Development and Characterization of Liquisolid Tablets Based on Mesoporous Clays or Silicas for Improving Glyburide Dissolution




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility Studies
2.3. Characterization of the Mesoporous Silicas and Clays
2.3.1. Apparent and Tapped Density
2.3.2. Powder Flowability
2.3.3. Powder Compactability
2.3.4. Specific Surface Area
2.4. Preparation of Liquisolid Systems
2.5. Characterization of Liquisolid Systems
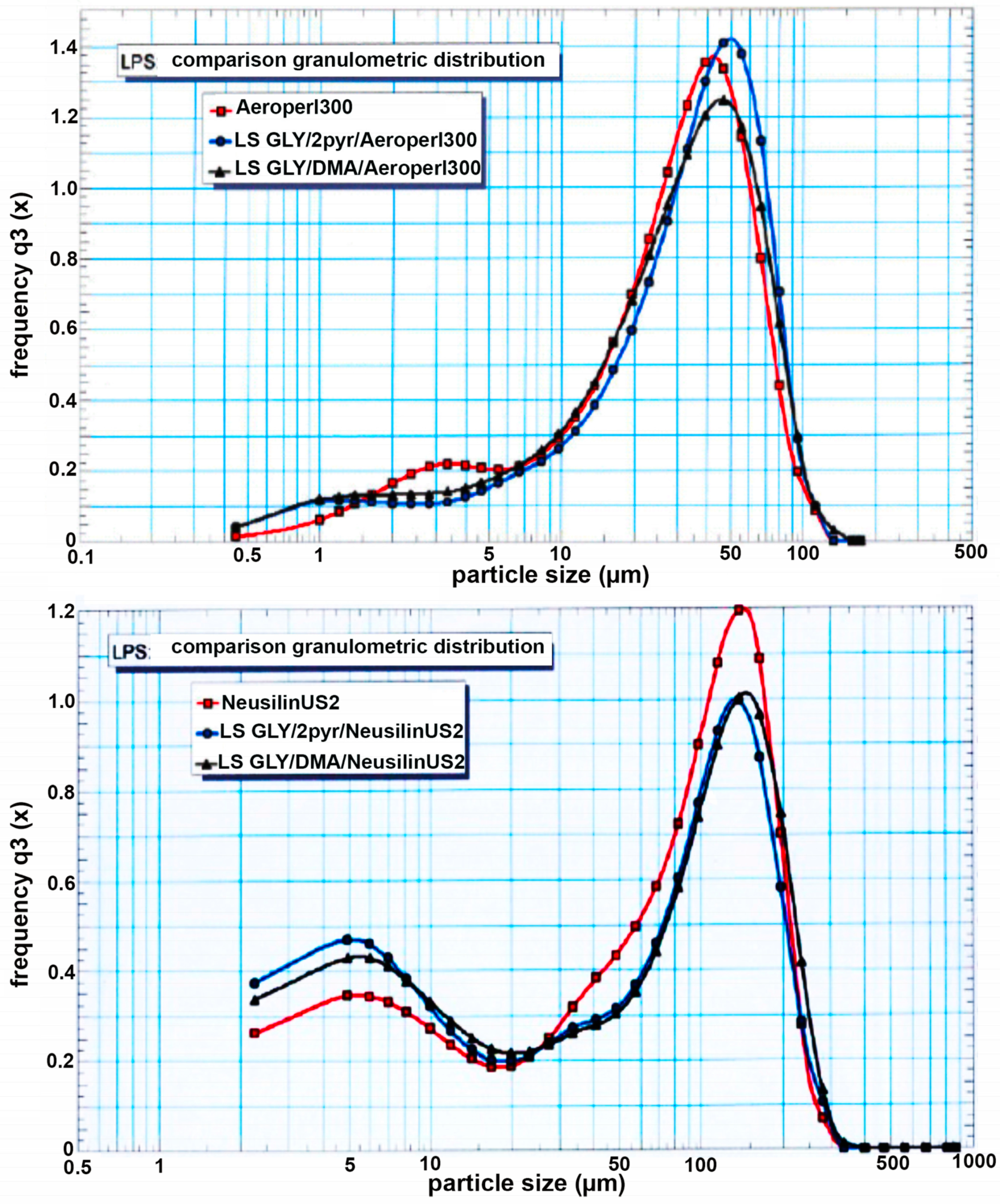
2.5.1. Particle Size Distribution
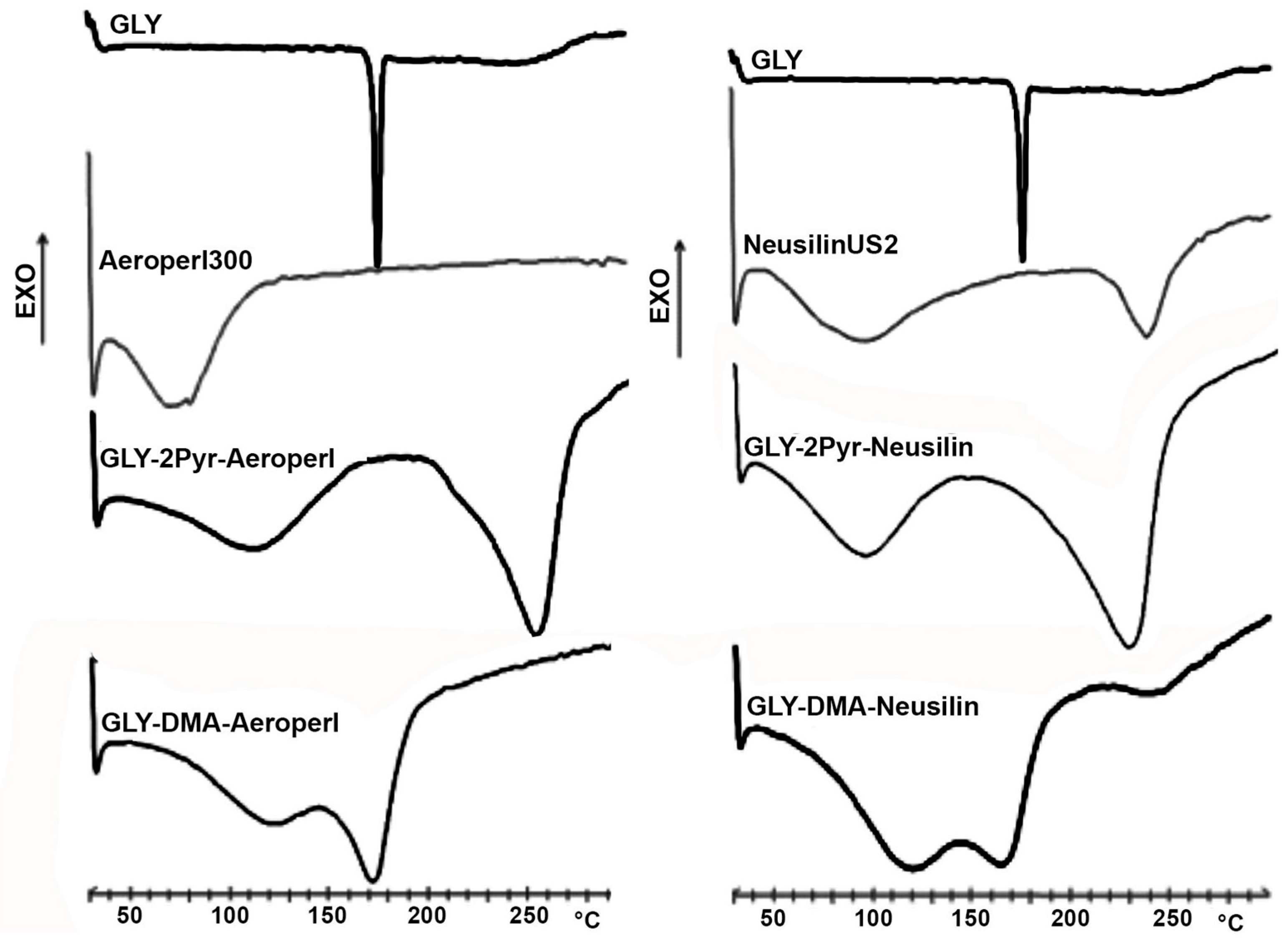
2.5.2. Differential Scanning Calorimetry (DSC)
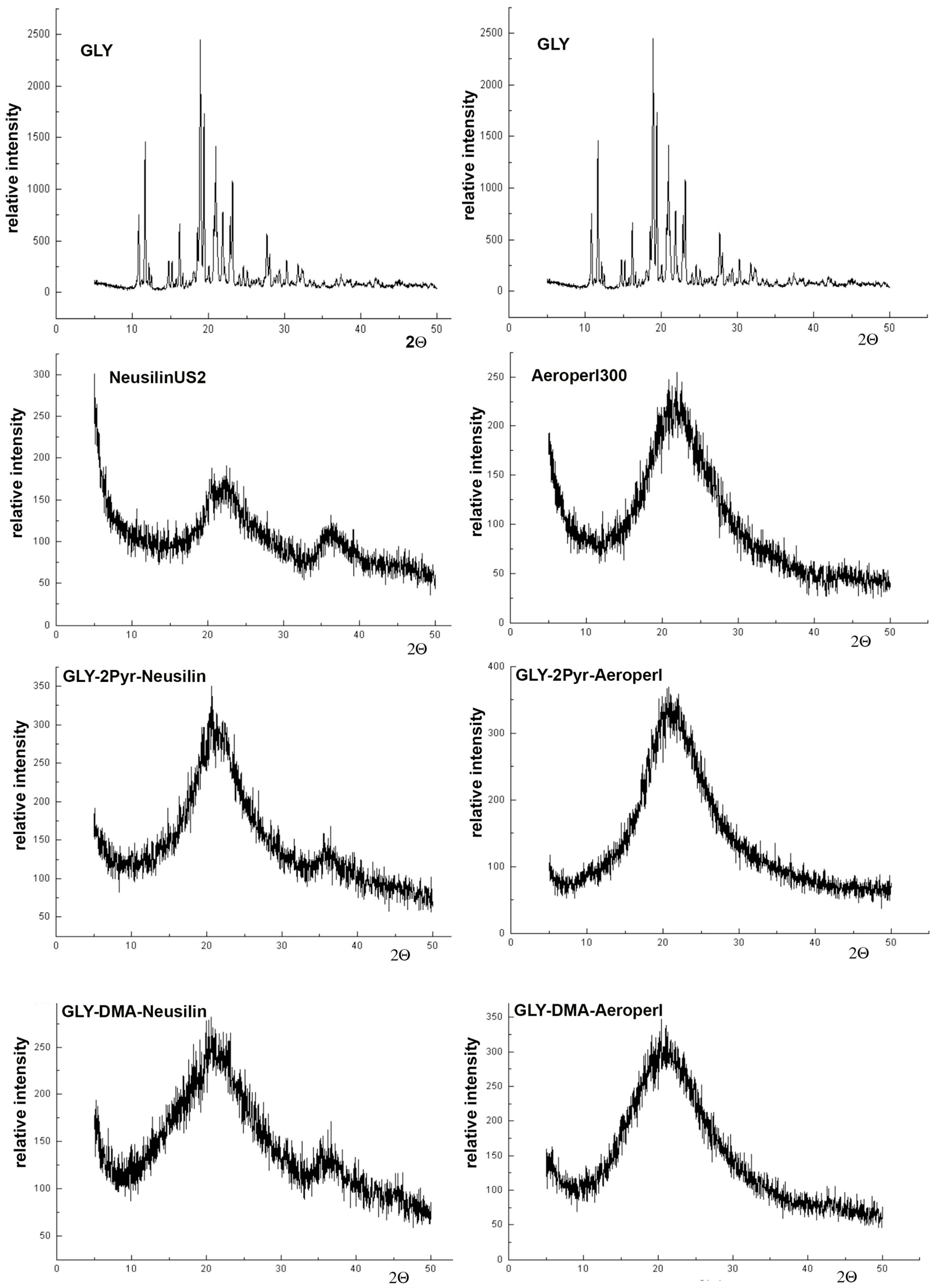
2.5.3. Powder X-Ray Diffractometry (PXRD)
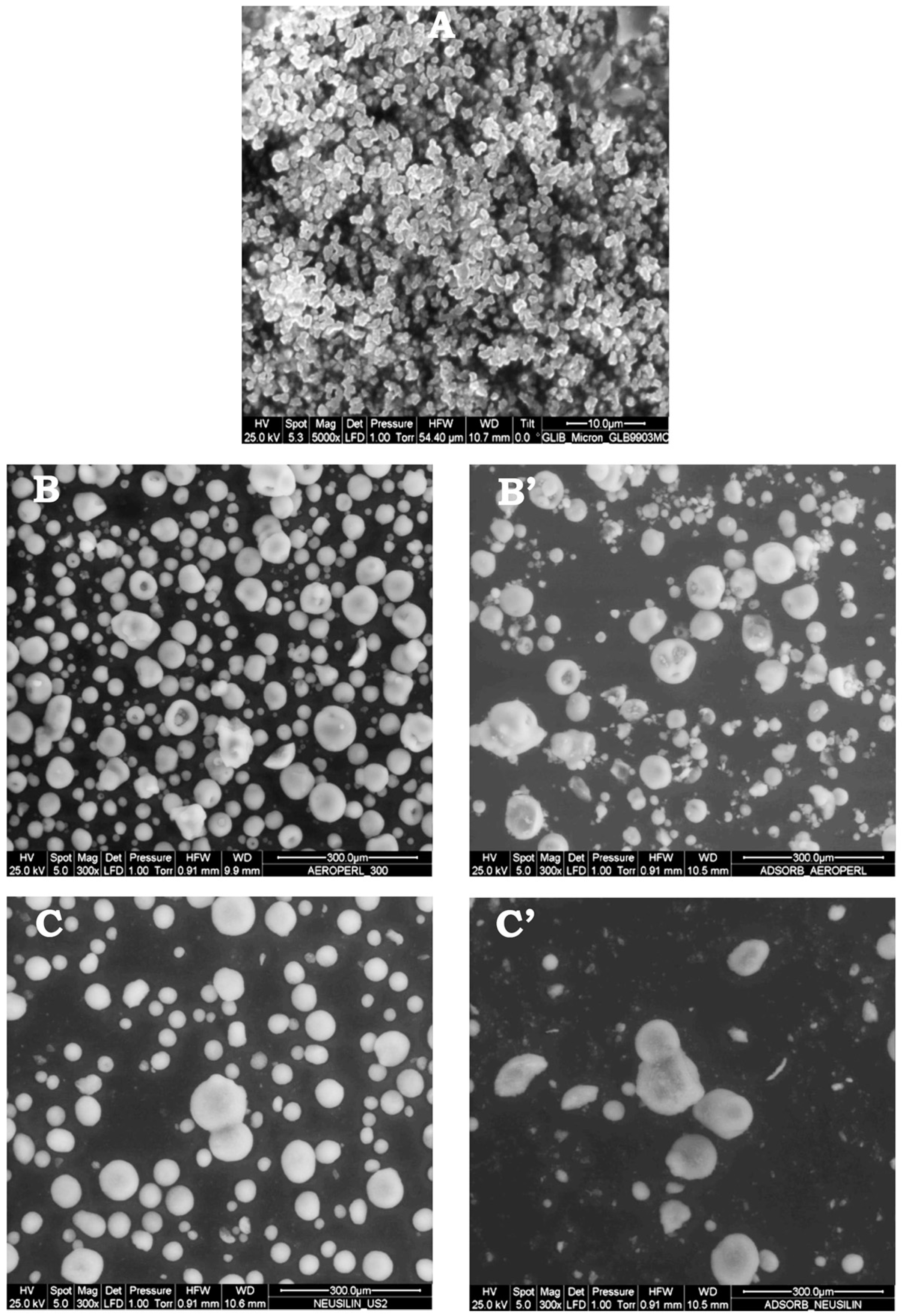
2.5.4. Environmental Scanning Electron Microscopy (ESEM)
2.5.5. Dissolution Test
2.6. Tablets Preparation
2.7. Tablets Characterization
3. Results and Discussion
3.1. Selection of the Solvents
3.2. Characterization and Selection of Mesoporous Carriers as Adsorbent Carrier
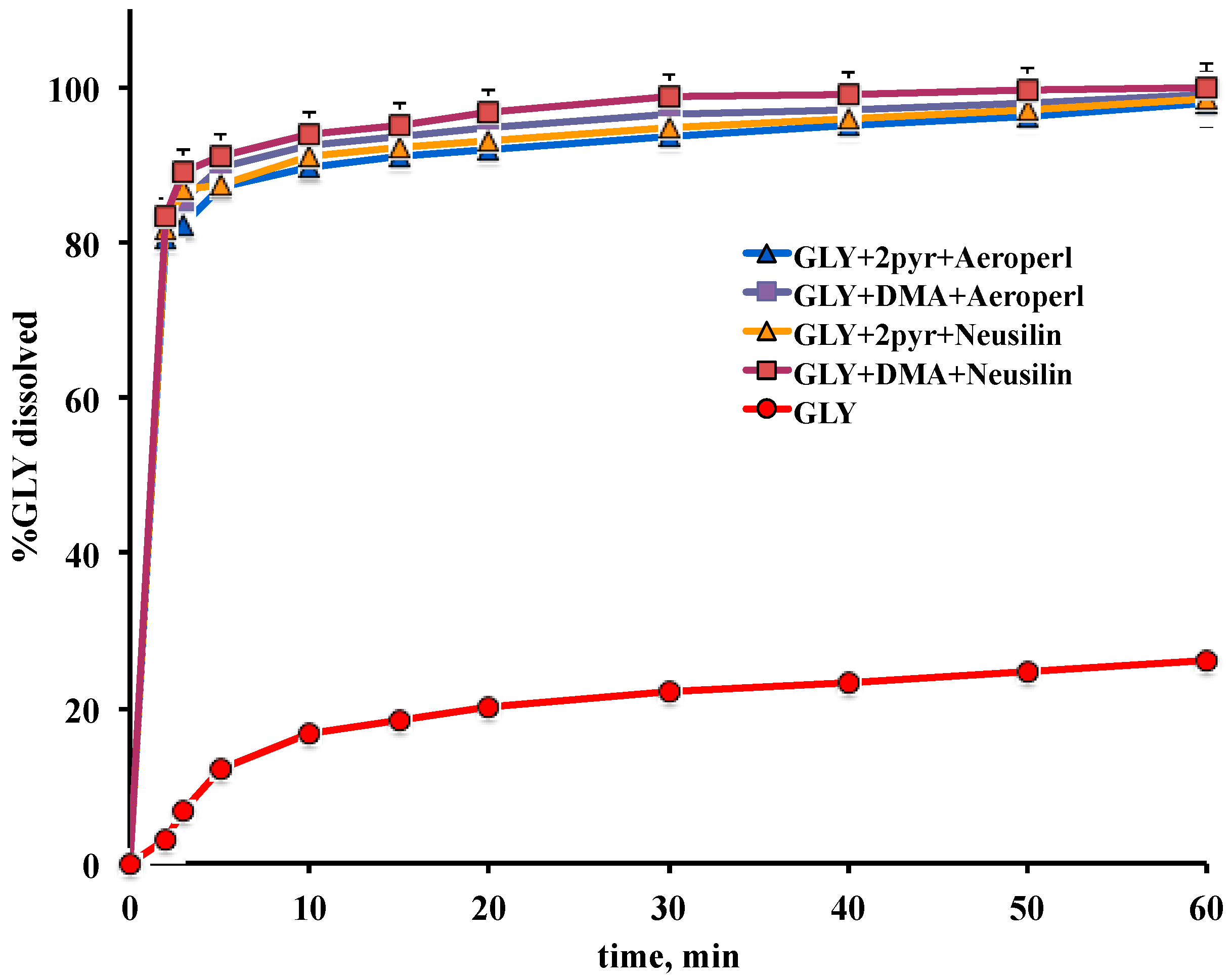
3.3. Preparation and Characterization of Liquisolid Systems
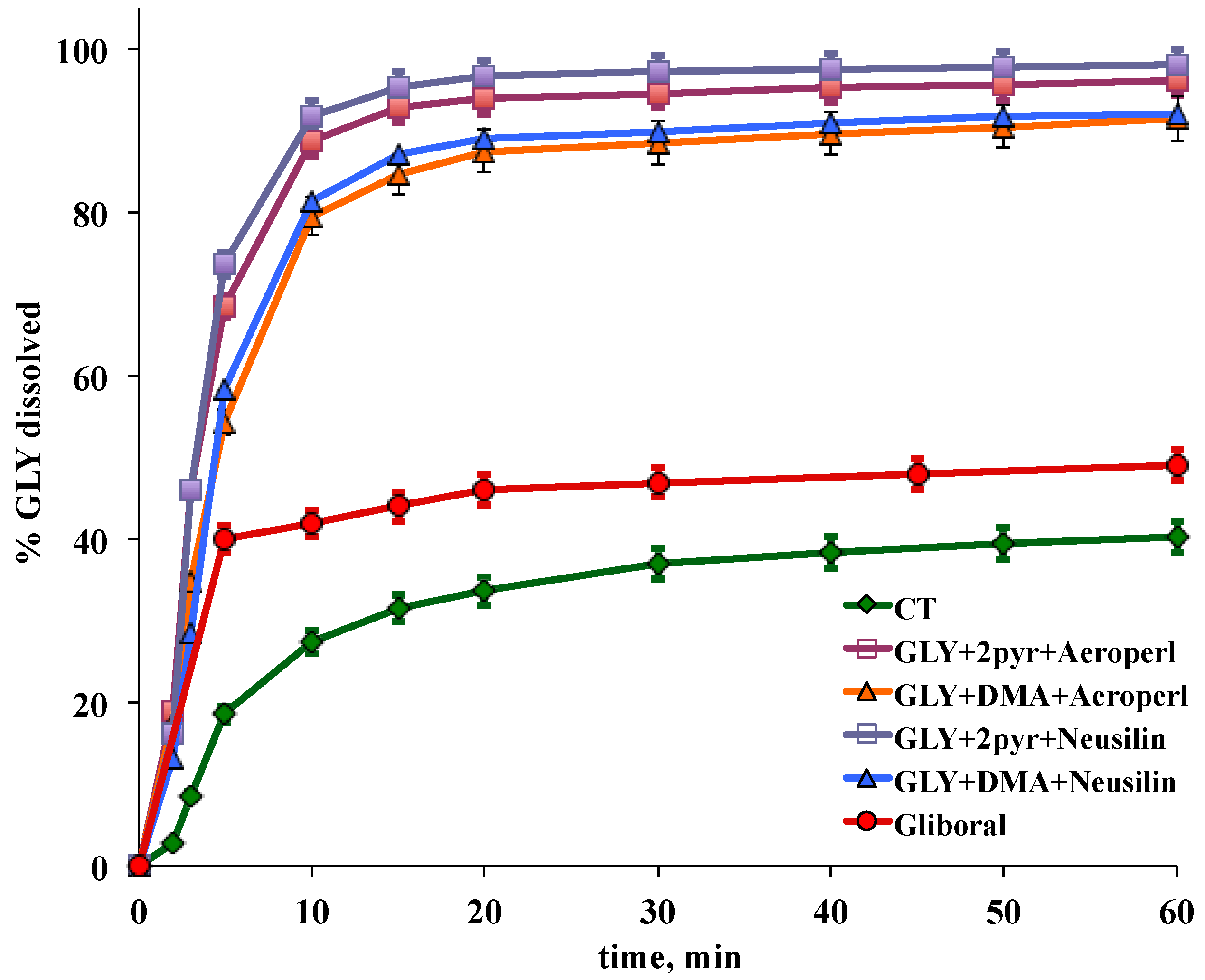
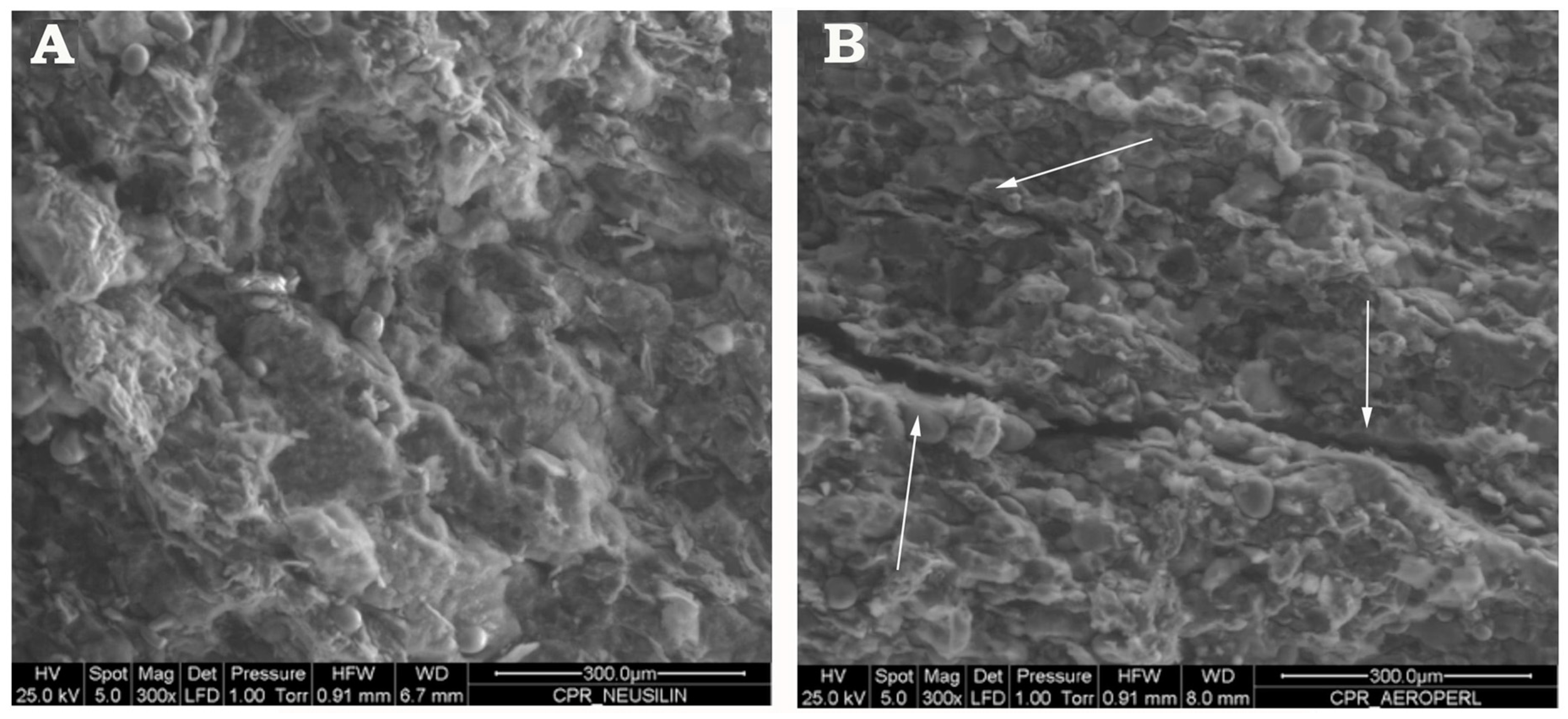
3.4. Preparation and Characterization of Liquisolid-Tablets
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pearson, J.G. Pharmacokinetics of glyburide. Am. J. Med. 1985, 79, 67–71. [Google Scholar] [CrossRef]
- Simard, J.M.; Sheth, K.N.; Kimberly, W.T.; Stern, B.J.; del Zoppo, G.J.; Jacobson, S.; Gerzanich, V. Glibenclamide in cerebral ischemia and stroke. Neurocrit. Care 2014, 20, 319–333. [Google Scholar] [CrossRef]
- Caffes, N.; Urland, D.B.; Gerzanich, V.; Simard, J.M. Glibenclamide for the treatment of ischemic and hemorrhagic stroke. Int. J. Mol. Sci. 2015, 16, 4973–4984. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Model List of Essential Medicines. 2015. Available online: http://www.who.int/medicines/publications/essentialmedicines/en/index.html (accessed on 24 April 2020).
- Lobenberg, R.; Kramer, J.; Shah, V.P.; Amidon, G.L.; Dressman, J.B. Dissolution testing as prognostic tool for oral drug absorption dissolution behavior of glibenclamide. Pharm. Res. 2000, 17, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Lobenberg, R.; Amidon, G.L. Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur. J. Pharm. Biopharm. 2000, 50, 3–12. [Google Scholar] [CrossRef]
- Galia, E.; Nicolaides, E.; Hörter, D.; Lobenberg, R.; Reppas, C.; Dressman, J.B. Evaluation of various dissolution media for predicting in vivo performance of class I and II drugs. Pharm. Res. 1998, 15, 698–705. [Google Scholar] [CrossRef]
- Chalk, J.B.; Patterson, M.; Smith, M.T.; Eadie, M.J. Correlations between in vitro dissolution, in vivo bioavailability and hypoglycaemic effect of oral glibenclamide. Eur. J. Clin. Pharmacol. 1986, 31, 177–187. [Google Scholar] [CrossRef]
- Blume, H.; Ali, S.L.; Siewert, M. Pharmaceutical quality of glibenclamide products: A multinational postmarket comparative study. Drug Dev. Ind. Pharm. 1993, 19, 2713–2741. [Google Scholar] [CrossRef]
- Neugebauer, G.; Betzien, G.; Hrstka, V.; Kaufmann, B.; von Mollendorff, E.; Abshagen, U. Absolute bioavailability and bioequivalence of glibenclamide. Int. J. Clin. Pharmacol. Ther. Toxicol. 1985, 23, 453–460. [Google Scholar]
- Mitrevej, A.; Sinchaipanid, N.; Juniaprasert, V.; Warintornuwat, L. Effect of grinding of β-cyclodextrin and glibenclamide on tablet properties. Drug Dev. Ind. Pharm. 1996, 22, 1237–1241. [Google Scholar] [CrossRef]
- Cirri, M.; Righi, F.; Maestrelli, F.; Mura, P. Development of glyburide fast-dissolving tablets base on the combined use of cyclodextrins and polymers. Drug Dev. Ind. Pharm. 2009, 35, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Wempe, M.F.; Zoeller, T.; Buchanan, N.L.; Lambert, J.L.; Ramsey, M.G.; Edgar, K.J.; Buchanan, C.M. Improving glyburide solubility and dissolution by complexation with hydroxybutyl-beta-cyclodextrin. J. Pharm. Pharmacol. 2009, 61, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Tashtoush, B.M.; Al-Oashi, Z.S.; Najib, N.M. In vitro and in vivo evaluation of glibenclamide in solid dispersion systems. Drug Dev. Ind. Pharm. 2004, 30, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Valleri, M.; Mura, P.; Maestrell, F.; Cirri, M.; Ballerini, R. Development and evaluation of glyburide fast dissolving tablets using solid dispersion technique. Drug Dev. Ind. Pharm. 2004, 30, 525–534. [Google Scholar] [CrossRef]
- Cirri, M.; Valleri, M.; Maestrelli, F.; Corti, G.; Mura, P. Fast-dissolving tablets of glyburide based on ternary solid dispersions with PEG 6000 and surfactants. Drug Deliv. 2007, 14, 247–255. [Google Scholar] [CrossRef]
- Seedher, N.; Kanojia, M. Micellar solubilization of some poorly soluble antidiabetic drugs. AAPS PharmSciTech 2008, 9, 431–436. [Google Scholar] [CrossRef]
- Bachhav, Y.G.; Patravale, V.B. SMEDDS of glyburide: Formulation, in vitro evaluation, and stability studies. AAPS PharmSciTech 2009, 10, 482–487. [Google Scholar] [CrossRef]
- Albertini, B.; Sabatino, M.D.; Melegari, C.; Passerini, N. Formulation of spray-congealed microparticles with self-emulsifying ability for enhanced glibenclamide dissolution performance. J. Microencapsul. 2015, 32, 181–192. [Google Scholar] [CrossRef]
- Cirri, M.; Roghi, A.; Valleri, M.; Mura, P. Development and characterization of fast-dissolving tablet formulations of glyburide based on solid self-microemulsifying systems. Eur. J. Pharm. Biopharm. 2016, 104, 19–29. [Google Scholar] [CrossRef]
- Spireas, S. Liquisolid Systems and Methods of Preparing Same. U.S. Patent 6,423,339 B1, 19 August 1998. [Google Scholar]
- Spireas, S.; Sadu, S. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int. J. Pharm. 1998, 166, 177–188. [Google Scholar] [CrossRef]
- Spireas, S.; Wang, T.; Grover, R. Effect of powder substrate on the dissolution properties of methychlothiazide liquisolid compacts. Drug Dev. Ind. Pharm. 1999, 25, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Nokhodchi, A.; Hentzschel, C.M.; Leopold, C.S. Drug release from liquisolid systems: Speed it up, slow it down. Expert Opin. Drug Deliv. 2011, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vraníková, B.; Niederquell, A.; Ditzingerb, F.; Šklubalová, Z.; Kuentz, M. Mechanistic aspects of drug loading in liquisolid systems with hydrophilic lipid-based. Int. J. Pharm. 2020, 578, 110999. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, R.H.; Kassem, M.A. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2008, 69, 993–1003. [Google Scholar] [CrossRef]
- Tiong, N.; Elkordy, A.A. Effects of liquisolid formulations on dissolution of naproxen. Eur. J. Pharm. Biopharm. 2009, 73, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Hentzschel, C.M.; Alnaief, M.; Smirnova, I.; Sakmann, A.; Leopold, C.S. Enhancement of griseofulvin release from liquisolid compacts. Eur. J. Pharm. Biopharm. 2012, 80, 130–135. [Google Scholar] [CrossRef]
- Chella, N.; Narra, N.; Rao, T.R. Preparation and characterization of liquisolid compacts for improved dissolution of telmisartan. J. Drug Deliv. 2014. [Google Scholar] [CrossRef]
- Azharshekoufeh, L.; Shokri, J.; Adibkiac, K.; Javadzadeh, Y. Liquisolid technology: What it can do for NSAIDs delivery? Colloids Surf. B 2015, 136, 185–191. [Google Scholar] [CrossRef]
- Khaled, K.A.; Asiri, Y.A.; El-Sayed, Y.M. In vivo evaluation of hydrochlorothiazide liquisolid tablets in beagle dogs. Int. J. Pharm. 2007, 222, 1–6. [Google Scholar] [CrossRef]
- El-Houssieny, B.M.; Wahman, L.F.; Arafa, N.M.S. Bioavailability and biological activity of liquisolid compact formula of repaglinide and its effect on glucose tolerance in rabbits. Biosci. Trends 2010, 4, 17–24. [Google Scholar]
- Sanka, K.; Poienti, S.; Bari Mohd, A.; Diwan, P.V. Improved oral delivery of clonazepam through liquisolid powder compact formulations: In-vitro and ex-vivo characterization. Powder Technol. 2014, 256, 336–344. [Google Scholar] [CrossRef]
- Manzano, M.; Colilla, M.; Vallet-Regí, M. Drug delivery from ordered mesoporous matrices. Expert Opin. Drug Deliv. 2009, 6, 1383–1400. [Google Scholar] [CrossRef] [PubMed]
- Mazanoab, M.; Vallet-Regí, M. New developments in ordered mesoporous materials for drug delivery. J. Mater. Chem. 2010, 20, 5593–5604. [Google Scholar]
- Huang, X.; Young, N.P.; Townley, H.E. Characterization and comparison of mesoporous silica particles for optimized drug delivery. Nanomater. Nanotechnol. 2014, 4. [Google Scholar] [CrossRef]
- Carretero, M.I.; Pozo, M. Clay and non-clay minerals in the pharmaceutical industry: Part I. Excipients and medical applications. Appl. Clay Sci. 2009, 46, 73–80. [Google Scholar] [CrossRef]
- Kim, M.H.; Choi, G.; Elzatahry, A.; Vinu, A.; Choy, Y.B.; Choy, J.H. Review of clay-drug hybrid materials for biomedical applications. Clays Clay Miner. 2016, 64, 115–130. [Google Scholar] [CrossRef]
- Aguzzi, C.; Cerezo, P.; Viseras, C.; Caramella, C. Use of clays as drug delivery systems: Possibilities and limitations. Appl. Clay Sci. 2007, 36, 22–36. [Google Scholar] [CrossRef]
- Viseras, C.; Cerezo, P.; Sanchez, R.; Salcedo, I.; Aguzzi, C. Current challenges in clay minerals for drug delivery. Appl. Clay Sci. 2010, 48, 291–295. [Google Scholar] [CrossRef]
- Hua, S.; Yang, H.; Wang, W.; Wang, A. Controlled release of ofloxacin from chitosan_montmorillonite hydrogel. Appl. Clay Sci. 2010, 50, 112–117. [Google Scholar] [CrossRef]
- Salcedo, I.; Aguzzi, C.; Sandri, G.; Bonferoni, M.C.; Mori, M.; Cerezo, P.; Sanchez, R.; Viseras, C.; Caramella, C. In vitro biocompatibility and mucoadhesion of montmorillonite chitosan nanocomposite: A new drug delivery. Appl. Clay Sci. 2012, 55, 131–137. [Google Scholar] [CrossRef]
- De Sousa Rodrigues, L.A.; Figueiras, A.; Veiga, F.; de Freitas, R.M.; Nunes, L.C.C.; da Silva Filho, E.C.; da Silva Filho, E.C.; da Silva Leite, C.M. The systems containing clays and clay minerals from modified drug release: A review. Colloids Surf. B 2013, 103, 642–651. [Google Scholar] [CrossRef]
- Choudhari, Y.; Hoefer, H.; Libanati, C.; Monsuur, F.; McCarthy, W. Mesoporous silica drug delivery systems. In Amorphous Solid Dispersions; Shah, N., Sandhu, H., Choi, D.S., Chokshi, H., Malick, A.W., Eds.; Springer: New York, NY, USA, 2014; pp. 665–693. [Google Scholar]
- Le, T.T.; Elyafi, A.K.E.; Mohammed, A.R.; Al-Khattawi, A. Delivery of poorly soluble drugs via mesoporous silica: Impact of drug overloading on release and thermal profile. Pharmaceutics 2019, 11, 269. [Google Scholar] [CrossRef]
- Mura, P.; Valleri, M.; Fabianelli, E.; Maestrelli, F.; Cirri, M. Characterization and evaluation of different mesoporous silica kinds as carriers for the development of effective oral dosage forms of glibenclamide. Int. J. Pharm. 2019, 563, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Javadzadeh, Y.; Jafari-Navimipour, B.; Nokhodchi, A. Liquisolid technique for dissolution rate enhancement of a high-dose water-insoluble drug (carbamazepine). Int. J. Pharm. 2007, 341, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.I. Powder flow properties: Compression properties. In Pharmaceutical Preformulation: The Physicochemical Properties of Drug Substances; Wiley, J., Ed.; Ellis Horwood: Chichester, UK, 1988; pp. 211–214. [Google Scholar]
- Washington, C. Particle Size Analysis in Pharmaceuticals and Other Industries; Taylor & Francis e-Library: Chichester, UK, 2005; pp. 1–19. [Google Scholar]
- Khan, K.A. The concept of dissolution efficiency. J. Pharm. Pharmacol. 1975, 27, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Yalkowsky, S.H. Solubilization of poorly soluble compounds using 2-pyrrolidone. Int. J. Pharm. 2007, 342, 1–5. [Google Scholar] [CrossRef]
- Kawakami, K.; Oda, N.; Miyoshi, K.; Funaki, T.; Ida, Y. Solubilization behavior of a poorly soluble drug under combined use of surfactants and cosolvents. Eur. J. Pharm. Sci. 2006, 28, 7–14. [Google Scholar] [CrossRef]
- Oechtering, D.; Boos, J.; Hempel, G. Monitoring of N,N-dimethylacetamide in children during i.v.-busulfan therapy by liquid chromatography-mass spectrometry. J. Chromatogr. B 2006, 838, 129–134. [Google Scholar] [CrossRef]
- Trame, M.N.; Bartelink, I.H.; Boos, J.; Boelens, J.J.; Hempel, G. Population pharmacokinetics of dimethylacetamide in children during standard and once-daily IV busulfan administration. Cancer Chemother. Pharmacol. 2013, 72, 1149–1155. [Google Scholar] [CrossRef]
- Bolhuis, G.K.; Van Der Voort, M.K.; Zuurman, K. Effect of magnesium stearate on bonding and porosity expansion of tablets produced from materials with different consolidation properties. Int. J. Pharm. 1999, 179, 107–115. [Google Scholar]
- Rojas, J.; Aristozabal, J.; Henao, M. Screening of several excipients for direct compression of tablets: A new perspective based on functional properties. J. Bas. Appl. Pharm. Sci. 2013, 34, 17–23. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Da (g/cm3) | DT (g/cm3) | CI % | HR | Flow (∅, mm) | Wells A (N) | Wells B (N) | Wells C (N) |
|---|---|---|---|---|---|---|---|---|
| Aeroperl®300 | 0.23 | 0.28 | 17.8 | 1.22 | 4 | 30 | 32 | 30 |
| Neusilin®US2 | 0.17 | 0.20 | 16.5 | 1.19 | 4 | 390 | 400 | 400 |
| Zeopharm®5170 | 0.32 | 0.34 | 6.2 | 1.07 | 4 | 15 | 16 | 16 |
| Tablet Code | Ingredients (mg) | Properties | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GLY | 2pyr | DMA | Neusilin | Aeroperl | Avicel | Ceolus | Explotab | Mg st | Compr. Force (kN) | Crush. Force (N) | Tens str (MPa) | Disin. Time (s) | |
| LS1 | 5 | 55 | -- | 55 | -- | 44 | 20 | 10 | 0.5 | 3.4 | 62 | 1.42 | 50 |
| LS2 | 5 | 55 | -- | -- | 55 | 44 | 20 | 10 | 0.5 | 15 | 50 | 1.28 | 90 |
| LS3 | 5 | -- | 47 | 47 | -- | 44 | 20 | 10 | 0.5 | 3.4 | 32 | 0.85 | 60 |
| LS4 | 5 | -- | 47 | -- | 47 | 44 | 20 | 10 | 0.5 | 15 | 29 | 0.79 | 85 |
| Tablet Code * | PD10 | PD60 | DE10 | DE60 |
|---|---|---|---|---|
| LS1 | 91.8 | 98.1 | 58.1 | 90.4 |
| LS2 | 88.6 | 96.3 | 55.9 | 88.1 |
| LS3 | 81.3 | 92.1 | 47.1 | 82.7 |
| LS4 | 79.6 | 91.4 | 46.8 | 81.5 |
| CT | 27.5 | 40.7 | 15.1 | 33.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirri, M.; Mura, P.; Valleri, M.; Brunetti, L. Development and Characterization of Liquisolid Tablets Based on Mesoporous Clays or Silicas for Improving Glyburide Dissolution. Pharmaceutics 2020, 12, 503. https://doi.org/10.3390/pharmaceutics12060503
Cirri M, Mura P, Valleri M, Brunetti L. Development and Characterization of Liquisolid Tablets Based on Mesoporous Clays or Silicas for Improving Glyburide Dissolution. Pharmaceutics. 2020; 12(6):503. https://doi.org/10.3390/pharmaceutics12060503
Chicago/Turabian StyleCirri, Marzia, Paola Mura, Maurizio Valleri, and Letizia Brunetti. 2020. "Development and Characterization of Liquisolid Tablets Based on Mesoporous Clays or Silicas for Improving Glyburide Dissolution" Pharmaceutics 12, no. 6: 503. https://doi.org/10.3390/pharmaceutics12060503
APA StyleCirri, M., Mura, P., Valleri, M., & Brunetti, L. (2020). Development and Characterization of Liquisolid Tablets Based on Mesoporous Clays or Silicas for Improving Glyburide Dissolution. Pharmaceutics, 12(6), 503. https://doi.org/10.3390/pharmaceutics12060503