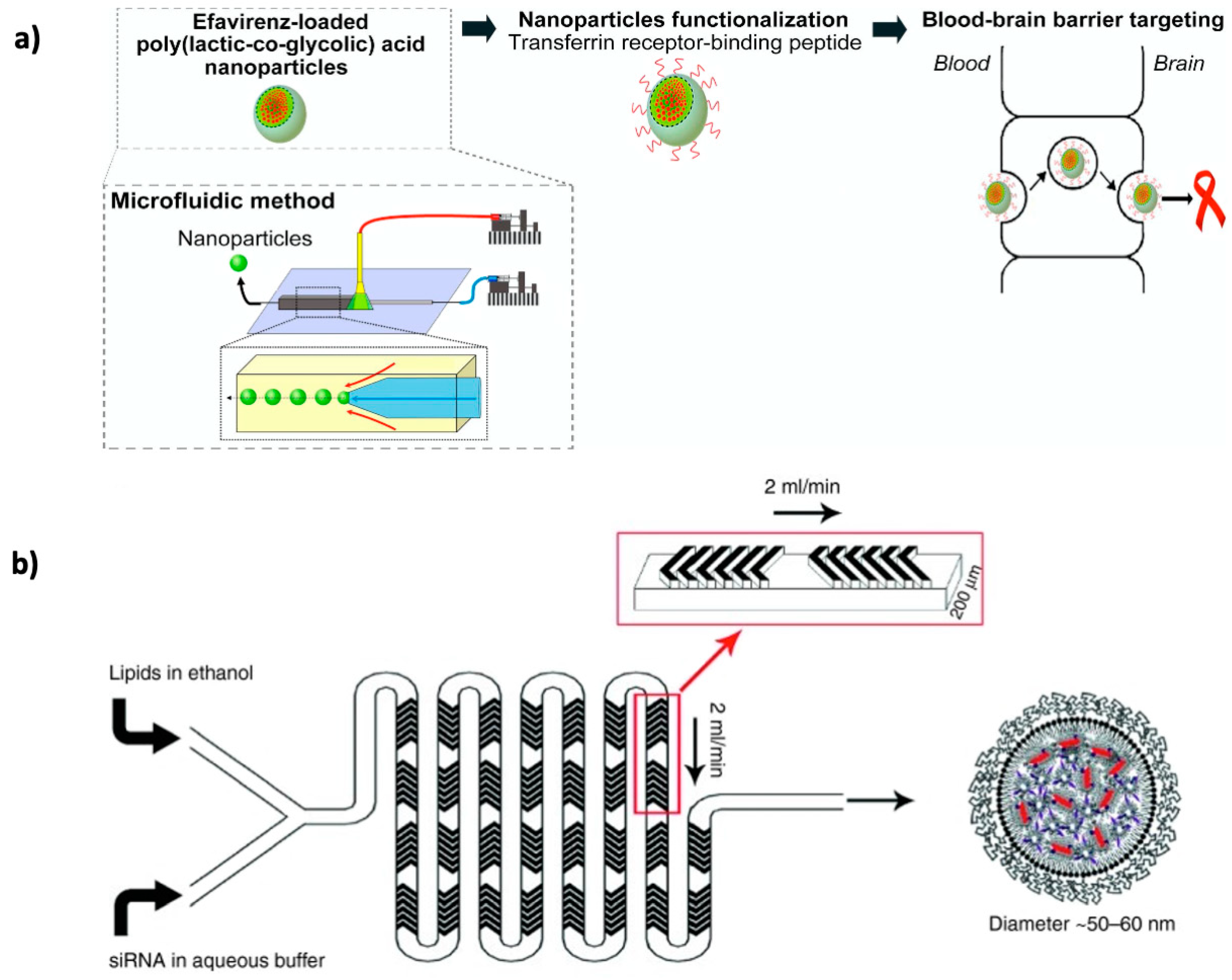{kind=link}
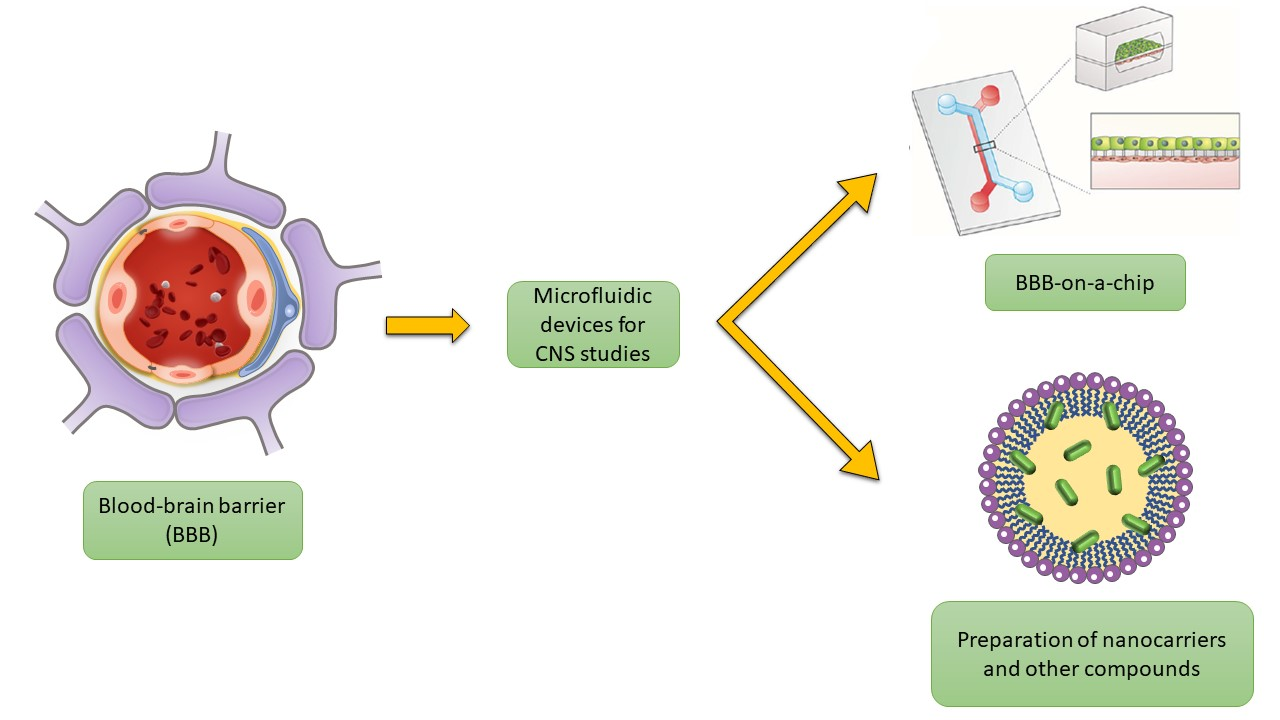
1. Introduction
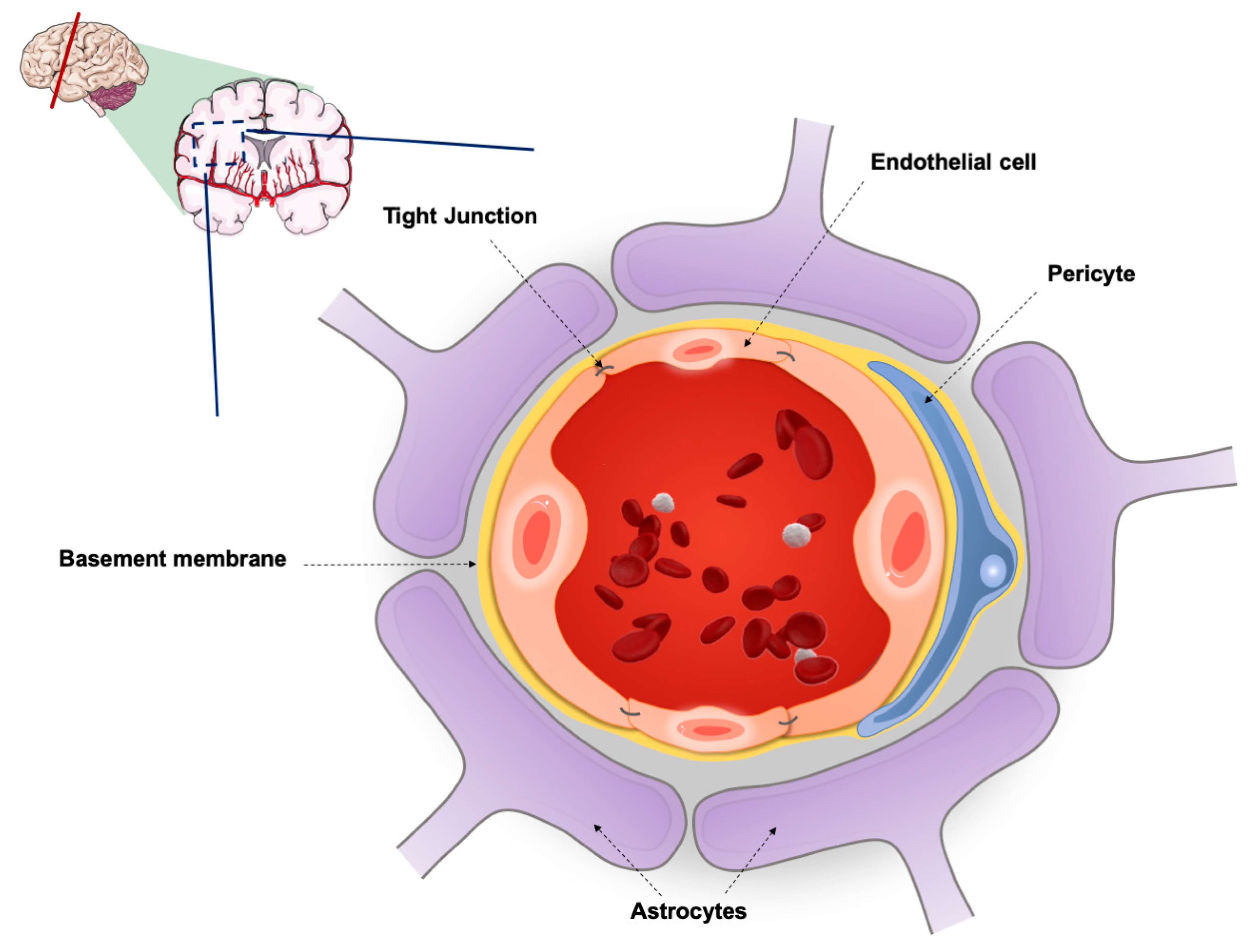
2. The Blood-Brain Barrier (BBB)
- Carrier-mediated transcytosis (binding of a molecule to a protein carrier on the apical side of the capillary wall, followed by its conformational change and transport of said molecule to the other side of the membrane).
- Receptor-mediated transcytosis (binding of a particular ligand to a specific receptor, such as transferrin or low-density lipoprotein receptors, with the formation of a ligand-receptor complex which is carried through the cytoplasm and released on the basolateral side of the BBB).
- Adsorptive transcytosis (electrostatic interaction between positively charged molecules and the negatively charged plasma membrane).
3. Experimental BBB Models
3.1. In Vivo Models
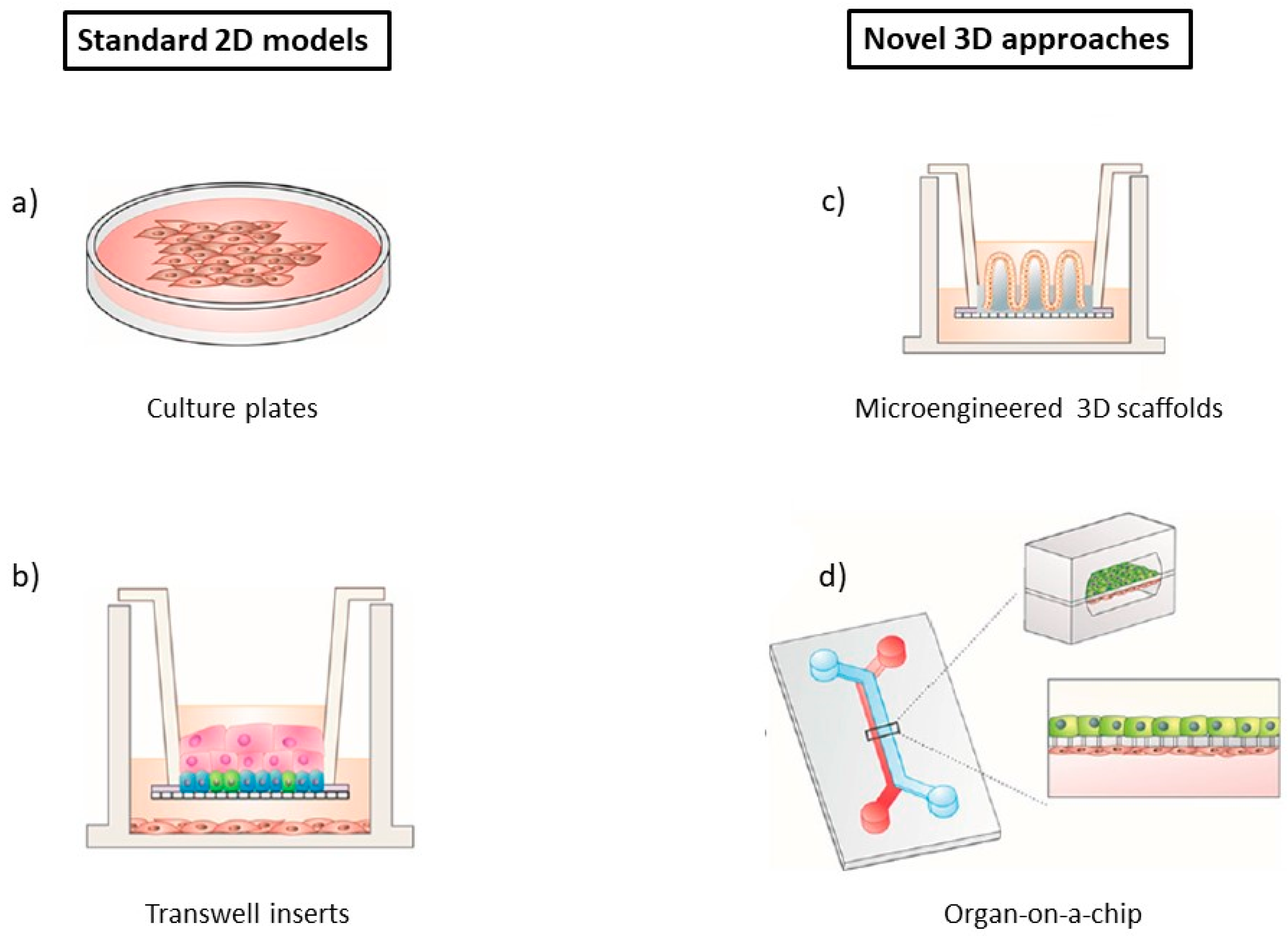
3.2. In Vitro Models
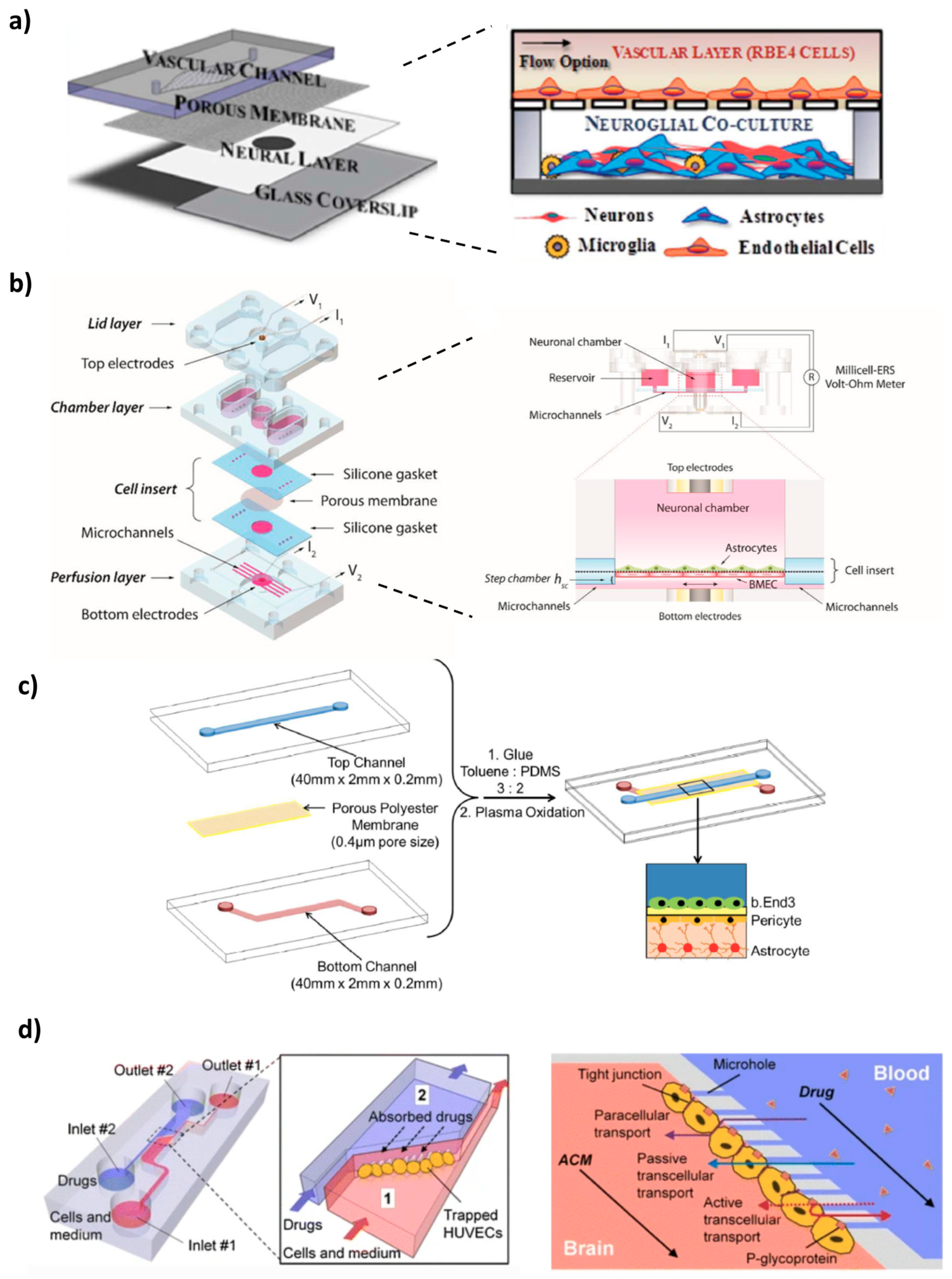
4. Microfluidic BBB In Vitro Models (BBB/Brain-on-a-Chip)
- (1)
- (2)
- (3)
- Continuous supply of nutrients and waste removal [40].
- (4)
- Schedule flexibility [40].
- (5)
- Reduced risk of contamination [47].
- (6)
- Possibility of multifunctional integration of analytical biosensors and other electronic apparatus (e.g., microscopy devices, microelectrode arrays (MEAs), etc.) for real-time/on-chip or downstream monitorization of cellular behaviour, detection of physiological parameters (e.g., biomarkers or chemokines levels) and in situ analysis of external stimuli, therefore decreasing analysis time [32,40,46,47].
- (7)
- High automation capability [47].
- (8)
- (9)
- Possibility to test in the same controlled environment both healthy and disease tissues [35].
4.1. 2D Microfluidic Systems
4.2. 3D Microfluidic Systems
5. Neurodegenerative Diseases Microfluidic Models
5.1. Alzheimer’s Disease
5.2. Parkinson’s Disease
5.3. Multiple Sclerosis
5.4. Amyotrophic Lateral Sclerosis
5.5. Huntington’s Disease
6. Microfluidic Synthesis of CNS-Targeted Nano/Microcarriers and Other Compounds
7. Challenges, Future Perspectives and Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Barnabas, W. Drug targeting strategies into the brain for treating neurological diseases. J Neurosci. Methods 2019, 311, 133–146. [Google Scholar] [CrossRef]
- Di Stefano, A.; Iannitelli, A.; Laserra, S.; Sozio, P. Drug delivery strategies for Alzheimer’s disease treatment. Expert Opin. Drug Deliv. 2011, 8, 581–603. [Google Scholar] [CrossRef]
- Squillaro, T.; Cimini, A.; Peluso, G.; Giordano, A.; Melone, M.A.B. Nano-delivery systems for encapsulation of dietary polyphenols: An experimental approach for neurodegenerative diseases and brain tumors. Biochem. Pharm. 2018, 154, 303–317. [Google Scholar] [CrossRef]
- Yacoubian, T.A. Chapter 1-neurodegenerative disorders: Why do we need new therapies. In Drug Discovery Approaches for the Treatment of Neurodegenerative Disorders; Adejare, A., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 1–16. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Qureshi, M.S.; Manan, A.; Pushparaj, P.N.; Asif, M.; Qazi, M.H.; Qazi, A.M.; Kamal, M.A.; Gan, S.H.; et al. Recent updates in the treatment of neurodegenerative disorders using natural compounds. Evid. Based Complement. Altern. Med. 2014, 2014, 7. [Google Scholar] [CrossRef]
- Choi, J.H.; Santhosh, M.; Choi, J.W. In vitro blood-brain barrier-integrated neurological disorder models using a microfluidic device. Micromachines 2019, 11, 21. [Google Scholar] [CrossRef]
- Oddo, A.; Peng, B.; Tong, Z.; Wei, Y.; Tong, W.Y.; Thissen, H.; Voelcker, N.H. Advances in microfluidic blood-brain barrier (BBB) models. Trends Biotechnol. 2019, 37, 1295–1314. [Google Scholar] [CrossRef]
- Martins, J.P.; Torrieri, G.; Santos, H.A. The importance of microfluidics for the preparation of nanoparticles as advanced drug delivery systems. Expert Opin. Drug Deliv. 2018, 15, 469–479. [Google Scholar] [CrossRef]
- Chiu, D.T.; deMello, A.J.; Di Carlo, D.; Doyle, P.S.; Hansen, C.; Maceiczyk, R.M.; Wootton, R.C.R. Small but perfectly formed? Successes, challenges, and opportunities for microfluidics in the chemical and biological sciences. Chem 2017, 2, 201–223. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Durães, F.; Pinto, M.; Sousa, E. Old drugs as new treatments for neurodegenerative diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmady, Z.S. Selective drug delivery approaches to lesioned brain through blood brain barrier disruption. Expert Opin. Drug Deliv. 2018, 15, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Karthivashan, G.; Ganesan, P.; Park, S.-Y.; Kim, J.-S.; Choi, D.-K. Therapeutic strategies and nano-drug delivery applications in management of ageing Alzheimer’s disease. Drug Deliv. 2018, 25, 307–320. [Google Scholar] [CrossRef]
- Moura, R.P.; Martins, C.; Pinto, S.; Sousa, F.; Sarmento, B. Blood-brain barrier receptors and transporters: An insight on their function and how to exploit them through nanotechnology. Expert Opin. Drug Deliv. 2019, 16, 271–285. [Google Scholar] [CrossRef]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar] [CrossRef]
- Bohr, A.; Colombo, S.; Jensen, H. Chapter 15-Future of microfluidics in research and in the market. In Microfluidics for Pharmaceutical Applications; Santos, H.A., Liu, D., Zhang, H., Eds.; William Andrew Publishing: Norwich, NY, USA, 2019; pp. 425–465. [Google Scholar] [CrossRef]
- Martins, C.; Araujo, F.; Gomes, M.J.; Fernandes, C.; Nunes, R.; Li, W.; Santos, H.A.; Borges, F.; Sarmento, B. Using microfluidic platforms to develop CNS-targeted polymeric nanoparticles for HIV therapy. Eur. J. Pharm. Biopharm. 2018. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, H.; Fontana, F.; Hirvonen, J.T.; Santos, H.A. Current developments and applications of microfluidic technology toward clinical translation of nanomedicines. Adv. Drug Deliv. Rev. 2018, 128, 54–83. [Google Scholar] [CrossRef]
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of current strategies for delivering Alzheimer’s disease drugs across the blood-brain barrier. Int. J. Mol. Sci. 2019, 20, 381. [Google Scholar] [CrossRef]
- Tang, W.; Fan, W.; Lau, J.; Deng, L.; Shen, Z.; Chen, X. Emerging blood-brain-barrier-crossing nanotechnology for brain cancer theranostics. Chem. Soc. Rev. 2019, 48, 2967–3014. [Google Scholar] [CrossRef]
- Alexander, A.; Agrawal, M.; Uddin, A.; Siddique, S.; Shehata, A.M.; Shaker, M.A.; Ata-Ur-Rahman, S.; Abdul, M.I.M.; Shaker, M.A. Recent expansions of novel strategies towards the drug targeting into the brain. Int. J. Nanomed. 2019, 14, 5895–5909. [Google Scholar] [CrossRef]
- Pan, Y.; Nicolazzo, J.A. Impact of aging, Alzheimer’s disease and Parkinson’s disease on the blood-brain barrier transport of therapeutics. Adv. Drug Deliv. Rev. 2018, 135, 62–74. [Google Scholar] [CrossRef]
- Kooij, G.; Mizee, M.R.; van Horssen, J.; Reijerkerk, A.; Witte, M.E.; Drexhage, J.A.; van der Pol, S.M.; van Het Hof, B.; Scheffer, G.; Scheper, R.; et al. Adenosine triphosphate-binding cassette transporters mediate chemokine (C-C motif) ligand 2 secretion from reactive astrocytes: Relevance to multiple sclerosis pathogenesis. Brain 2011, 134, 555–570. [Google Scholar] [CrossRef]
- Jablonski, M.R.; Jacob, D.A.; Campos, C.; Miller, D.S.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol. Dis. 2012, 47, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Erdo, F.; Krajcsi, P. Age-related functional and expressional changes in efflux pathways at the blood-brain barrier. Front. Aging Neurosci. 2019, 11, 196. [Google Scholar] [CrossRef] [PubMed]
- Erdő, F.; Denes, L.; de Lange, E. Age-associated physiological and pathological changes at the blood-brain barrier: A review. J. Cereb. Blood Flow Metab. 2017, 37, 4–24. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease. Epilepsia 2012, 53, 1–6. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxidative Med. Cell. Longev. 2019, 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Osaki, T.; Shin, Y.; Sivathanu, V.; Campisi, M.; Kamm, R.D. In vitro microfluidic models for neurodegenerative disorders. Adv. Healthc. Mater. 2018, 7, 1700489. [Google Scholar] [CrossRef]
- Gubert, C.; Kong, G.; Renoir, T.; Hannan, A.J. Exercise, diet and stress as modulators of gut microbiota: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 134, 104621. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.; Goh, E. Chapter 9-Blood-brain barrier on a chip. In Methods in Cell Biology; Doh, J., Fletcher, D., Piel, M., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 146, pp. 159–182. [Google Scholar]
- Van der Helm, M.W.; van der Meer, A.D.; Eijkel, J.C.T.; van den Berg, A.; Segerink, L.I. Microfluidic organ-on-chip technology for blood-brain barrier research. Tissue Barriers 2016, 4, e1142493. [Google Scholar] [CrossRef]
- Ugolini, G.S.; Cruz-Moreira, D.; Visone, R.; Redaelli, A.; Rasponi, M. Microfabricated physiological models for in vitro drug screening applications. Micromachines 2016, 7, 233. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, S.; Zheng, J.; Li, Y.; Huang, H. Recent progress in microfluidic models of the blood-brain barrier. Micromachines 2019, 10, 375. [Google Scholar] [CrossRef]
- Haring, A.P.; Sontheimer, H.; Johnson, B.N. Microphysiological human brain and neural systems-on-a-chip: Potential alternatives to small animal models and emerging platforms for drug discovery and personalized medicine. Stem. Cell Rev. Rep. 2017, 13, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Park, J.; Lim, J.; Lee, C.J.; Lee, S.-H. Central nervous system and its disease models on a chip. Trends Biotechnol. 2015, 33, 762–776. [Google Scholar] [CrossRef]
- Coluccio, M.L.; Perozziello, G.; Malara, N.; Parrotta, E.; Zhang, P.; Gentile, F.; Limongi, T.; Raj, P.M.; Cuda, G.; Candeloro, P.; et al. Microfluidic platforms for cell cultures and investigations. Microelectron. Eng. 2019, 208, 14–28. [Google Scholar] [CrossRef]
- Geraili, A.; Jafari, P.; Hassani, M.S.; Araghi, B.H.; Mohammadi, M.H.; Ghafari, A.M.; Tamrin, S.H.; Modarres, H.P.; Kolahchi, A.R.; Ahadian, S.; et al. Controlling differentiation of stem cells for developing personalized organ-on-chip platforms. Adv. Healthc. Mater. 2018, 7, 1700426. [Google Scholar] [CrossRef]
- Karimi, M.; Bahrami, S.; Mirshekari, H.; Basri, S.M.; Nik, A.B.; Aref, A.R.; Akbari, M.; Hamblin, M.R. Microfluidic systems for stem cell-based neural tissue engineering. Lab Chip 2016, 16, 2551–2571. [Google Scholar] [CrossRef]
- Mendes, B.; Marques, C.; Carvalho, I.; Costa, P.; Martins, S.; Ferreira, D.; Sarmento, B. Influence of glioma cells on a new co-culture in vitro blood-brain barrier model for characterization and validation of permeability. Int. J. Pharm. 2015, 490, 94–101. [Google Scholar] [CrossRef]
- Torras, N.; García-Díaz, M.; Fernández-Majada, V.; Martínez, E. Mimicking epithelial tissues in three-dimensional cell culture models. Front. Bioeng. Biotechnol. 2018, 6, 197. [Google Scholar] [CrossRef]
- Xu, H.; Li, Z.; Yu, Y.; Sizdahkhani, S.; Ho, W.S.; Yin, F.; Wang, L.; Zhu, G.; Zhang, M.; Jiang, L.; et al. A dynamic in vivo-like organotypic blood-brain barrier model to probe metastatic brain tumors. Sci. Rep. 2016, 6, 36670. [Google Scholar] [CrossRef]
- Convery, N.; Gadegaard, N. 30 years of microfluidics. Micro Nano Eng. 2019, 2, 76–91. [Google Scholar] [CrossRef]
- Sosa-Hernández, J.E.; Villalba-Rodríguez, A.M.; Romero-Castillo, K.D.; Aguilar-Aguila-Isaías, M.A.; García-Reyes, I.E.; Hernández-Antonio, A.; Ahmed, I.; Sharma, A.; Parra-Saldívar, R.; Iqbal, H.M.N. Organs-on-a-chip module: A review from the development and applications perspective. Micromachines 2018, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Cavero, I.; Guillon, J.-M.; Holzgrefe, H.H. Human organotypic bioconstructs from organ-on-chip devices for human-predictive biological insights on drug candidates. Expert Opin. Drug Saf. 2019, 18, 651–677. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M.T. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef]
- Jeong, S.; Kim, S.; Buonocore, J.; Park, J.; Welsh, C.J.; Li, J.; Han, A. A three-dimensional arrayed microfluidic blood-brain barrier model with integrated electrical sensor array. IEEE Trans. Biomed. Eng. 2018, 65, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Griep, L.M.; Wolbers, F.; de Wagenaar, B.; ter Braak, P.M.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Vermes, I.; van der Meer, A.D.; van den Berg, A. BBB ON CHIP: Microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed. Microdevices 2013, 15, 145–150. [Google Scholar] [CrossRef]
- Achyuta, A.K.H.; Conway, A.J.; Crouse, R.B.; Bannister, E.C.; Lee, R.N.; Katnik, C.P.; Behensky, A.A.; Cuevas, J.; Sundaram, S.S. A modular approach to create a neurovascular unit-on-a-chip. Lab Chip 2013, 13, 542–553. [Google Scholar] [CrossRef]
- Wang, Y.I.; Abaci, H.E.; Shuler, M.L. Microfluidic blood-brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol. Bioeng. 2017, 114, 184–194. [Google Scholar] [CrossRef]
- Walter, F.R.; Valkai, S.; Kincses, A.; Petneházi, A.; Czeller, T.; Veszelka, S.; Ormos, P.; Deli, M.A.; Dér, A. A versatile lab-on-a-chip tool for modeling biological barriers. Sens. Actuators B Chem. 2016, 222, 1209–1219. [Google Scholar] [CrossRef]
- Wang, J.D.; Khafagy, E.-S.; Khanafer, K.; Takayama, S.; ElSayed, M.E.H. Organization of endothelial cells, pericytes, and astrocytes into a 3D microfluidic in vitro model of the blood-brain barrier. Mol. Pharm. 2016, 13, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Yeon, J.H.; Na, D.; Choi, K.; Ryu, S.-W.; Choi, C.; Park, J.-K. Reliable permeability assay system in a microfluidic device mimicking cerebral vasculatures. Biomed. Microdevices 2012, 14, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Papademetriou, I.; Vedula, E.; Charest, J.; Porter, T. Effect of flow on targeting and penetration of angiopep-decorated nanoparticles in a microfluidic model blood-brain barrier. PLoS ONE 2018, 13, e0205158. [Google Scholar] [CrossRef] [PubMed]
- Briuglia, M.-L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef]
- Park, T.-E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-enhanced blood-brain barrier chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef]
- Falanga, A.P.; Pitingolo, G.; Celentano, M.; Cosentino, A.; Melone, P.; Vecchione, R.; Guarnieri, D.; Netti, P.A. Shuttle-mediated nanoparticle transport across an in vitro brain endothelium under flow conditions. Biotechnol. Bioeng. 2017, 114, 1087–1095. [Google Scholar] [CrossRef]
- Prabhakarpandian, B.; Shen, M.-C.; Nichols, J.B.; Mills, I.R.; Sidoryk-Wegrzynowicz, M.; Aschner, M.; Pant, K. SyM-BBB: A microfluidic blood brain barrier model. Lab Chip 2013, 13, 1093–1101. [Google Scholar] [CrossRef]
- Marino, A.; Tricinci, O.; Battaglini, M.; Filippeschi, C.; Mattoli, V.; Sinibaldi, E.; Ciofani, G. A 3D real-scale, biomimetic, and biohybrid model of the blood-brain barrier fabricated through two-photon lithography. Small 2018, 14, 1702959. [Google Scholar] [CrossRef]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood-brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef]
- Maoz, B.M.; Herland, A.; FitzGerald, E.A.; Grevesse, T.; Vidoudez, C.; Pacheco, A.R.; Sheehy, S.P.; Park, T.-E.; Dauth, S.; Mannix, R.; et al. A linked organ-on-chip model of the human neurovascular unit reveals the metabolic coupling of endothelial and neuronal cells. Nat. Biotechnol. 2018, 36, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Lee, S.-R.; Ko, J.; Son, K.; Tahk, D.; Ahn, J.; Im, C.; Jeon, N.L. A low permeability microfluidic blood-brain barrier platform with direct contact between perfusable vascular network and astrocytes. Sci. Rep. 2017, 7, 8083. [Google Scholar] [CrossRef] [PubMed]
- Partyka, P.P.; Godsey, G.A.; Galie, J.R.; Kosciuk, M.C.; Acharya, N.K.; Nagele, R.G.; Galie, P.A. Mechanical stress regulates transport in a compliant 3D model of the blood-brain barrier. Biomaterials 2017, 115, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Deosarkar, S.P.; Prabhakarpandian, B.; Wang, B.; Sheffield, J.B.; Krynska, B.; Kiani, M.F. A novel dynamic neonatal blood-brain barrier on a chip. PLoS ONE 2015, 10, e0142725. [Google Scholar] [CrossRef]
- Adriani, G.; Ma, D.; Pavesi, A.; Kamm, R.D.; Goh, E.L.K. A 3D neurovascular microfluidic model consisting of neurons, astrocytes and cerebral endothelial cells as a blood-brain barrier. Lab Chip 2017, 17, 448–459. [Google Scholar] [CrossRef]
- Campisi, M.; Shin, Y.; Osaki, T.; Hajal, C.; Chiono, V.; Kamm, R.D. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials 2018, 180, 117–129. [Google Scholar] [CrossRef]
- Jamieson, J.J.; Linville, R.M.; Ding, Y.Y.; Gerecht, S.; Searson, P.C. Role of iPSC-derived pericytes on barrier function of iPSC-derived brain microvascular endothelial cells in 2D and 3D. Fluids Barriers CNS 2019, 16, 15. [Google Scholar] [CrossRef]
- Linville, R.M.; DeStefano, J.G.; Sklar, M.B.; Xu, Z.; Farrell, A.M.; Bogorad, M.I.; Chu, C.; Walczak, P.; Cheng, L.; Mahairaki, V.; et al. Human iPSC-derived blood-brain barrier microvessels: Validation of barrier function and endothelial cell behavior. Biomaterials 2019, 190, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Motallebnejad, P.; Thomas, A.; Swisher, S.L.; Azarin, S.M. An isogenic hiPSC-derived BBB-on-a-chip. Biomicrofluidics 2019, 13. [Google Scholar] [CrossRef]
- Canfield, S.G.; Stebbins, M.J.; Faubion, M.G.; Gastfriend, B.D.; Palecek, S.P.; Shusta, E.V. An isogenic neurovascular unit model comprised of human induced pluripotent stem cell-derived brain microvascular endothelial cells, pericytes, astrocytes, and neurons. Fluids Barriers CNS 2019, 16, 25. [Google Scholar] [CrossRef]
- Stebbins, M.J.; Gastfriend, B.D.; Canfield, S.G.; Lee, M.-S.; Richards, D.; Faubion, M.G.; Li, W.-J.; Daneman, R.; Palecek, S.P.; Shusta, E.V. Human pluripotent stem cell-derived brain pericyte-like cells induce blood-brain barrier properties. Sci. Adv. 2019, 5, eaau7375. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chung, M.; Lee, S.-R.; Jeon, N.L. 3D brain angiogenesis model to reconstitute functional human blood-brain barrier in vitro. Biotechnol. Bioeng. 2020, 117, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Guo, Y.; Zhu, Y.; Qin, J. Engineering stem cell-derived 3D brain organoids in a perfusable organ-on-a-chip system. RSC Adv. 2018, 8, 1677–1685. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Zhu, Y.; Qin, J. Human brain organoid-on-a-chip to model prenatal nicotine exposure. Lab Chip 2018, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Seo, J.H.; Wong, K.H.K.; Terasaki, Y.; Park, J.; Bong, K.; Arai, K.; Lo, E.H.; Irimia, D. Three-dimensional blood-brain barrier model for in vitro studies of neurovascular pathology. Sci. Rep. 2015, 5, 15222. [Google Scholar] [CrossRef] [PubMed]
- Parimalam, S.S.; Badilescu, S.; Sonenberg, N.; Bhat, R.; Packirisamy, M. Lab-on-a-chip for the development of pro-/anti-angiogenic nanomedicines to treat brain diseases. Int. J. Mol. Sci. 2019, 20, 6126. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Zverova, M. Clinical aspects of Alzheimer’s disease. Clin. Biochem. 2019, 72, 3–6. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T.; Vogel, M. A vaccine against Alzheimer’s disease: Anything left but faith? Expert. Opin. Biol. 2019, 19, 73–78. [Google Scholar] [CrossRef]
- Davtyan, H.; Hovakimyan, A.; Kiani Shabestari, S.; Antonyan, T.; Coburn, M.A.; Zagorski, K.; Chailyan, G.; Petrushina, I.; Svystun, O.; Danhash, E.; et al. Testing a MultiTEP-based combination vaccine to reduce abeta and tau pathology in Tau22/5xFAD bigenic mice. Alzheimers Res. 2019, 11, 107. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Alzheimer’s disease: Key developments support promising perspectives for therapy. Pharm. Res. 2019, 146, 104316. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, B.K.; Jeong, G.S.; Hyun, J.K.; Lee, C.J.; Lee, S.H. Three-dimensional brain-on-a-chip with an interstitial level of flow and its application as an in vitro model of Alzheimer’s disease. Lab Chip 2015, 15, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Hashimoto, T.; Wong, E.; Hori, Y.; Wood, L.B.; Zhao, L.; Haigis, K.M.; Hyman, B.T.; Irimia, D. Microfluidic chemotaxis platform for differentiating the roles of soluble and bound amyloid-β on microglial accumulation. Sci. Rep. 2013, 3, 1823. [Google Scholar] [CrossRef] [PubMed]
- Song, H.L.; Shim, S.; Kim, D.H.; Won, S.H.; Joo, S.; Kim, S.; Jeon, N.L.; Yoon, S.Y. β-Amyloid is transmitted via neuronal connections along axonal membranes. Ann. Neurol. 2014, 75, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Deleglise, B.; Magnifico, S.; Duplus, E.; Vaur, P.; Soubeyre, V.; Belle, M.; Vignes, M.; Viovy, J.-L.; Jacotot, E.; Peyrin, J.-M.; et al. β-amyloid induces a dying-back process and remote trans-synaptic alterations in a microfluidic-based reconstructed neuronal network. Acta Neuropathol. Commun. 2014, 2, 145. [Google Scholar] [CrossRef]
- Choi, Y.J.; Chae, S.; Kim, J.H.; Barald, K.F.; Park, J.Y.; Lee, S.H. Neurotoxic amyloid beta oligomeric assemblies recreated in microfluidic platform with interstitial level of slow flow. Sci. Rep. 2013, 3, 1921. [Google Scholar] [CrossRef]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- McGregor, M.M.; Nelson, A.B. Circuit mechanisms of Parkinson’s disease. Neuron 2019, 101, 1042–1056. [Google Scholar] [CrossRef]
- Teixeira, M.I.; Lopes, C.M.; Amaral, M.H.; Costa, P.C. Current insights on lipid nanocarrier-assisted drug delivery in the treatment of neurodegenerative diseases. Eur. J. Pharm. Biopharm. 2020, 149, 192–217. [Google Scholar] [CrossRef]
- Moreno, E.L.; Hachi, S.; Hemmer, K.; Trietsch, S.J.; Baumuratov, A.S.; Hankemeier, T.; Vulto, P.; Schwamborn, J.C.; Fleming, R.M. Differentiation of neuroepithelial stem cells into functional dopaminergic neurons in 3D microfluidic cell culture. Lab Chip 2015, 15, 2419–2428. [Google Scholar] [CrossRef] [PubMed]
- Kane, K.I.W.; Moreno, E.L.; Hachi, S.; Walter, M.; Jarazo, J.; Oliveira, M.A.P.; Hankemeier, T.; Vulto, P.; Schwamborn, J.C.; Thoma, M.; et al. Automated microfluidic cell culture of stem cell derived dopaminergic neurons. Sci. Rep. 2019, 9, 1796. [Google Scholar] [CrossRef] [PubMed]
- Bolognin, S.; Fossépré, M.; Qing, X.; Jarazo, J.; Ščančar, J.; Moreno, E.L.; Nickels, S.L.; Wasner, K.; Ouzren, N.; Walter, J.; et al. 3D cultures of Parkinson’s disease-specific dopaminergic neurons for high content phenotyping and drug testing. Adv. Sci. 2019, 6, 1800927. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.T.; Chutna, O.; Chu, V.; Conde, J.P.; Outeiro, T.F. A novel microfluidic cell co-culture platform for the study of the molecular mechanisms of Parkinson’s disease and other synucleinopathies. Front. Neurosci. 2016, 10, 511. [Google Scholar] [CrossRef]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef]
- Lu, X.; Kim-Han, J.S.; O’Malley, K.L.; Sakiyama-Elbert, S.E. A microdevice platform for visualizing mitochondrial transport in aligned dopaminergic axons. J. Neurosci. Methods 2012. [Google Scholar] [CrossRef]
- Kerman, B.E.; Kim, H.J.; Padmanabhan, K.; Mei, A.; Georges, S.; Joens, M.S.; Fitzpatrick, J.A.; Jappelli, R.; Chandross, K.J.; August, P.; et al. In vitro myelin formation using embryonic stem cells. Development 2015, 142, 2213–2225. [Google Scholar] [CrossRef]
- Hosmane, S.; Tegenge, M.A.; Rajbhandari, L.; Uapinyoying, P.; Kumar, G.N.; Thakor, N.; Venkatesan, A. Toll/interleukin-1 receptor domain-containing adapter inducing interferon-β mediates microglial phagocytosis of degenerating axons. J. Neurosci. 2012, 32, 7745–7757. [Google Scholar] [CrossRef]
- Machado, C.B.; Pluchon, P.; Harley, P.; Rigby, M.; Sabater, V.G.; Stevenson, D.C.; Hynes, S.; Lowe, A.; Burrone, J.; Viasnoff, V.; et al. In vitro modeling of nerve–muscle connectivity in a compartmentalized tissue culture device. Adv. Biosyst. 2019, 3, 1800307. [Google Scholar] [CrossRef] [PubMed]
- Southam, K.A.; King, A.E.; Blizzard, C.A.; McCormack, G.H.; Dickson, T.C. Microfluidic primary culture model of the lower motor neuron–neuromuscular junction circuit. J. Neurosci. Methods 2013, 218, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, A.; Zahavi, E.E.; Gradus, T.; Ben-Yaakov, K.; Perlson, E. Compartmental microfluidic system for studying muscle–neuron communication and neuromuscular junction maintenance. Eur. J. Cell Biol. 2016, 95, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Kunze, A.; Lengacher, S.; Dirren, E.; Aebischer, P.; Magistretti, P.J.; Renaud, P. Astrocyte-neuron co-culture on microchips based on the model of SOD mutation to mimic ALS. Integr. Biol. 2013, 5, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Sances, S.; Ho, R.; Vatine, G.; West, D.; Laperle, A.; Meyer, A.; Godoy, M.; Kay, P.S.; Mandefro, B.; Hatata, S.; et al. Human iPSC-derived endothelial cells and microengineered organ-chip enhance neuronal development. Stem Cell Rep. 2018, 10, 1222–1236. [Google Scholar] [CrossRef]
- Osaki, T.; Uzel, S.G.M.; Kamm, R.D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 2018, 4, eaat5847. [Google Scholar] [CrossRef]
- Tan, V.X.; Lassus, B.; Lim, C.K.; Tixador, P.; Courte, J.; Bessede, A.; Guillemin, G.J.; Peyrin, J.M. Neurotoxicity of the cyanotoxin BMAA through axonal degeneration and intercellular spreading. Neurotox. Res. 2018, 33, 62–75. [Google Scholar] [CrossRef]
- Virlogeux, A.; Moutaux, E.; Christaller, W.; Genoux, A.; Bruyère, J.; Fino, E.; Charlot, B.; Cazorla, M.; Saudou, F. Reconstituting corticostriatal network on-a-chip reveals the contribution of the presynaptic compartment to Huntington’s disease. Cell Rep. 2018, 22, 110–122. [Google Scholar] [CrossRef]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Barthakur, S.; Kasendra, M.; Lucchesi, C.; Kerns, J.; et al. Human iPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem. Cell 2019, 24, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Feng, Z.; Hu, D.; Xu, S.; Middha, E.; Pan, Y.; Liu, C.; Zheng, H.; Qian, J.; Sheng, Z.; et al. Precise deciphering of brain vasculatures and microscopic tumors with dual NIR-II fluorescence and photoacoustic imaging. Adv. Mater. 2019, 31, 1902504. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Chen, J.; Chen, N.; Middha, E.; Xu, S.; Pan, Y.; Wu, M.; Li, K.; Liu, C.; Liu, B. High-resolution 3D NIR-II photoacoustic imaging of cerebral and tumor vasculatures using conjugated polymer nanoparticles as contrast agent. Adv. Mater. 2019, 31, 1808355. [Google Scholar] [CrossRef] [PubMed]
- Jgamadze, D.; Liu, L.; Vogler, S.; Chu, L.Y.; Pautot, S. Thermoswitching microgel carriers improve neuronal cell growth and cell release for cell transplantation. Tissue Eng. Part C Methods 2015, 21, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Walgrave, H.; Salta, E.; Álvarez, D.M.; Castro-López, V.; Loza, M.; Alonso, M.J. Nose-to-brain delivery of enveloped RNA-cell permeating peptide nanocomplexes for the treatment of neurodegenerative diseases. Biomaterials 2020, 230, 119657. [Google Scholar] [CrossRef] [PubMed]
- Rungta, R.L.; Choi, H.B.; Lin, P.J.; Ko, R.W.; Ashby, D.; Nair, J.; Manoharan, M.; Cullis, P.R.; Macvicar, B.A. Lipid nanoparticle delivery of siRNA to silence neuronal gene expression in the brain. Mol. Nucleic Acids 2013, 2, e136. [Google Scholar] [CrossRef]
- Yu, D.; Khan, O.F.; Suvà, M.L.; Dong, B.; Panek, W.K.; Xiao, T.; Wu, M.; Han, Y.; Ahmed, A.U.; Balyasnikova, I.V.; et al. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. USA 2017, 114, E6147–E6156. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, F.; Knapp, K.A.; Nickels, M.L.; Manning, H.C.; Bellan, L.M. A simple microfluidic platform for rapid and efficient production of the radiotracer [(18)F] fallypride. Lab Chip 2018, 18, 1369–1377. [Google Scholar] [CrossRef]
- Probst, C.; Schneider, S.; Loskill, P. High-throughput organ-on-a-chip systems: Current status and remaining challenges. Curr. Opin. Biomed. Eng. 2018, 6, 33–41. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-chip: Recent breakthroughs and future prospects. Biomed. Eng. Online 2020, 19, 9. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, M.I.; Amaral, M.H.; Costa, P.C.; Lopes, C.M.; Lamprou, D.A. Recent Developments in Microfluidic Technologies for Central Nervous System Targeted Studies. Pharmaceutics 2020, 12, 542. https://doi.org/10.3390/pharmaceutics12060542
Teixeira MI, Amaral MH, Costa PC, Lopes CM, Lamprou DA. Recent Developments in Microfluidic Technologies for Central Nervous System Targeted Studies. Pharmaceutics. 2020; 12(6):542. https://doi.org/10.3390/pharmaceutics12060542
Chicago/Turabian StyleTeixeira, Maria Inês, Maria Helena Amaral, Paulo C. Costa, Carla M. Lopes, and Dimitrios A. Lamprou. 2020. "Recent Developments in Microfluidic Technologies for Central Nervous System Targeted Studies" Pharmaceutics 12, no. 6: 542. https://doi.org/10.3390/pharmaceutics12060542
APA StyleTeixeira, M. I., Amaral, M. H., Costa, P. C., Lopes, C. M., & Lamprou, D. A. (2020). Recent Developments in Microfluidic Technologies for Central Nervous System Targeted Studies. Pharmaceutics, 12(6), 542. https://doi.org/10.3390/pharmaceutics12060542