Design of Polymeric Nanocapsules for Intranasal Vaccination against Mycobacterium Tuberculosis: Influence of the Polymeric Shell and Antigen Positioning

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials for the Synthesis of the Nanocapsules (NCs)
2.2. Nanocapsules’ Synthesis and Characterization
2.3. Adsorption of the Antigen onto the Nanocapsules and Quantification of the Adsorbed Protein
2.4. Materials for Protein, Cell, and Animal Studies
2.5. Cells and Culture Conditions
2.6. Evaluation of Bacterial or Endotoxin Contamination
2.7. Toxicity Studies on Macrophages and Lung Epithelial Cells
2.8. Production of Reactive Oxygen Species (ROS)
2.9. Analysis of Complement Activation
2.10. Cytokine Profile Evaluation
2.11. Immunization Experiments
2.12. Detection of Specific Antibodies
2.13. Statistical Analysis
3. Results
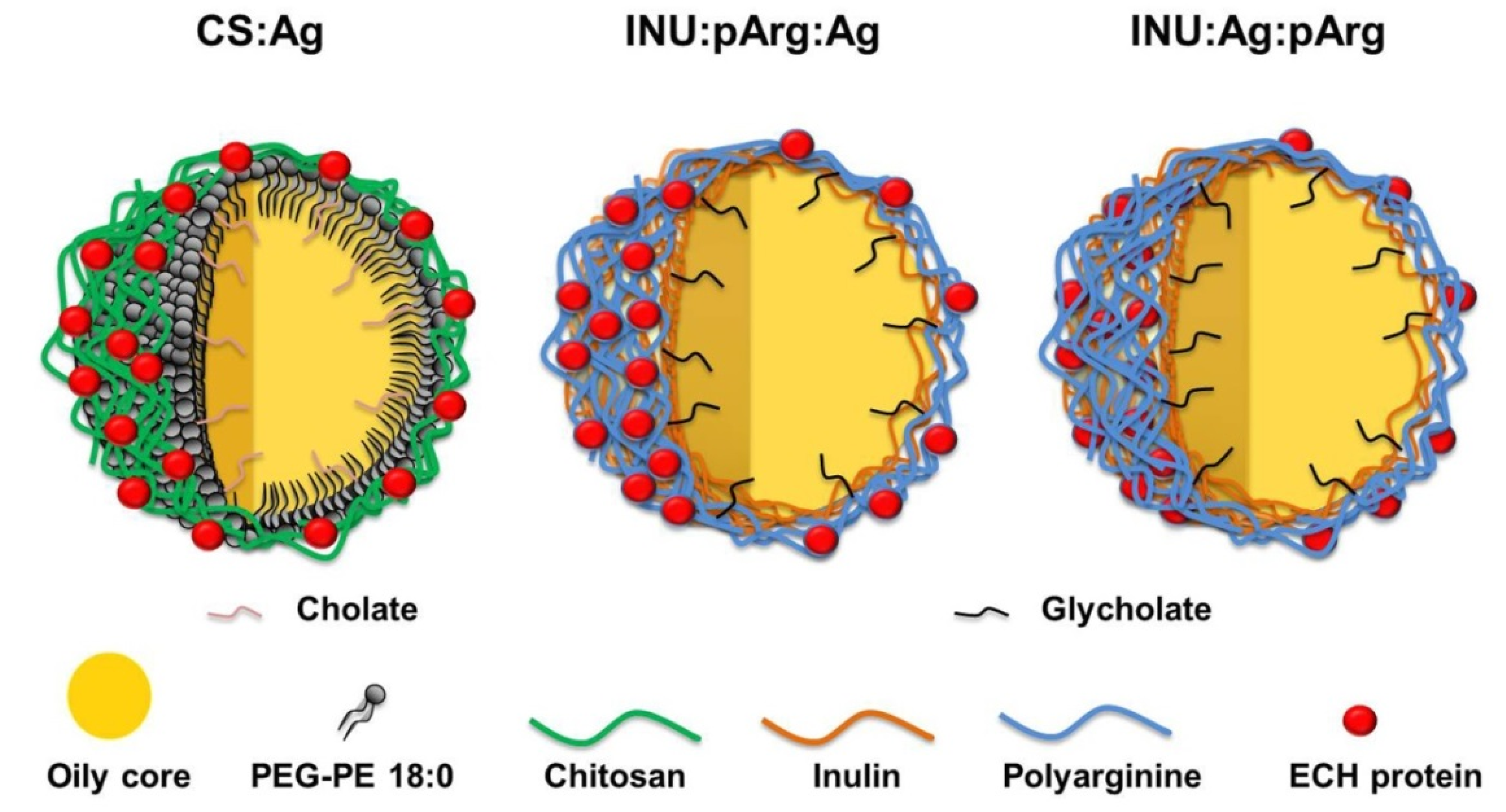
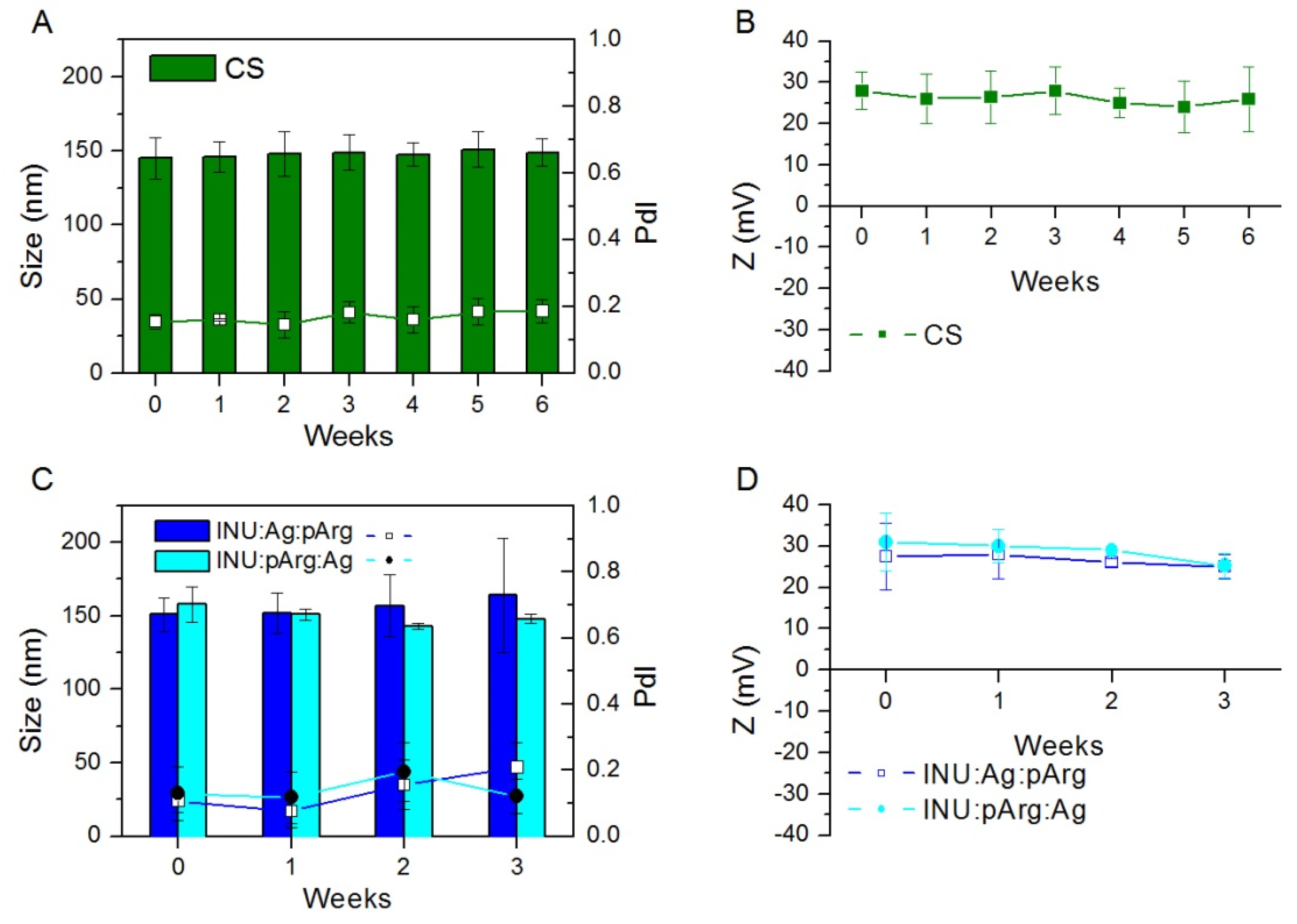
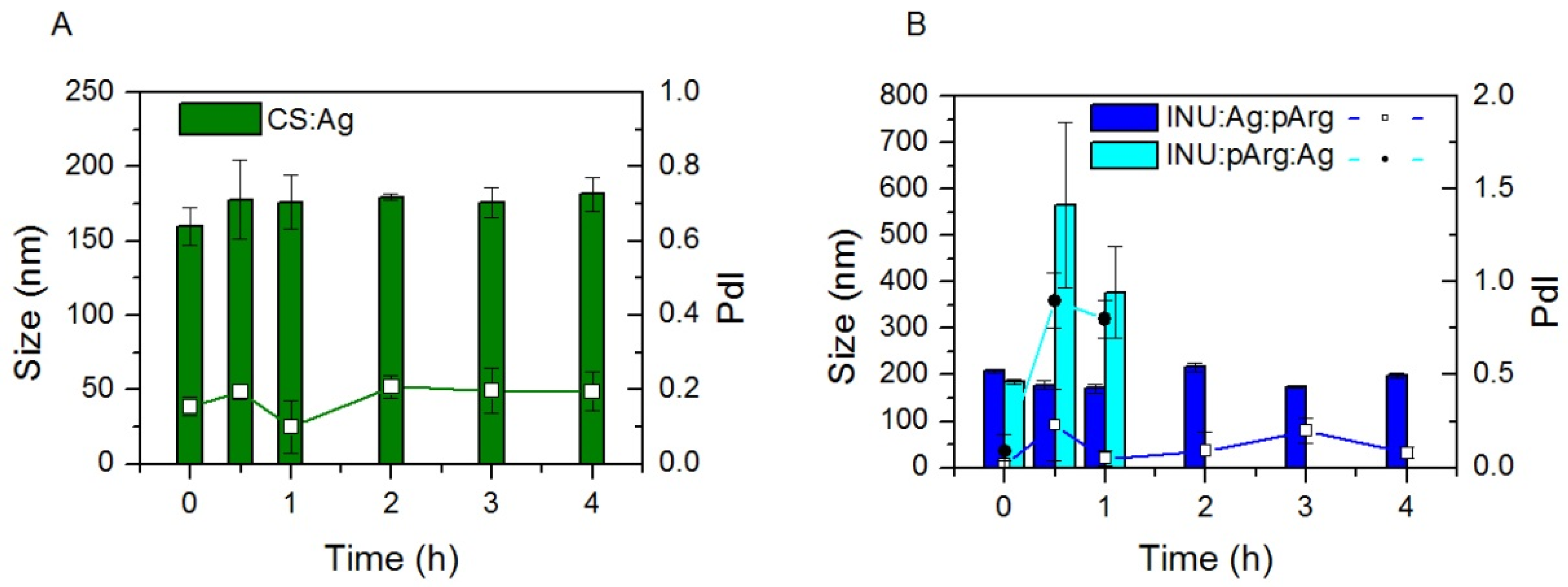
3.1. Physicochemical Characterization and Stability of the Nanocapsules
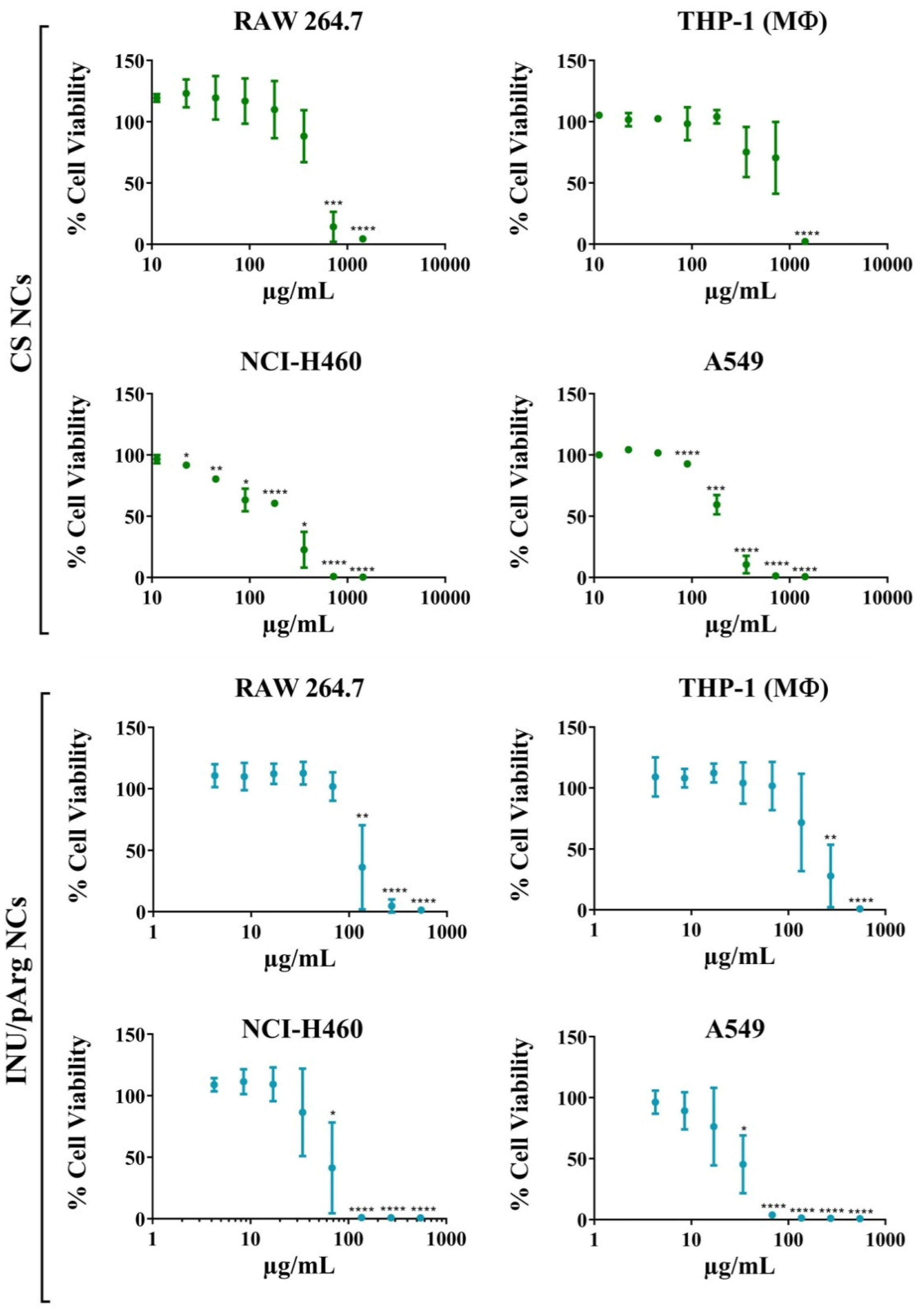
3.2. Cytocompatibility of the Polymeric Nanocapsules in Different Cell Lines
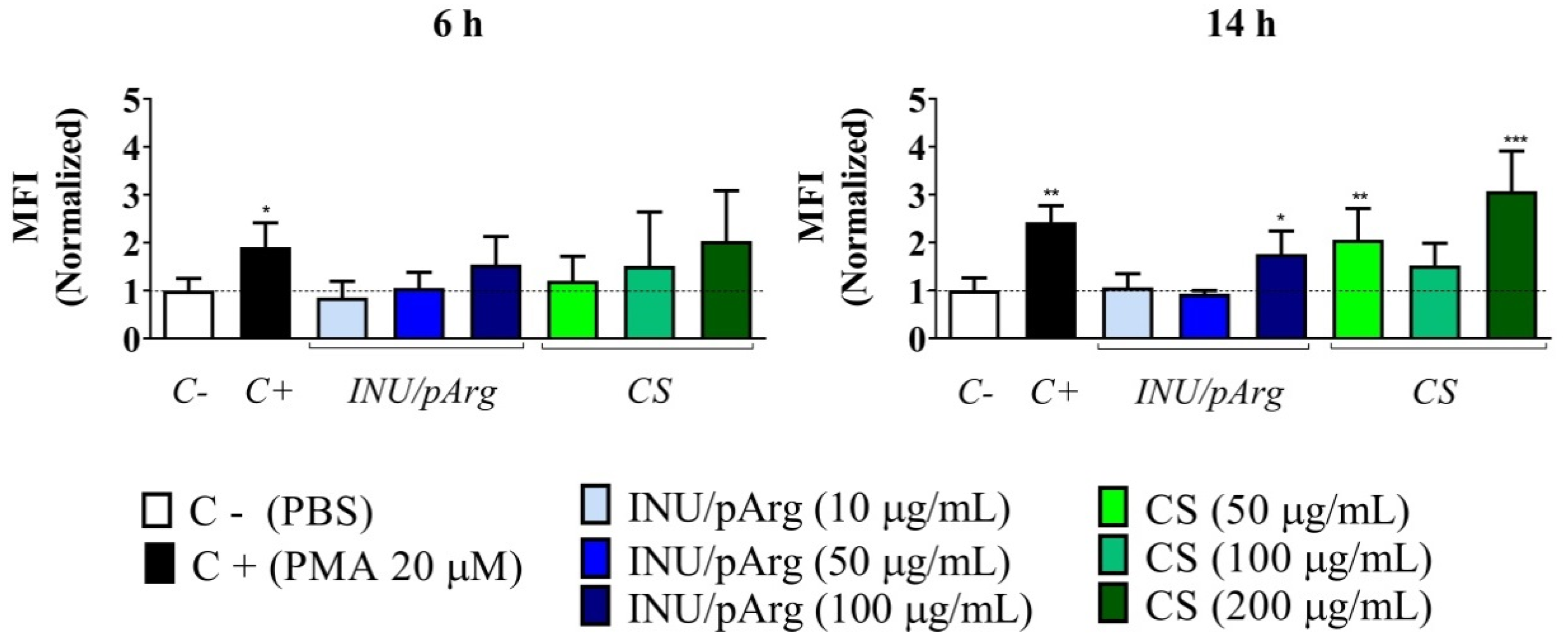
3.3. Reactive Oxygen Species’ Release in HL-60 Cells upon Exposure to the Nanocapsules
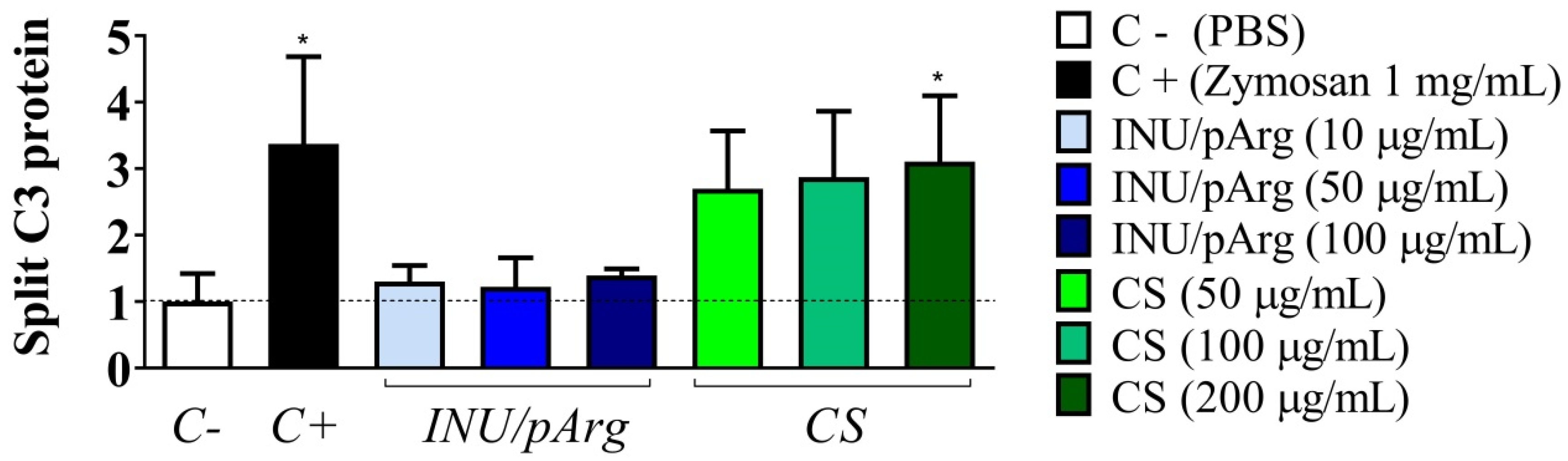
3.4. Complement Activation in Human Plasma Induced by the Polymeric Nanocapsules
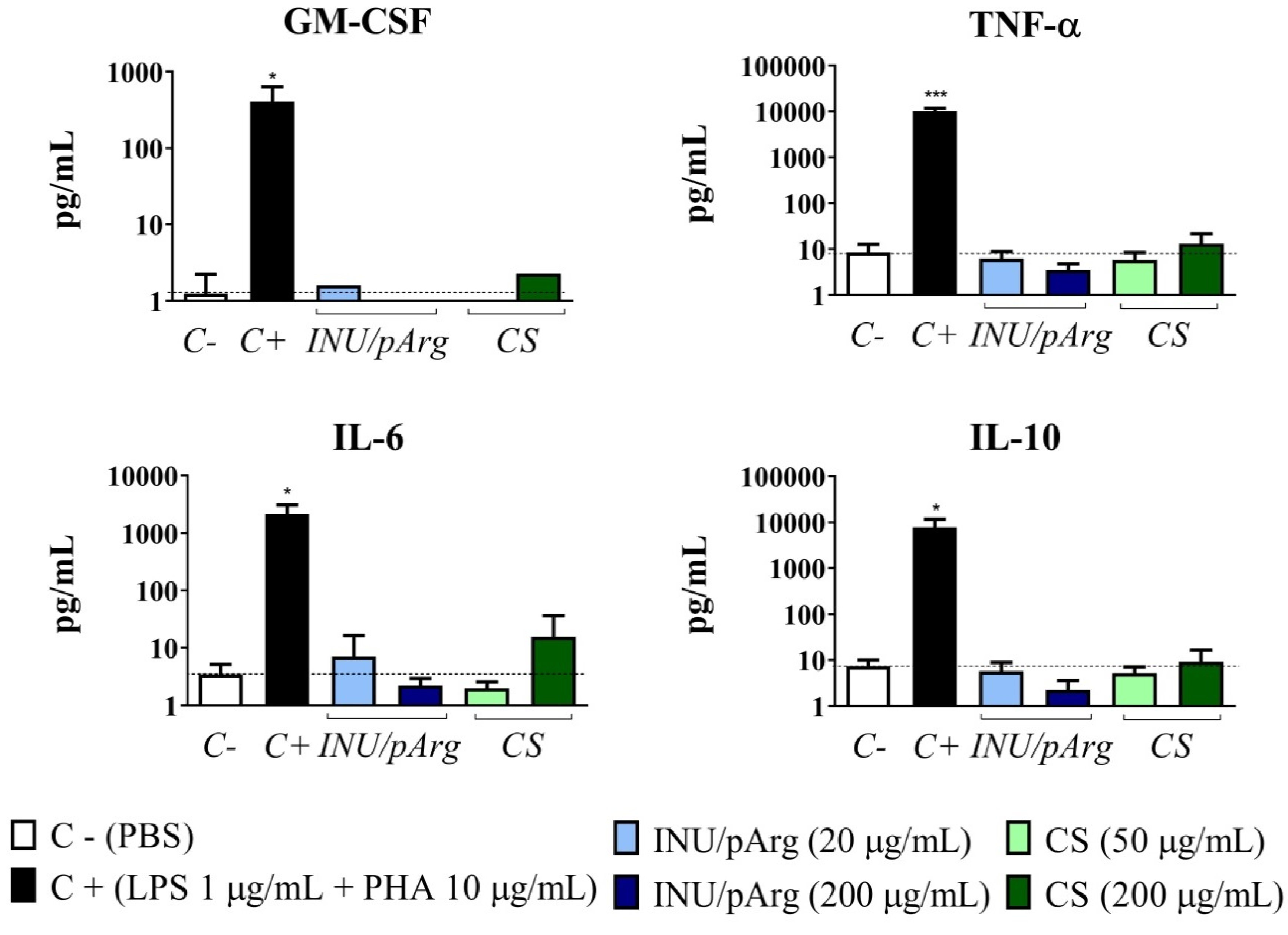
3.5. Cytokine Release Induced by the Polymeric Nanocapsules
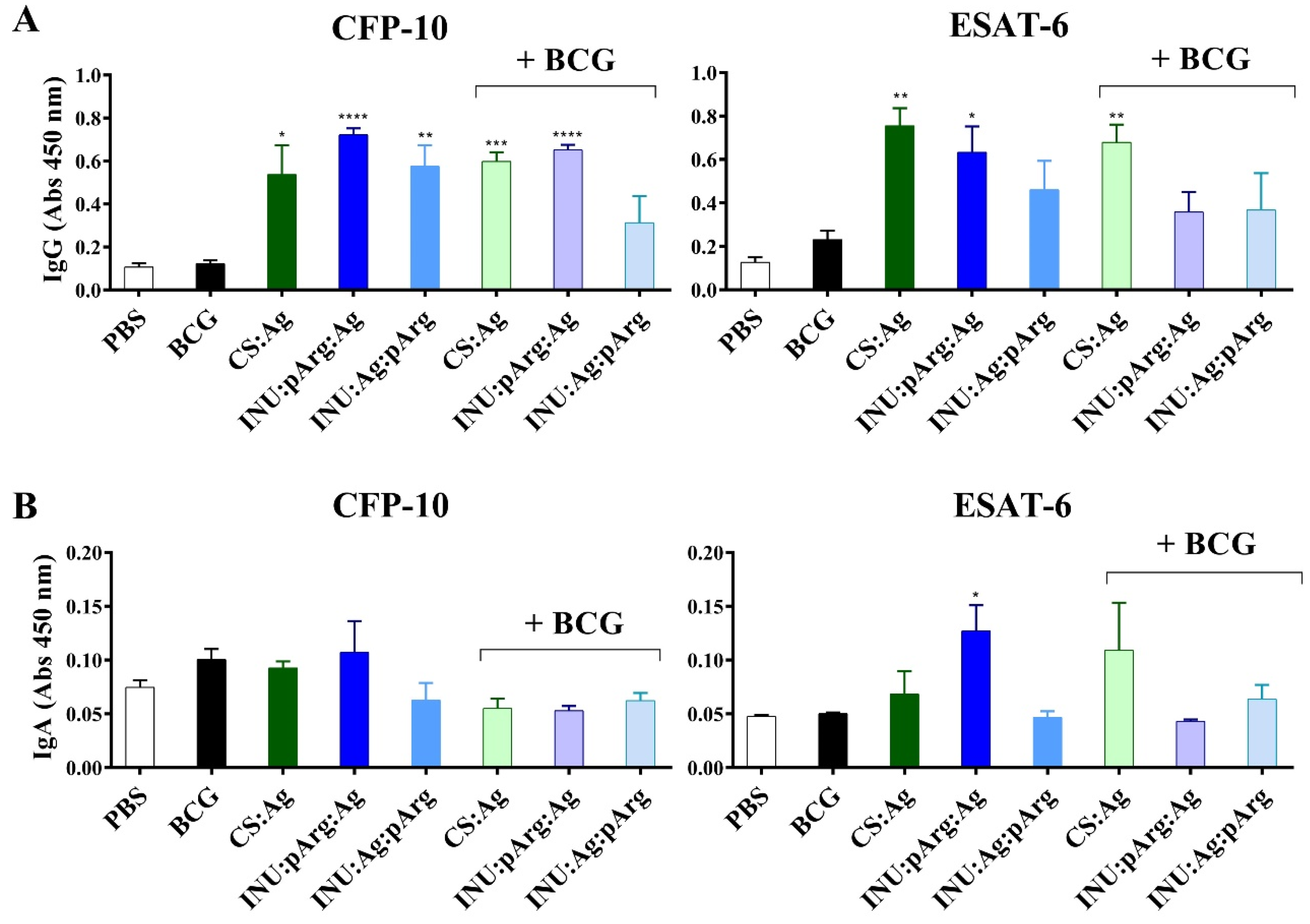
3.6. Systemic Immune Response against the ESAT-6/CFP-10 Fusion Protein after Intranasal Immunization with the Polymeric Nanocapsules
3.6.1. Characterization of Specific IgG and IgA Antibodies against ESAT-6/CFP-10
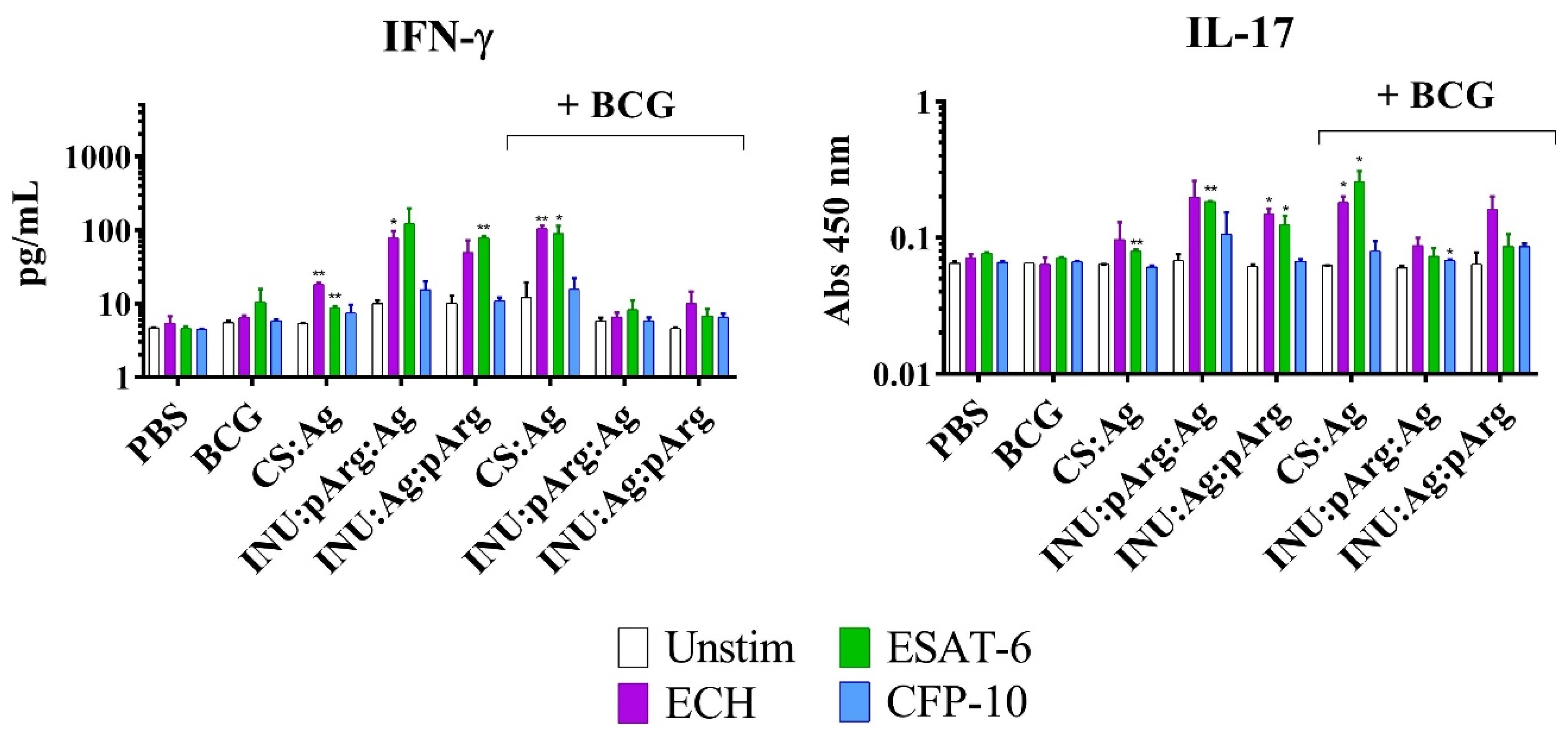
3.6.2. Characterization of the Cellular Immune Response in Splenocytes from Immunized Animals
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gengenbacher, M.; Kaufmann, S.H.E. Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 2012, 36, 514–532. [Google Scholar] [CrossRef]
- Fine, P.E.M. Bcg: The challenge continues. Scand. J. Infect. Dis. 2001, 33, 58–60. [Google Scholar] [CrossRef]
- Vartak, A.; Sucheck, S.J. Recent advances in subunit vaccine carriers. Vaccines 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Bowen, W.S.; Svrivastava, A.K.; Batra, L.; Barsoumian, H.; Shirwan, H. Current challenges for cancer vaccine adjuvant development. Expert Rev. Vaccines 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Peleteiro, M.; Presas, E.; Gonzalez-Aramundiz, J.V.; Sanchez-Correa, B.; Simon-Vazquez, R.; Csaba, N.; Alonso, M.J.; Gonzalez-Fernandez, A. Polymeric nanocapsules for vaccine delivery: Influence of the polymeric shell on the interaction with the immune system. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Dacoba, T.G.; Olivera, A.; Torres, D.; Crecente-Campo, J.; Alonso, M.J. Modulating the immune system through nanotechnology. Semin. Immunol. 2017, 34, 78–102. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Vicente, S.; Peleteiro, M.; Díaz-Freitas, B.; Sanchez, A.; González-Fernández, Á.; Alonso, M.J. Co-delivery of viral proteins and a tlr7 agonist from polysaccharide nanocapsules: A needle-free vaccination strategy. J. Control. Release 2013, 172, 773–781. [Google Scholar] [CrossRef]
- González-Aramundiz, J.V.; Peleteiro, M.; González-Fernández, Á.; Alonso, M.J.; Csaba, N.S. Protamine nanocapsules for the development of thermostable adjuvanted nanovaccines. Mol. Pharm. 2018, 15, 5653–5664. [Google Scholar] [CrossRef]
- Vicente, S.; Peleteiro, M.; Gonzalez-Aramundiz, J.V.; Díaz-Freitas, B.; Martínez-Pulgarín, S.; Neissa, J.I.; Escribano, J.M.; Sanchez, A.; González-Fernández, Á.; Alonso, M.J. Highly versatile immunostimulating nanocapsules for specific immune potentiation. Nanomedicine-Uk 2014, 9, 2273–2289. [Google Scholar] [CrossRef]
- Crecente-Campo, J.; Lorenzo-Abalde, S.; Mora, A.; Marzoa, J.; Csaba, N.; Blanco, J.; González-Fernández, Á.; Alonso, M.J. Bilayer polymeric nanocapsules: A formulation approach for a thermostable and adjuvanted e. Coli antigen vaccine. J. Control. Release 2018, 286, 20–32. [Google Scholar] [CrossRef] [PubMed]
- González-Aramundiz, J.V.; Presas, E.; Dalmau-Mena, I.; Martínez-Pulgarín, S.; Alonso, C.; Escribano, J.M.; Alonso, M.J.; Csaba, N.S. Rational design of protamine nanocapsules as antigen delivery carriers. J. Control. Release 2017, 245, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Vicente, S.; Diaz-Freitas, B.; Peleteiro, M.; Sanchez, A.; Pascual, D.W.; Gonzalez-Fernandez, A.; Alonso, M.J. A polymer/oil based nanovaccine as a single-dose immunization approach. PLoS ONE 2013, 8, e62500. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-rna nanoparticles generate protective immunity against lethal ebola, h1n1 influenza, and toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef]
- Boyaka, P.N. Inducing mucosal iga: A challenge for vaccine adjuvants and delivery systems. J. Immunol. 2017, 199, 9–16. [Google Scholar] [CrossRef]
- Prego, C.; Torres, D.; Alonso, M.J. Chitosan nanocapsules: A new carrier for nasal peptide delivery. J. Drug Deliv. Sci. Technol. 2006, 16, 331–337. [Google Scholar] [CrossRef]
- Crecente-Campo, J.; Alonso, M.J. Engineering, on-demand manufacturing, and scaling-up of polymeric nanocapsules. Bioeng. Transl. Med. 2019, 4, 38–50. [Google Scholar] [CrossRef]
- Prego, C.; Paolicelli, P.; Díaz, B.; Vicente, S.; Sánchez, A.; González-Fernández, Á.; Alonso, M.J. Chitosan-based nanoparticles for improving immunization against hepatitis b infection. Vaccine 2010, 28, 2607–2614. [Google Scholar] [CrossRef]
- Xu, W.; Shen, Y.; Jiang, Z.; Wang, Y.; Chu, Y.; Xiong, S. Intranasal delivery of chitosan–DNA vaccine generates mucosal siga and anti-cvb3 protection. Vaccine 2004, 22, 3603–3612. [Google Scholar] [CrossRef]
- Amidi, M.; Romeijn, S.G.; Verhoef, J.C.; Junginger, H.E.; Bungener, L.; Huckriede, A.; Crommelin, D.J.A.; Jiskoot, W. N-trimethyl chitosan (tmc) nanoparticles loaded with influenza subunit antigen for intranasal vaccination: Biological properties and immunogenicity in a mouse model. Vaccine 2007, 25, 144–153. [Google Scholar] [CrossRef]
- Yuhong, J.; Man, L.; Zhirong, Z.; Tao, G.; Xun, S. Cell-penetrating peptides as delivery enhancers for vaccine. Curr. Pharm. Biotechnol. 2014, 15, 256–266. [Google Scholar]
- Guo, S.; Xue, R.; Li, Y.; Wang, S.M.; Ren, L.; Xu, J.J. The cfp10/esat6 complex of mycobacterium tuberculosis may function as a regulator of macrophage cell death at different stages of tuberculosis infection. Med. Hypotheses 2012, 78, 389–392. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Hickey, A.J. Plga microparticles in respirable sizes enhance an in vitro t cell response to recombinant mycobacterium tuberculosis antigen tb10.4-ag85b. Pharm. Res. 2010, 27, 350–360. [Google Scholar] [CrossRef]
- Cordeiro, A.S.; Crecente-Campo, J.; Bouzo, B.L.; González, S.F.; de la Fuente, M.; Alonso, M.J. Engineering polymeric nanocapsules for an efficient drainage and biodistribution in the lymphatic system. J. Drug Target. 2019, 27, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. Ros function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Kotsias, F.; Hoffmann, E.; Amigorena, S.; Savina, A. Reactive oxygen species production in the phagosome: Impact on antigen presentation in dendritic cells. Antioxid. Redox. Signal. 2012, 18, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chen, W.; Huang, L. Reactive oxygen species play a central role in the activity of cationic liposome based cancer vaccine. J. Control. Release 2008, 130, 22–28. [Google Scholar] [CrossRef]
- Teufelhofer, O.; Weiss, R.M.; Parzefall, W.; Schulte-Hermann, R.; Micksche, M.; Berger, W.; Elbling, L. Promyelocytic hl60 cells express nadph oxidase and are excellent targets in a rapid spectrophotometric microplate assay for extracellular superoxide. Toxicol. Sci. 2003, 76, 376–383. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Sureda, A.; Mestre, A.; Tur, J.A.; Pons, A. The double edge of reactive oxygen species as damaging and signaling molecules in hl60 cell culture. Cell. Physiol. Biochem. 2010, 25, 241–252. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yin, Y.; Wang, L.; Zhang, W.; Chen, X.; Yang, X.; Xu, J.; Ma, G. Engineering biomaterial-associated complement activation to improve vaccine efficacy. Biomacromolecules 2013, 14, 3321–3328. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Carrillo, J.L.; Contreras-Cordero, J.F.; Gutiérrez-Coronado, O.; Villalobos-Gutiérrez, P.T.; Ramos-Gracia, L.G.; Hernández-Reyes, V.E. Cytokine profiling plays a crucial role in activating immune system to clear infectious pathogens. In Immune Response Activation and Immunomodulation, 1st ed.; Tyagi, R., Singh Bisen, P., Eds.; IntechOpen: London, UK, 2019; pp. 1–30. [Google Scholar]
- Iezzi, G.; Scotet, E.; Scheidegger, D.; Lanzavecchia, A. The interplay between the duration of tcr and cytokine signaling determines t cell polarization. Eur. J. Immunol. 1999, 29, 4092–4101. [Google Scholar] [CrossRef]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.; Tato, C.M.; Siegel, R. 10—Cytokines and cytokine receptors. In Clinical Immunology, 3rd ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Mosby: Edinburgh, UK, 2008; pp. 139–171. [Google Scholar]
- Stanley, M.A. Imiquimod and the imidazoquinolones: Mechanism of action and therapeutic potential. Clin. Exp. Dermatol. 2002, 27, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Olmsted, S.S.; Padgett, J.L.; Yudin, A.I.; Whaley, K.J.; Moench, T.R.; Cone, R.A. Diffusion of macromolecules and virus-like particles in human cervical mucus. Biophys. J. 2001, 81, 1930–1937. [Google Scholar] [CrossRef]
- Desai, M.P.; Labhasetwar, V.; Amidon, G.L.; Levy, R.J. Gastrointestinal uptake of biodegradable microparticles: Effect of particle size. Pharm. Res. 1996, 13, 1838–1845. [Google Scholar] [CrossRef]
- Shakweh, M.; Ponchel, G.; Fattal, E. Particle uptake by peyer’s patches: A pathway for drug and vaccine delivery. Expert Opin. Drug Deliv. 2004, 1, 141–163. [Google Scholar] [CrossRef]
- Oyewumi, M.O.; Kumar, A.; Cui, Z. Nano-microparticles as immune adjuvants: Correlating particle sizes and the resultant immune responses. Expert Rev. Vaccines 2010, 9, 1095–1107. [Google Scholar] [CrossRef]
- Vila, A.; Gill, H.; McCallion, O.; Alonso, M.J. Transport of pla-peg particles across the nasal mucosa: Effect of particle size and peg coating density. J. Control. Release 2004, 98, 231–244. [Google Scholar] [CrossRef]
- Bernasconi, V.; Norling, K.; Bally, M.; Höök, F.; Lycke, N.Y. Mucosal vaccine development based on liposome technology. J. Immunol. Res. 2016, 2016, 5482087. [Google Scholar] [CrossRef] [PubMed]
- Stano, A.; Nembrini, C.; Swartz, M.A.; Hubbell, J.A.; Simeoni, E. Nanoparticle size influences the magnitude and quality of mucosal immune responses after intranasal immunization. Vaccine 2012, 30, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Tobío, M.; Gref, R.; Sánchez, A.; Langer, R.; Alonso, M.J. Stealth pla-peg nanoparticles as protein carriers for nasal administration. Pharm. Res. 1998, 15, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.K.; O’Hanlon, D.E.; Harrold, S.; Man, S.T.; Wang, Y.-Y.; Cone, R.; Hanes, J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc. Natl. Acad. Sci. USA 2007, 104, 1482–1487. [Google Scholar] [CrossRef]
- Lai, S.K.; Wang, Y.-Y.; Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef]
- Foged, C.; Sundblad, A.; Hovgaard, L. Targeting vaccines to dendritic cells. Pharm. Res. 2002, 19, 229–238. [Google Scholar] [CrossRef]
- Fromen, C.A.; Robbins, G.R.; Shen, T.W.; Kai, M.P.; Ting, J.P.Y.; DeSimone, J.M. Controlled analysis of nanoparticle charge on mucosal and systemic antibody responses following pulmonary immunization. Proc. Natl. Acad. Sci. USA 2015, 112, 488–493. [Google Scholar] [CrossRef]
- Namvarpour, M.; Tebianian, M.; Mansouri, R.; Ebrahimi, S.M.; Kashkooli, S. Comparison of different immunization routes on the immune responses induced by mycobacterium tuberculosis esat-6/cfp-10 recombinant protein. Biologicals 2019, 59, 6–11. [Google Scholar] [CrossRef]
- Lerm, M.; Netea, M.G. Trained immunity: A new avenue for tuberculosis vaccine development. J. Intern. Med. 2016, 279, 337–346. [Google Scholar] [CrossRef]
- Crecente-Campo, J.; Guerra-Varela, J.; Peleteiro, M.; Gutiérrez-Lovera, C.; Fernández-Mariño, I.; Diéguez-Docampo, A.; González-Fernández, Á.; Sánchez, L.; Alonso, M.J. The size and composition of polymeric nanocapsules dictate their interaction with macrophages and biodistribution in zebrafish. J. Control. Release 2019, 308, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Crecente-Campo, J.; Virgilio, T.; Morone, D.; Calviño-Sampedro, C.; Fernández-Mariño, I.; Olivera, A.; Varela-Calvino, R.; González, S.F.; Alonso, M.J. Design of polymeric nanocapsules to improve their lympho-targeting capacity. Nanomedicine-Uk 2019, 14, 3013–3033. [Google Scholar] [CrossRef]
- Temchura, V.V.; Kozlova, D.; Sokolova, V.; Überla, K.; Epple, M. Targeting and activation of antigen-specific b-cells by calcium phosphate nanoparticles loaded with protein antigen. Biomaterials 2014, 35, 6098–6105. [Google Scholar] [CrossRef]
- Moon, J.J.; Suh, H.; Li, A.V.; Ockenhouse, C.F.; Yadava, A.; Irvine, D.J. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand tfh cells and promote germinal center induction. Proc. Natl. Acad. Sci. USA 2012, 109, 1080–1085. [Google Scholar] [CrossRef] [PubMed]
- Neutra, M.R.; Kozlowski, P.A. Mucosal vaccines: The promise and the challenge. Nat. Rev. Immunol. 2006, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late developmental plasticity in the t helper 17 lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef]
- Nikitina, I.Y.; Panteleev, A.V.; Kosmiadi, G.A.; Serdyuk, Y.V.; Nenasheva, T.A.; Nikolaev, A.A.; Gorelova, L.A.; Radaeva, T.V.; Kiseleva, Y.Y.; Bozhenko, V.K.; et al. Th1, th17, and th1th17 lymphocytes during tuberculosis: Th1 lymphocytes predominate and appear as low-differentiated cxcr3+ccr6+ cells in the blood and highly differentiated cxcr3+/−ccr6− cells in the lungs. J. Immunol. 2018, 200, 2090–2103. [Google Scholar] [CrossRef]
- Ong, E.; He, Y.; Yang, Z. Epitope promiscuity and population coverage of mycobacterium tuberculosis protein antigens in current subunit vaccines under development. Infect. Genet. Evol. 2020, 80, 104186. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Composition | CS NCs | INU/pArg NCs |
|---|---|---|
| Oil | Miglyol/linoleic acid | Miglyol/linoleic acid/glycerin |
| Adjuvant | Imiquimod | |
| Polymer | Chitosan | Modified inulin |
| Surfactant | 18:0 PE-PEG1000 | |
| Co-surfactant | Sodium cholate | Sodium glycocholate |
| Second polymeric shell | - | Polyarginine |
| Antigen disposition | Surface | Surface/Polymer bilayer |
| Nanocapsules | Size (nm) | Z (mV) | IMQ %EE | %AA |
|---|---|---|---|---|
| CS | 145 ± 14 | +28 ± 4.5 | 60 ± 4 | n.a. |
| CS:Ag | 152 ± 15 | +8 ± 10 | n.a. | 60 ± 6 |
| INU | 158 ± 7 | −37 ± 4 | 69 ± 6 | n.a. |
| INU:Ag | 136 ± 7 | −36 ± 6 | - | n.d. |
| INU/pArg | 158 ± 22 | +20 ± 1 | - | n.a. |
| INU:Ag:pArg | 151 ± 11 | +27 ± 8 | - | 55 ± 5 |
| INU:pArg:Ag | 158 ± 12 | +31 ± 7 | - | 30 ± 10 |
| Cell Line | IC50 (µg/mL) | |||
|---|---|---|---|---|
| CS | INU/pArg | |||
| NCs | IMQ | NCs | IMQ | |
| RAW 264.7 | 455 | 2.96 | 117 | 2.05 |
| THP-1 | 810 | 5.26 | 192 | 3.36 |
| NCI H460 | 226 | 1.47 | 57 | 1.00 |
| A549 | 197 | 1.28 | 31 | 0.54 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diego-González, L.; Crecente-Campo, J.; Paul, M.J.; Singh, M.; Reljic, R.; Alonso, M.J.; González-Fernández, Á.; Simón-Vázquez, R. Design of Polymeric Nanocapsules for Intranasal Vaccination against Mycobacterium Tuberculosis: Influence of the Polymeric Shell and Antigen Positioning. Pharmaceutics 2020, 12, 489. https://doi.org/10.3390/pharmaceutics12060489
Diego-González L, Crecente-Campo J, Paul MJ, Singh M, Reljic R, Alonso MJ, González-Fernández Á, Simón-Vázquez R. Design of Polymeric Nanocapsules for Intranasal Vaccination against Mycobacterium Tuberculosis: Influence of the Polymeric Shell and Antigen Positioning. Pharmaceutics. 2020; 12(6):489. https://doi.org/10.3390/pharmaceutics12060489
Chicago/Turabian StyleDiego-González, Lara, José Crecente-Campo, Matthew John Paul, Mahavir Singh, Rajko Reljic, María José Alonso, África González-Fernández, and Rosana Simón-Vázquez. 2020. "Design of Polymeric Nanocapsules for Intranasal Vaccination against Mycobacterium Tuberculosis: Influence of the Polymeric Shell and Antigen Positioning" Pharmaceutics 12, no. 6: 489. https://doi.org/10.3390/pharmaceutics12060489
APA StyleDiego-González, L., Crecente-Campo, J., Paul, M. J., Singh, M., Reljic, R., Alonso, M. J., González-Fernández, Á., & Simón-Vázquez, R. (2020). Design of Polymeric Nanocapsules for Intranasal Vaccination against Mycobacterium Tuberculosis: Influence of the Polymeric Shell and Antigen Positioning. Pharmaceutics, 12(6), 489. https://doi.org/10.3390/pharmaceutics12060489