Predictive Value of Precision-Cut Kidney Slices as an Ex Vivo Screening Platform for Therapeutics in Human Renal Fibrosis

 , , , , ,
, , , , , 
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Chemicals
2.3. Human Material
2.4. Experimental Animals and Surgical Procedures
2.5. Preparation, Incubation, and Treatment of Precision-Cut Kidney Slices (PCKS)
2.6. PCKS Viability
2.7. PCKS RNA Isolation and RT-qPCR
2.8. PCKS Histology and Immunohistochemistry
2.9. Pro-collagen Iα1 ELISA Measurement in Human PCKS
2.10. Cell Culture, Macromolecular Crowding and Treatments
2.11. RNA Isolation and RT-qPCR (HRFs)
2.12. Immunocytochemistry (HRFs)
2.13. Statistical Analysis
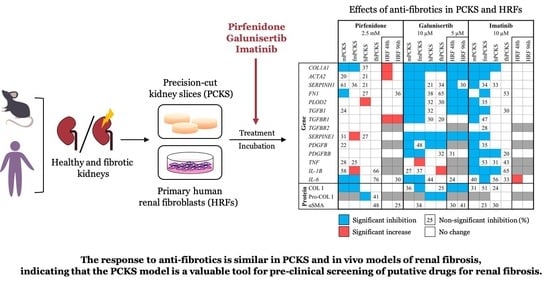
3. Results
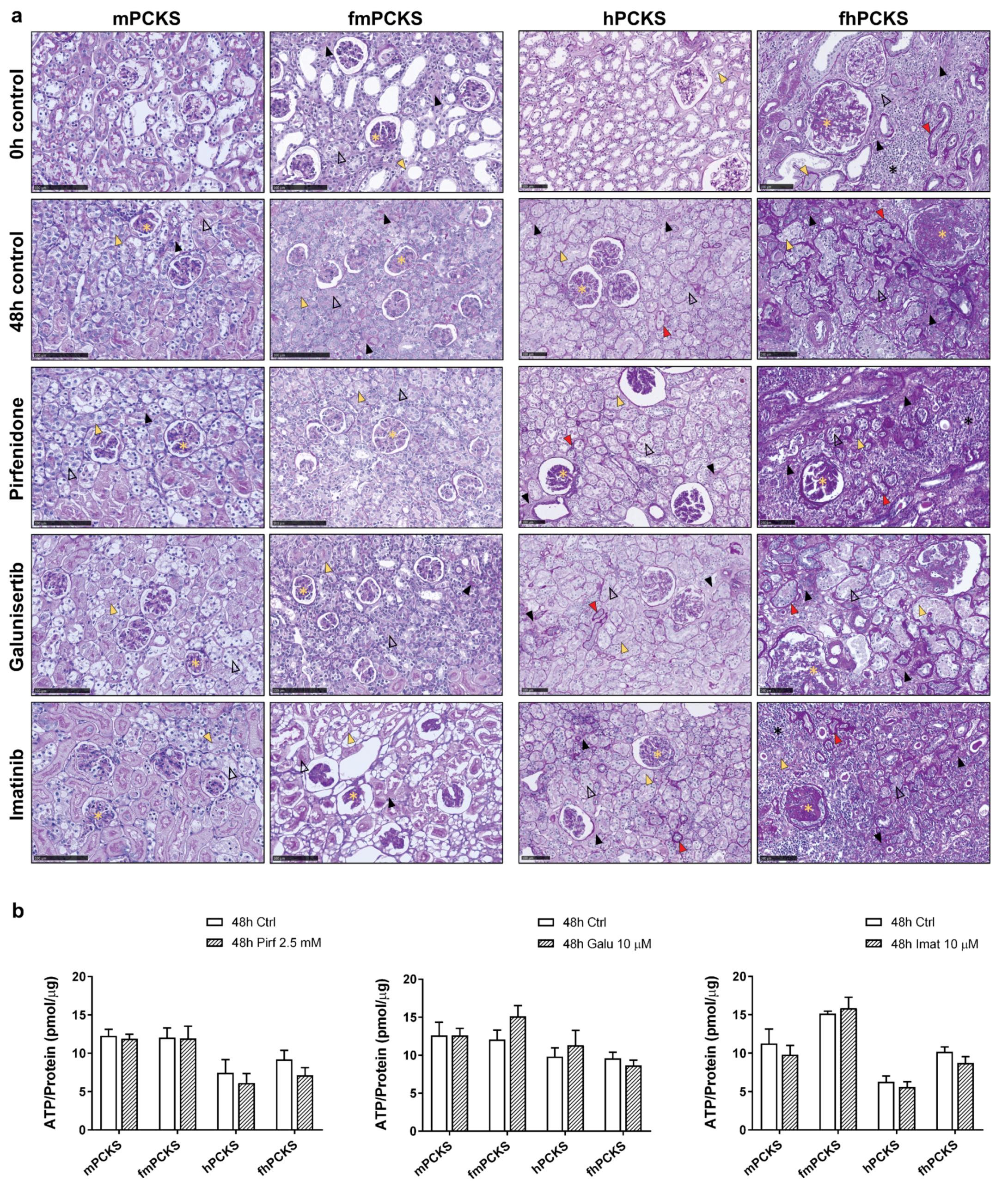
3.1. Tissue Viability in Response to Ex Vivo Culture and Pharmacological Intervention
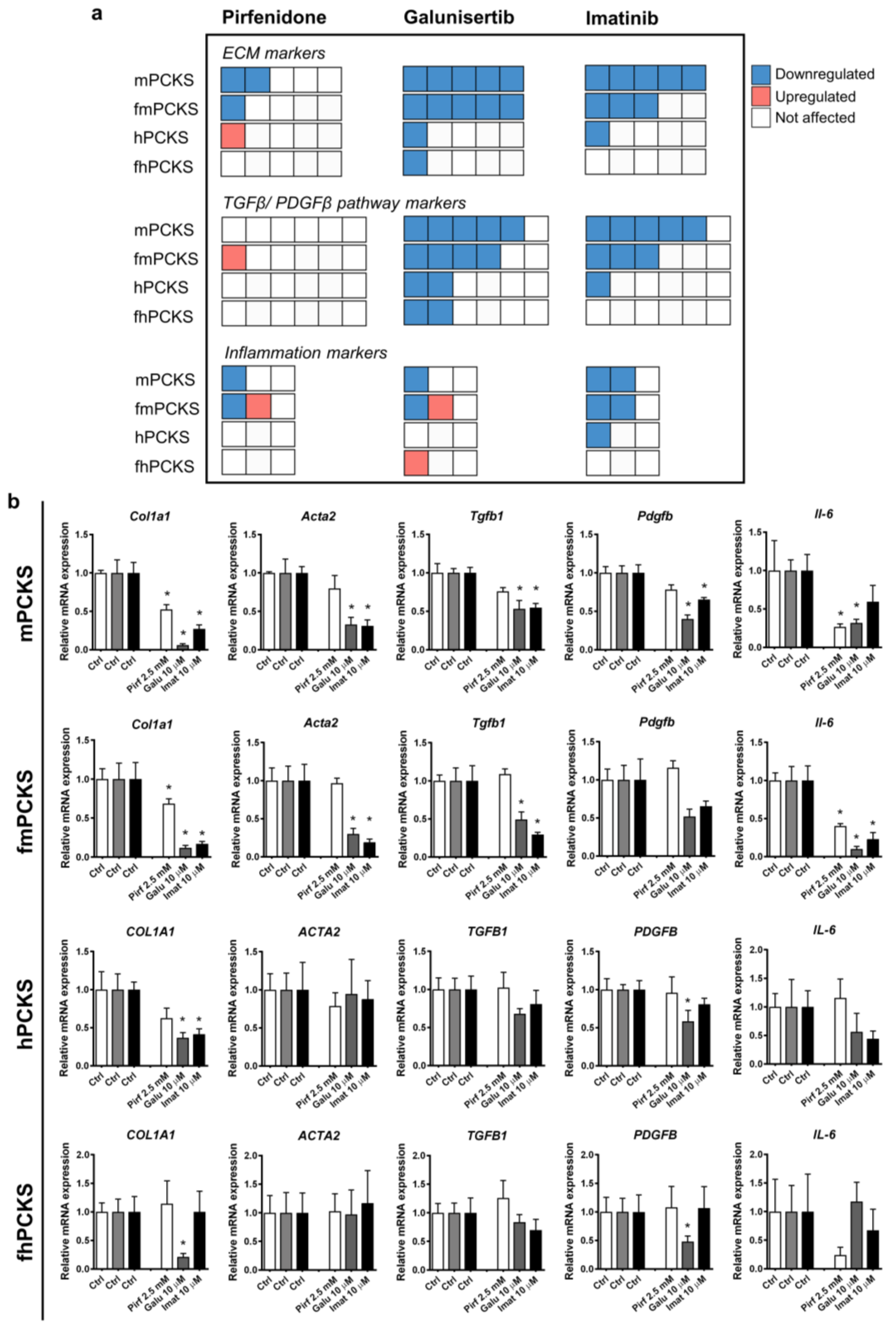
3.2. Culture-Driven Inflammatory and Fibrogenic State of PCKS
3.3. Ex Vivo Effects of Pirfenidone, Galunisertib and Imatinib in PCKS
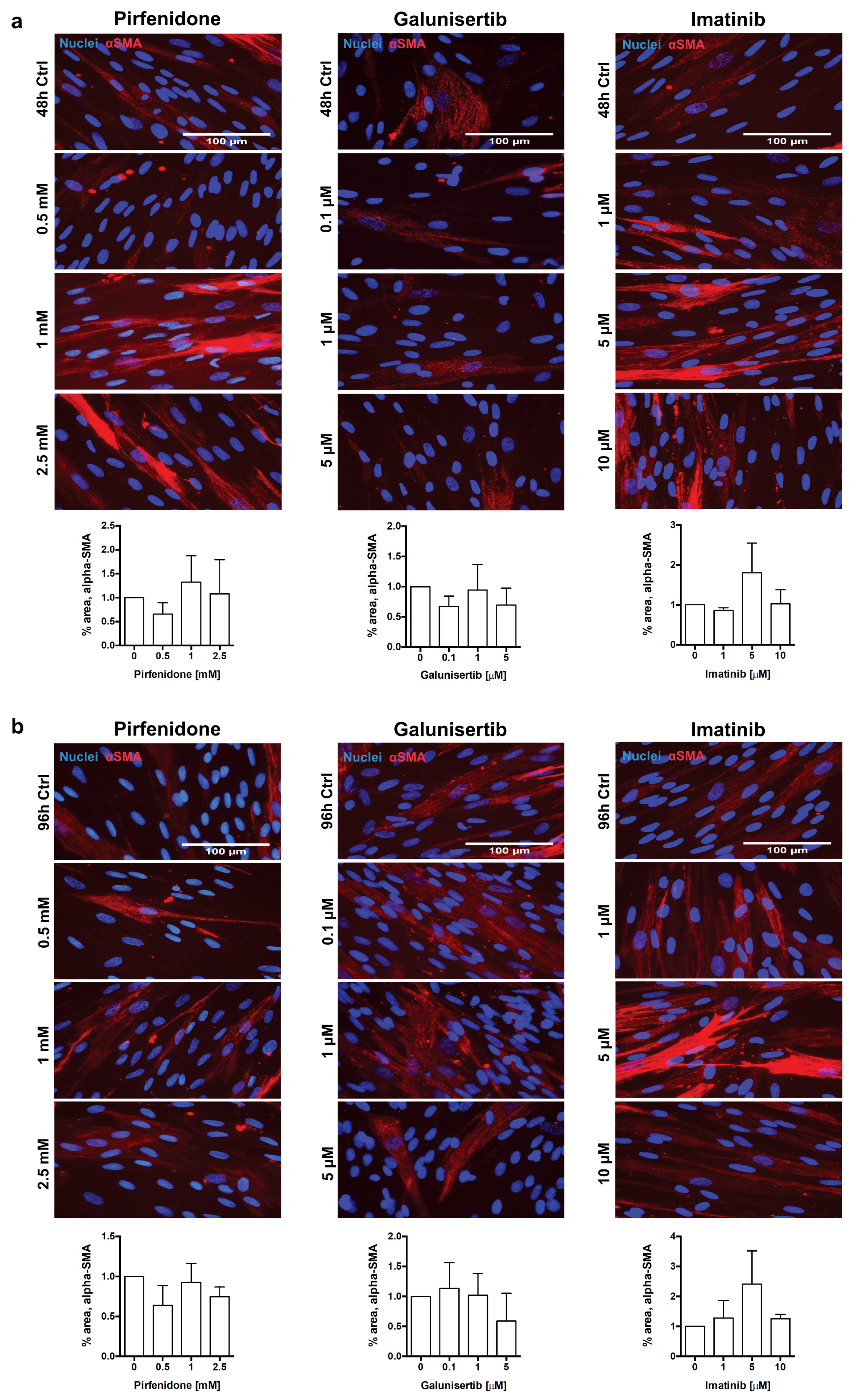
3.4. In Vitro Effects of Pirfenidone, Galunisertib and Imatinib Treatment in Primary Human Renal Fibroblasts
4. Discussion
4.1. PCKS Injurious Responses to the Culturing
4.2. PCKS Responses to Pharmacological Treatment in Perspective
4.2.1. Pirfenidone
4.2.2. Imatinib
4.2.3. Galunisertib
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, H.-C.; Zuo, Y.; Fogo, A.B. Models of chronic kidney disease. Drug Discov. Today Dis. Model 2010, 7, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Schwartz, W.B.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Schanstra, J.P.; Zürbig, P.; Alkhalaf, A.; Argiles, A.; Bakker, S.J.L.; Beige, J.; Bilo, H.J.G.; Chatzikyrkou, C.; Dakna, M.; Dawson, J.; et al. Diagnosis and prediction of CKD progression by assessment of urinary peptides. J. Am. Soc. Nephrol. 2015, 26, 1999–2010. [Google Scholar] [CrossRef]
- Breyer, M.D.; Susztak, K. The next generation of therapeutics for chronic kidney disease. Nat. Rev. Drug Discov. 2016, 15, 568–588. [Google Scholar] [CrossRef] [PubMed]
- Boor, P.; Ostendorf, T.; Floege, J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets. Nat. Rev. Nephrol. 2010, 6, 643–656. [Google Scholar] [CrossRef]
- Fligny, C.; Duffield, J.S. Activation of pericytes. Curr. Opin. Rheumatol. 2013, 25, 78–86. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Lin, S.-L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate Tracing Reveals the Pericyte and Not Epithelial Origin of Myofibroblasts in Kidney Fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Hu, H.-H.; Chen, D.-Q.; Wang, Y.-N.; Feng, Y.-L.; Cao, G.; Vaziri, N.D.; Zhao, Y.-Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef]
- Ostendorf, T.; Eitner, F.; Floege, J. The PDGF family in renal fibrosis. Pediatr. Nephrol. 2012, 27, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Floege, J.; Boor, P. PDGF in organ fibrosis. Mol. Asp. Med. 2018, 62, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Györfi, A.H.; Matei, A.-E.; Distler, J.H.W. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef]
- Kok, H.M.; Falke, L.L.; Goldschmeding, R.; Nguyen, T.Q. Targeting CTGF, EGF and PDGF pathways to prevent progression of kidney disease. Nat. Rev. Nephrol. 2014, 10, 700–711. [Google Scholar] [CrossRef]
- Klinkhammer, B.M.; Goldschmeding, R.; Floege, J.; Boor, P. Treatment of Renal Fibrosis—Turning Challenges into Opportunities. Adv. Chronic Kidney Dis. 2017, 24, 117–129. [Google Scholar] [CrossRef]
- Poosti, F.; Pham, B.T.; Oosterhuis, D.; Poelstra, K.; van Goor, H.; Olinga, P.; Hillebrands, J.-L. Precision-cut kidney slices (PCKS) to study development of renal fibrosis and efficacy of drug targeting ex vivo. Dis. Models Mech. 2015, 8, 1227–1236. [Google Scholar] [CrossRef]
- Genovese, F.; Kàrpàti, Z.S.; Nielsen, S.H.; Karsdal, M.A. Precision-Cut Kidney Slices as a Tool to Understand the Dynamics of Extracellular Matrix Remodeling in Renal Fibrosis. Biomark. Insights 2016, 11, 77–84. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Q.; Xiao, J.; Lei, J.; Liu, Y.; Xu, H.; Hong, Z. Molecular validation of the precision-cut kidney slice (PCKS) model of renal fibrosis through assessment of TGF-β1-induced Smad and p38/ERK signaling. Int. Immunopharmacol. 2016, 34, 32–36. [Google Scholar] [CrossRef]
- Stribos, E.G.D.; Hillebrands, J.-L.; Olinga, P.; Mutsaers, H.A.M. Renal fibrosis in precision-cut kidney slices. Eur. J. Pharmacol. 2016, 790, 57–61. [Google Scholar] [CrossRef]
- Jensen, M.S.; Mutsaers, H.A.M.; Tingskov, S.J.; Christensen, M.; Madsen, M.G.; Olinga, P.; Kwon, T.; Nørregaard, R. Activation of the prostaglandin E2 EP2 receptor attenuates renal fibrosis in unilateral ureteral obstructed mice and human kidney slices. Acta Physiol. 2019, 227, e13291. [Google Scholar] [CrossRef] [PubMed]
- Stribos, E.G.D.; Luangmonkong, T.; Leliveld, A.M.; de Jong, I.J.; van Son, W.J.; Hillebrands, J.-L.; Seelen, M.A.; van Goor, H.; Olinga, P.; Mutsaers, H.A.M. Precision-cut human kidney slices as a model to elucidate the process of renal fibrosis. Transl. Res. 2016, 170, 8–16.e1. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar]
- Luangmonkong, T.; Suriguga, S.; Bigaeva, E.; Boersema, M.; Oosterhuis, D.; de Jong, K.P.; Schuppan, D.; Mutsaers, H.A.M.; Olinga, P. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharmacol. 2017, 174, 3107–3117. [Google Scholar] [CrossRef]
- Hammad, S.; Cavalcanti, E.; Werle, J.; Caruso, M.L.; Dropmann, A.; Ignazzi, A.; Ebert, M.P.; Dooley, S.; Giannelli, G. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch. Toxicol. 2018, 92, 2297–2309. [Google Scholar] [CrossRef]
- Masuda, A.; Nakamura, T.; Abe, M.; Iwamoto, H.; Sakaue, T.; Tanaka, T.; Suzuki, H.; Koga, H.; Torimura, T. Promotion of liver regeneration and anti-fibrotic effects of the TGF-β receptor kinase inhibitor galunisertib in CCl4-treated mice. Int. J. Mol. Med. 2020, 46, 427–438. [Google Scholar] [CrossRef]
- Buchdunger, E.; O’Reilly, T.; Wood, J. Pharmacology of imatinib (STI571). Eur. J. Cancer 2002, 38 (Suppl. 5), S28–S36. [Google Scholar] [CrossRef]
- Yoshiji, H.; Noguchi, R.; Kuriyama, S.; Ikenaka, Y.; Yoshii, J.; Yanase, K.; Namisaki, T.; Kitade, M.; Masaki, T.; Fukui, H.; et al. Imatinib mesylate (STI-571) attenuates liver fibrosis development in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, 907–913. [Google Scholar] [CrossRef]
- Yoshiji, H.; Kuriyama, S.; Noguchi, R.; Ikenaka, Y.; Yoshii, J.; Yanase, K.; Namisaki, T.; Kitade, M.; Yamazaki, M.; Asada, K.; et al. Amelioration of liver fibrogenesis by dual inhibition of PDGF and TGF-β with a combination of imatinib mesylate and ACE inhibitor in rats. Int. J. Mol. Med. 2006, 17, 899–904. [Google Scholar] [CrossRef]
- Boor, P.; Sebekova, K.; Ostendorf, T.; Floege, J. Treatment targets in renal fibrosis. Nephrol. Dial. Transplant. 2007, 22, 3391–3407. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Eitner, F.; Alpers, C.E. A New Look at Platelet-Derived Growth Factor in Renal Disease. J. Am. Soc. Nephrol. 2008, 19, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Peng, Y.X.; Wang, Z.B.; Fish, P.V.; Kaar, J.L.; Koepsel, R.R.; Russell, A.J.; Lareu, R.R.; Raghunath, M. The Scar-in-a-Jar: Studying potential antifibrotic compounds from the epigenetic to extracellular level in a single wellb. Br. J. Pharmacol. 2009, 158, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, I.A.M.; Olinga, P.; de Jager, M.H.; Merema, M.T.; de Kanter, R.; van de Kerkhof, E.G.; Groothuis, G.M.M. Preparation and incubation of precision-cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat. Protoc. 2010, 5, 1540–1551. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ruigrok, M.J.R.; Tomar, J.; Frijlink, H.W.; Melgert, B.N.; Hinrichs, W.L.J.; Olinga, P. The effects of oxygen concentration on cell death, anti-oxidant transcription, acute inflammation, and cell proliferation in precision-cut lung slices. Sci. Rep. 2019, 9, 16239. [Google Scholar] [CrossRef]
- Rashid, R.; Lim, N.S.J.; Chee, S.M.L.; Png, S.N.; Wohland, T.; Raghunath, M. Novel Use for Polyvinylpyrrolidone as a Macromolecular Crowder for Enhanced Extracellular Matrix Deposition and Cell Proliferation. Tissue Eng. Part C Methods 2014, 20, 994–1002. [Google Scholar] [CrossRef]
- Nakayama, S.; Mukae, H.; Sakamoto, N.; Kakugawa, T.; Yoshioka, S.; Soda, H.; Oku, H.; Urata, Y.; Kondo, T.; Kubota, H.; et al. Pirfenidone inhibits the expression of HSP47 in TGF-β1-stimulated human lung fibroblasts. Life Sci. 2008, 82, 210–217. [Google Scholar] [CrossRef]
- Molina-Molina, M.; Machahua-Huamani, C.; Vicens-Zygmunt, V.; Llatjós, R.; Escobar, I.; Sala-Llinas, E.; Luburich-Hernaiz, P.; Dorca, J.; Montes-Worboys, A. Anti-fibrotic effects of pirfenidone and rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm. Med. 2018, 18, 63–75. [Google Scholar] [CrossRef]
- Kim, Y.; Fiel, M.I.; Albanis, E.; Chou, H.I.; Zhang, W.; Khitrov, G.; Friedman, S.L. Anti-fibrotic activity and enhanced interleukin-6 production by hepatic stellate cells in response to imatinib mesylate. Liver Int. 2012, 32, 1008–1017. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ pathway with galunisertib, a TGFβRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Stribos, E.G.D.; Seelen, M.A.; van Goor, H.; Olinga, P.; Mutsaers, H.A.M. Murine Precision-Cut Kidney Slices as an ex vivo Model to Evaluate the Role of Transforming Growth Factor-β1 Signaling in the Onset of Renal Fibrosis. Front. Physiol. 2017, 8, 1026. [Google Scholar] [CrossRef] [PubMed]
- Sands, J.M. Translating Kidney Fibrosis: Role of the EP2 Receptor. Acta Physiol. 2019, 227, e13318. [Google Scholar] [CrossRef]
- Bigaeva, E.; Gore, E.; Simon, E.; Zwick, M.; Oldenburger, A.; de Jong, K.P.; Hofker, H.S.; Schlepütz, M.; Nicklin, P.; Boersema, M.; et al. Transcriptomic characterization of culture-associated changes in murine and human precision-cut tissue slices. Arch. Toxicol. 2019, 93, 3549–3583. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 2007, 117, 524–529. [Google Scholar] [CrossRef]
- Hinz, B. Myofibroblasts. Exp. Eye Res. 2015, 142, 56–70. [Google Scholar] [CrossRef]
- Sun, K.-H.; Chang, Y.; Reed, N.I.; Sheppard, D. α-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGFβ activation or collagen production across multiple models of organ fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L824–L836. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M. Renal Fibroblasts and Myofibroblasts in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2006, 17, 2992–2998. [Google Scholar] [CrossRef]
- Campanholle, G.; Ligresti, G.; Gharib, S.A.; Duffield, J.S. Cellular Mechanisms of Tissue Fibrosis. 3. Novel mechanisms of kidney fibrosis. Am. J. Physiol Cell Physiol. 2013, 304, C591–C603. [Google Scholar] [CrossRef]
- Iyer, S.N.; Wild, J.S.; Schiedt, M.J.; Hyde, D.M.; Margolin, S.B.; Giri, S.N. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J. Lab. Clin. Med. 1995, 125, 779–785. [Google Scholar]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of Pirfenidone on Procollagen Gene Expression at the Transcriptional Level in Bleomycin Hamster Model of Lung Fibrosis. J. Pharmacol. Exp. Ther. 1999, 289, 211–218. [Google Scholar] [PubMed]
- Shimizu, F.; Fukagawa, M.; Yamauchi, S.; Taniyama, M.; Komemushi, S.; Margolin, S.B.; Kurokawa, K. Pirfenidone prevents the progression of irreversible glomerular sclerotic lesions in rats. Nephrology 1997, 3, 315–322. [Google Scholar] [CrossRef]
- Leh, S.; Vaagnes, Ø.; Margolin, S.B.; Iversen, B.M.; Forslund, T. Pirfenidone and candesartan ameliorate morphological damage in mild chronic anti-GBM nephritis in rats. Nephrol. Dial. Transplant. 2005, 20, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Ni, H.-F.; Pan, M.-M.; Liu, H.; Xu, M.; Zhang, M.-H.; Liu, B.-C. Pirfenidone inhibits macrophage infiltration in 5/6 nephrectomized rats. Am. J. Physiol. Ren. Physiol. 2013, 304, 676–685. [Google Scholar] [CrossRef]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone Slows Renal Function Decline in Patients with Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef]
- Cho, M.E.; Kopp, J.B. Pirfenidone: An anti-fibrotic therapy for progressive kidney disease. Expert Opin. Investig. Drugs 2010, 19, 275–283. [Google Scholar] [CrossRef]
- Sharma, K.; Ix, J.H.; Mathew, A.V.; Cho, M.; Pflueger, A.; Dunn, S.R.; Francos, B.; Sharma, S.; Falkner, B.; McGowan, T.A.; et al. Pirfenidone for Diabetic Nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1144–1151. [Google Scholar] [CrossRef]
- Allinovi, M.; De Chiara, L.; Angelotti, M.L.; Becherucci, F.; Romagnani, P. Anti-fibrotic treatments: A review of clinical evidence. Matrix Biol. 2018, 68–69, 333–354. [Google Scholar] [CrossRef]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-Î2-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef]
- Takakura, K.; Tahara, A.; Sanagi, M.; Itoh, H.; Tomura, Y. Antifibrotic Effects of Pirfenidone in Rat Proximal Tubular Epithelial Cells. Ren. Fail. 2012, 34, 1309–1316. [Google Scholar] [CrossRef]
- Yuan, Q.; Wang, L.; Zhang, F.; Wang, R.; Fu, X.; Peng, Z.; Ning, W.; Hu, G.; Wang, Z.; Tao, L.; et al. Fluorofenidone suppresses epithelial-mesenchymal transition and the expression of connective tissue growth factor via inhibiting TGF-/Smads signaling in human proximal tubular epithelial cells. Pharmazie 2011, 66, 961–967. [Google Scholar] [PubMed]
- Miric, G.; Dallemagne, C.; Endre, Z.; Margolin, S.; Taylor, S.M.; Brown, L. Reversal of cardiac and renal fibrosis by pirfenidone and spironolactone in streptozotocin-diabetic rats. Br. J. Pharmacol. 2001, 133, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Pirfenidone Treatment Decreases Transforming Growth Factor-beta1 and Matrix Proteins and Ameliorates Fibrosis in Chronic Cyclosporine Nephrotoxicity. Am. J. Transplant. 2002, 2, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Brook, N.R.; Waller, J.R.; Bicknell, G.R.; Nicholson, M.L. The Novel Antifibrotic Agent Pirfenidone Attenuates the Profibrotic Environment Generated by Calcineurin Inhibitors in the Rat Salt-Depletion Model. Transplant. Proc. 2005, 37, 130–133. [Google Scholar] [CrossRef]
- RamachandraRao, S.P.; Zhu, Y.; Ravasi, T.; McGowan, T.A.; Toh, I.; Dunn, S.R.; Okada, S.; Shaw, M.A.; Sharma, K. Pirfenidone Is Renoprotective in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2009, 20, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Bao, L.; Kim, Y.J.; Cho, I.H.; Lee, C.H.; Hyun, B.H.; Margolin, S.; Park, Y.H. Pirfenidone Suppressed the Development of Glomerulosclerosis in the FGS/Kist mouse. J. Korean Med. Sci. 2003, 18, 527–533. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukagawa, M.; Kuroda, T.; Hata, S.; Iwasaki, Y.; Nemoto, M.; Shirai, K.; Yamauchi, S.; Margolin, S.B.; Shimizu, F.; et al. Pirfenidone prevents collagen accumulation in the remnant kidney in rats with partial nephrectomy. Kidney Int. Suppl. 1997, 63, S239–S243. [Google Scholar]
- Takakuta, K.; Fujimori, A.; Chikanishi, T.; Tanokura, A.; Iwatsuki, Y.; Yamamoto, M.; Nakajima, H.; Okada, M.; Itoh, H. Renoprotective properties of pirfenidone in subtotally nephrectomized rats. Eur. J. Pharmacol. 2010, 629, 118–124. [Google Scholar] [CrossRef]
- Shimizu, T.; Kuroda, T.; Hata, S.; Fukagawa, M.; Margolin, S.B.; Kurokawa, K. Pirfenidone improves renal function and fibrosis in the post-obstructed kidney. Kidney Int. 1998, 54, 99–109. [Google Scholar] [CrossRef]
- Ostendorf, T.; Boor, P.; van Roeyen, C.R.C.; Floege, J. Platelet-derived growth factors (PDGFs) in glomerular and tubulointerstitial fibrosis. Kidney Int. Suppl. 2014, 4, 65–69. [Google Scholar] [CrossRef]
- Bigaeva, E.; Stribos, E.G.D.; Mutsaers, H.A.M.; Piersma, B.; Leliveld, A.M.; de Jong, I.J.; Bank, R.A.; Seelen, M.A.; van Goor, H.; Wollin, L.; et al. Inhibition of tyrosine kinase receptor signaling attenuates fibrogenesis in an ex vivo model of human renal fibrosis. Am. J. Physiol. Physiol. 2020, 318, F117–F134. [Google Scholar] [CrossRef] [PubMed]
- Lassila, M.; Jandeleit-Dahm, K.; Seah, K.K.; Smith, C.M.; Calkin, A.C.; Allen, T.J.; Cooper, M.E. Imatinib Attenuates Diabetic Nephropathy in Apolipoprotein E-Knockout Mice. J. Am. Soc. Nephrol. 2005, 16, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Iyoda, M.; Shibata, T.; Wada, Y.; Kuno, Y.; Shindo-Hirai, Y.; Matsumoto, K.; Akizawa, T. Long-and short-term treatment with imatinib attenuates the development of chronic kidney disease in experimental anti-glomerular basement membrane nephritis. Nephrol. Dial. Transpl. 2013, 28, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Dewar, A.L.; Cambareri, A.C.; Zannettino, A.C.W.; Miller, B.L.; Doherty, K.V.; Hughes, T.P.; Lyons, A.B. Macrophage colony-stimulating factor receptor c-fms is a novel target of imatinib. Blood 2005, 105, 3127–3132. [Google Scholar] [CrossRef] [PubMed]
- Iyoda, M.; Shibata, T.; Kawaguchi, M.; Yamaoka, T.; Akizawa, T. Preventive and therapeutic effects of imatinib in Wistar-Kyoto rats with anti-glomerular basement membrane glomerulonephritis. Kidney Int. 2009, 75, 1060–1070. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wallace, E.; Gewin, L. Imatinib: Novel Treatment of Immune-Mediated Kidney Injury. J. Am. Soc. Nephrol. 2013, 24, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.P.; Yates, K.M.; Sumner, E.L.; Stone, W.J.; Wang, Y.; Koury, M.J.; Fogo, A.B.; Zent, R. Chronic myeloid leukemia-associated membranoproliferative glomerulonephritis that responded to imatinib mesylate therapy. Clin. Nephrol. 2007, 67, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.; Fogo, A.B.; Schulman, G. Imatinib Therapy for Non–Infection-Related Type II Cryoglobulinemia With Membranoproliferative Glomerulonephritis. Am. J. Kidney Dis. 2011, 59, 122–125. [Google Scholar] [CrossRef]
- Neef, M.; Ledermann, M.; Saegesser, H.; Schneider, V.; Widmer, N.; Decosterd, L.A.; Rochat, B.; Reichen, J. Oral imatinib treatment reduces early fibrogenesis but does not prevent progression in the long term. J. Hepatol. 2006, 44, 167–175. [Google Scholar] [CrossRef]
- Wang, S.; Wilkes, M.C.; Leof, E.B.; Hirschberg, R. Imatinib mesylate blocks a non-Smad TGF-β pathway and reduces renal fibrogenesis in vivo. FASEB J. 2005, 19, 1–11. [Google Scholar] [CrossRef]
- Nechemia-Arbely, Y.; Barkan, D.; Pizov, G.; Shriki, A.; Rose-John, S.; Galun, E.; Axelrod, J.H. IL-6/IL-6R axis plays a critical role in acute kidney injury. J. Am. Soc. Nephrol. 2008, 19, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.E.; Kelly, D.J.; Mckay, T.; Chadban, S.; Hill, P.A.; Cooper, M.E.; Atkins, R.C.; Nikolic-Paterson, D.J. PDGF signal transduction inhibition ameliorates experimental mesangial proliferative glomerulonephritis. Kidney Int. 2001, 59, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Savikko, J.; Taskinen, E.; von Willebrand, E. Chronic allograft nephropathy is prevented by inhibition of platelet-derived growth factor receptor: Tyrosine kinase inhibitors as a potential therapy. Transplantation 2003, 75, 1147–1153. [Google Scholar] [CrossRef]
- Sadanaga, A.; Nakashima, H.; Masutani, K.; Miyake, K.; Shimizu, S.; Igawa, T.; Sugiyama, N.; Niiro, H.; Hirakata, H.; Harada, M. Amelioration of autoimmune nephritis by imatinib in MRL/lpr mice. Arthritis Rheumatol. 2005, 52, 3987–3996. [Google Scholar] [CrossRef]
- Zoja, C.; Corna, D.; Rottoli, D.; Zanchi, C.; Abbate, M.; Remuzzi, G. Imatinib ameliorates renal disease and survival in murine lupus autoimmune disease. Kidney Int. 2006, 70, 97–103. [Google Scholar] [CrossRef]
- Iyoda, M.; Hudkins, K.L.; Becker-Herman, S.; Wietecha, T.A.; Banas, M.C.; Guo, S.; Meyer-Bahlburg, A.; Kowalewska, J.; Liu, G.; Ziegler, S.F.; et al. Imatinib Suppresses Cryoglobulinemia and Secondary Membranoproliferative Glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 68–77. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Chang, F.-C.; Wu, C.-F.; Chou, Y.-H.; Hsu, H.-L.; Chiang, W.-C.; Shen, J.; Chen, Y.-M.; Wu, K.-D.; Tsai, T.-J.; et al. Platelet-derived growth factor receptor signaling activates pericyte—myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011, 80, 1170–1181. [Google Scholar] [CrossRef]
- Yingling, J.M.; McMillen, W.T.; Yan, L.; Huang, H.; Sawyer, J.S.; Graff, J.; Clawson, D.K.; Britt, K.S.; Anderson, B.D.; Beight, D.W.; et al. Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-β receptor type I inhibitor. Oncotarget 2018, 9, 6659–6677. [Google Scholar] [CrossRef]
- Ding, H.; Zhou, Y.; Huang, H. MiR-101a ameliorates AngII-mediated hypertensive nephropathy by blockade of TGFβ/Smad3 and NF-κB signalling in a mouse model of hypertension. Clin. Exp. Pharmacol. Physiol. 2019, 46, 246–254. [Google Scholar] [CrossRef]
- Bigaeva, E.; Gore, E.; Mutsaers, H.A.M.; Oosterhuis, D.; Kim, Y.O.; Schuppan, D.; Bank, R.A.; Boersema, M.; Olinga, P. Exploring organ-specific features of fibrogenesis using murine precision-cut tissue slices. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165582. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Healthy Renal Tissue (n = 16) | Fibrotic Renal Tissue (n = 9) |
|---|---|---|
| Gender (% male) | 56 | 33 |
| Age (in years) | 61 ± 14 | 41 ± 12 |
| Serum creatinine before nephrectomy (umol/L) | 76.5 ± 24.2 | 420.0 ± 407.6 |
| eGFR before nephrectomy (ml/min/1.73 m2) * | 86.6 ± 19.3 | NA |
| Model of Renal Fibrosis | Intervention | Main Reported Effects on Renal Fibrosis | Ref |
|---|---|---|---|
| Mesangial proliferative (anti-Thy1) glomerulonephritis (Wistar rats) | 500 mg/kg; daily, mixed in the food, for 42 days | - Pirfenidone reduced mesangial matrix expansion and interstitial fibrosis (evaluated by HE, PAS and PAM staining); - reduced protein expression of type I collagen and α-SMA in glomeruli, affecting mesangial cell transdifferentiation to myofibroblasts (evaluated by immunofluorescence); - suppressed accumulation of ED-1 positive macrophages; - attenuated interstitial inflammatory cell infiltration. | [52] |
| Streptozotocin-induced diabetes (Wistar rats) | 200 mg/kg, daily, mixed in drinking water, for 4 weeks (treatment started 2 weeks after ablation) | - Pirfenidone attenuated fibrotic changes in the kidneys of diabetic rats; - reduced collagen deposition (evaluated by % Sirius red staining); - decreased renal and plasma concentration of fibronectin; | [62] |
| Salt-depletion model with ciclosporin-induced renal fibrosis (Sprague-Dawley rats) | salt-depleted diet for 7 days prior to the 28-day treatment with PFD at 250 mg/kg alone or in combination with CSA; daily treatment; 0.5% PFD mixed in the low-salt diet | - Pirfenidone attenuated CSA-induced tubulointerstitial fibrosis (by 48%) and tubular atrophy (histological score on PAS stained sections); - reduced renal cortical TGFβ1 mRNA and protein expression in CSA-treated rats; - reduced CSA-induced mRNA expression of PAI-1 and biglycan. | [63] |
| Salt-depletion model with ciclosporin-induced renal fibrosis (Sprague-Dawley rats) | salt-depleted diet for 7 days prior to the 28-day treatment with PFD at 250 mg/kg, 500 mg/kg or 750 mg/kg in combination with CSA or tacrolimus (FK). Daily treatment; PFD was mixed in the low-salt diet. | - Pirfenidone did not significantly affect CSA-induced mRNA expression of TGFβ1; - reduced CSA-induced mRNA expression of type III collagen and TIMP-1; - restored CSA-inhibited mRNA expression of MMP2, and showed no effect on MMP9; - this model failed to display histological tubulointerstitial fibrosis with the addition of CSA or FK, so any effect of pirfenidone on structural parameters could not be seen. | [64] |
| Diabetic nephropathy (C57BL6 db/db mice) | 25 mg/day; 0.5% PFD mixed in the food; for 4 weeks total from week 17 to week 21 | - Pirfenidone reduced mesangial matrix expansion; - reduced the diabetic stimulation of renal type I collagen, type IV collagen, and fibronectin gene expression to control levels. | [65] |
| Anti-glomerular basement membrane glomerulonephritis (Wistar rats) | 500 mg/kg; daily by oral gavage, for 8 weeks | - Pirfenidone attenuated glomerular segmental sclerosis and tubular degeneration (evaluated by histological score on PAS); only minor interstitial fibrosis was observed; - reduced podocyte injury; - reduced mRNA expression of type I collagen; - showed increased number of ED-1 positive glomerular macrophages, but reduced number of ED-1 positive interstitial cells. | [53] |
| Focal segmental glomerulosclerosis (FGS/Kist mice) | 50 mg/kg; daily, 0.5% PFD mixed in the food; for up to 3 months | - Pirfenidone attenuated the development of glomerulosclerosis and tubulointerstitial fibrosis after 3 months of treatment (histological score); - reduced mRNA expression of TGFβ1 after 2 months; - showed no clear effect on mRNA expression of TIMP-1 and MMP3. | [66] |
| Subtotal nephrectomy (rats) | 500 mg/kg; daily, mixed in the food, for 12 weeks | - Pirfenidone reduced collagen accumulation (detected by hydroxyproline content) in the cortex of the remnant kidney; - reduced mRNA expression of type IV and type I collagen - suppressed the increased mRNA expression of TGFβ1. | [67] |
| Subtotal nephrectomy (Wistar rats) | 700 mg/kg; daily, 1% PFD mixed in the food, for 8 weeks | - Pirfenidone attenuated interstitial fibrosis in the cortex (evaluated by Azan staining), although did not show prominent effects on glomerular and tubular degeneration; - reduced mRNA expression of TGFβ1 and protein expression of fibronectin; - had little effect on the expression of α-SMA (i.e., myofibroblast differentiation); - showed no effect on infiltration of inflammatory cells in the cortical interstitium. | [68] |
| Subtotal nephrectomy (Sprague-Dawley rats) | 500 mg/kg; once a day by oral gavage for 12 weeks | - Pirfenidone attenuated interstitial fibrosis in the cortex and reduced the scores of glomerulosclerosis (evaluated by PAS and Masson staining); - decreased protein expression of TGFβ1, CTGF, α-SMA, fibronectin, and fibroblast-specific protein-1 (FSP-1); - inhibited mRNA and protein expression of chemokines MCP-1 and MIP-1α, as well as TNFα, IL-6 and iNOS expressed by M1 macrophages, and CD206 and CD86 expressed by M2 macrophages; - decreased the infiltration of inflammatory cells in the cortical interstitium; - decreased tubulointerstitial and glomerular macrophage infiltration. | [54] |
| UUO model (Sprague-Dawley rats) | 500 mg/kg; daily, mixed in the food, for up to 21 days | - Pirfenidone reduced collagen accumulation (detected by hydroxyproline content) at 14 and 21 days of UUO; - reduced type IV collagen mRNA expression at 14 and 21 days of UUO (and not after 7 days); - reduced type I collagen mRNA expression at 21 days UUO; - inhibited mRNA expression of MMP2 at 14 and 21 days; - inhibited increases in TGFβ1 mRNA after 7 days; - attenuated fibrotic lesions; - pirfenidone does not reverse the increased collagen accumulation in the acute phase of UUO (i.e., 3 and 7 days). | [69] |
| Model of Renal Fibrosis | Intervention | Main Reported Effects on Renal Fibrosis | Ref |
|---|---|---|---|
| Mesangial proliferative (anti-Thy-1) glomerulonephritis (Wistar rats) | 50 mg/kg; daily by intraperitoneal injections, for 6 days | - Imatinib ameliorated glomerular hypercellularity; - reduced mesangial cell proliferation (ED-1 and BrdU double IHC); - reduced activation of mesangial cells (α-SMA IHC) and type IV collagen deposition; - no effect on glomerular macrophage infiltration and proliferation. | [83] |
| Chronic allograft nephropathy (allografts from Dark Agouti to Wistar-Furth rats) | 10 mg/kg; daily by oral gavage for 5 or 90 days | - Imatinib inhibited the development of fibrosis associated with chronic allograft nephropathy (Masson trichrome staining); - reduced protein expression of ligands PDGFA and PDGFB, and receptors PDGFRα and PDGFRβ; - inhibited infiltration of macrophages and CD4+ T cells into the grafts. | [84] |
| Diabetic nephropathy (C57BL6 apolipoprotein E–knockout mice) | 10 mg/kg; daily by oral gavage for 20 weeks | - Imatinib reduced glomerular injury (attenuated increased glomerular size and mesangial area) and tubulointerstitial area; - prevented the increase in α-SMA–positive cell expression in the glomeruli and in the tubulointerstitium in diabetic mice; - reduced accumulation of collagen type I and type IV; - reduced expression of Ki-67–positive cells; - reduced PDGFB protein and gene expression; - reduced PDGFRβ gene expression; - reduced TGFβ1 protein expression, but not gene expression; - reduced CTGF protein and gene expression; - decreased macrophage infiltration. | [72] |
| Lupus nephritis (MRL/lpr mice) | 10 mg/kg or 50 mg/kg; daily by oral gavage 4 times per week up to 8 weeks | Imatinib at 50 mg/kg: - ameliorated glomerulonephritis (reduced proliferation of glomerular cells, infiltrating inflammatory cells and reduced mesangial matrix); - reduced proportional area of glomeruli staining positive for PDGFRβ; - reduced expression mRNA levels of PDGFRβ and TGFβ1. - Treatment with 10 mg/kg imatinib did not affect PDGF signaling, and thus failed to ameliorate the nephritis. | [85] |
| Lupus nephritis (New Zealand Black/White (NZB/W) F1 hybrid mice) | 50 mg/kg; twice a day by oral gavage for 3 months (or longer for survival studies) | - Imatinib limited glomerular hypercellularity, IgG deposits, and tubulointerstitial damage; - reduced interstitial expression of α-SMA in fibroblasts; - reduced mRNA expression of TGFβ1; - reduced infiltrates of monocytes/macrophages. | [86] |
| Cryoglobulinemic MPGN (TSLP-transgenic mice) | 50 mg/kg; daily by intraperitoneal injection for 2, 4 or 8 weeks | - Imatinib decreased accumulation of collagen IV; - reduced mesangial cell activation (measured by % GTA occupied by α-SMA–positive cells); - parenchymal injury and fibrosis were attenuated in a time-dependent manner. - reduced the infiltration of inflammatory cells; - inhibited B cell development; | [87] |
| Anti-glomerular basement nephritis (Wistar-Kyoto rats) | 50 mg/kg; intraperitoneal injection, from one day before up to 13 days (early treatment) or from day 7 to 20 (late treatment) | - Imatinib reduced glomerular fibrin deposition; - ameliorated glomerular injury (reduced crescents formation and necrosis evaluated by morphological analysis); - attenuated the PDGFRβ mRNA upregulation; - attenuated the c-fms and M-CSF mRNA expression; - reduced glomerular macrophage accumulation, along with an inhibition of IL-1b or MCP-1 expression. | [75] |
| Anti-glomerular basement nephritis (Wistar-Kyoto rats) | 25 mg/kg; daily by intraperitoneal injection, from day 7 to day 49 (long-term treatment) or from day 7 to 13 (short-term treatment) | - Imatinib reduced glomerulosclerosis and improved tubulointerstitial damage in rats with NTS nephritis (evaluated by histological analysis); - decreased collagen type I gene expression; - reduced TGFβ1 gene and protein expression. | [73] |
| UUO model (Sprague-Dawley rats) | dose-accelerating schedule for 7 days (days 1-2: 50 mg/kg; days 3-4: 100 mg/kg; days 5–7: 150 mg/kg), once daily by intraperitoneal injection | - Imatinib blocked TGFβ-stimulated c-abl activity in fibroblasts in obstructed kidneys; - did not block increased PDGFB expression, neither affected increased PAI-1 levels; - reduced proliferation of vimentin-expressing fibroblasts in obstructed kidneys, although induction of α-SMA in fibroblasts was not mitigated; - reduced fibrogenesis, as indicated by reduced accumulation of collagen type III and IV and fibronectin in the renal interstitium; - did not reduce interstitial macrophage infiltration. | [80] |
| UUO model (C57BL6 wild-type or Coll-GFP mice) | 50 mg/kg; 2h before the surgery, then twice a day until sacrifice on day 4 or 14 | - Imatinib decreased PDGFRα and PDGFRβ phosphorylation in pericytes in UUO kidneys; - decreased the expanded population of myofibroblasts by inhibiting cell proliferation; - attenuated fibrosis development in UUO kidneys (measured by % Sirius red area); - attenuated macrophage infiltration. | [88] |
| Unilateral IRI model (C57BL6 wild-type or Coll-GFP mice) | 50 mg/kg; 2h before the surgery, then twice a day until sacrifice on day 4 or 14 | - Imatinib inhibited pericyte activation and expansion of myofibroblasts in IRI kidneys on days 4 and 14 post IRI; - attenuated interstitial fibrosis on day 14 post IRI (measured by Sirius red staining). | [88] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bigaeva, E.; Puerta Cavanzo, N.; Stribos, E.G.D.; de Jong, A.J.; Biel, C.; Mutsaers, H.A.M.; Jensen, M.S.; Nørregaard, R.; Leliveld, A.M.; de Jong, I.J.; et al. Predictive Value of Precision-Cut Kidney Slices as an Ex Vivo Screening Platform for Therapeutics in Human Renal Fibrosis. Pharmaceutics 2020, 12, 459. https://doi.org/10.3390/pharmaceutics12050459
Bigaeva E, Puerta Cavanzo N, Stribos EGD, de Jong AJ, Biel C, Mutsaers HAM, Jensen MS, Nørregaard R, Leliveld AM, de Jong IJ, et al. Predictive Value of Precision-Cut Kidney Slices as an Ex Vivo Screening Platform for Therapeutics in Human Renal Fibrosis. Pharmaceutics. 2020; 12(5):459. https://doi.org/10.3390/pharmaceutics12050459
Chicago/Turabian StyleBigaeva, Emilia, Nataly Puerta Cavanzo, Elisabeth G. D. Stribos, Amos J. de Jong, Carin Biel, Henricus A. M. Mutsaers, Michael S. Jensen, Rikke Nørregaard, Anna M. Leliveld, Igle J. de Jong, and et al. 2020. "Predictive Value of Precision-Cut Kidney Slices as an Ex Vivo Screening Platform for Therapeutics in Human Renal Fibrosis" Pharmaceutics 12, no. 5: 459. https://doi.org/10.3390/pharmaceutics12050459
APA StyleBigaeva, E., Puerta Cavanzo, N., Stribos, E. G. D., de Jong, A. J., Biel, C., Mutsaers, H. A. M., Jensen, M. S., Nørregaard, R., Leliveld, A. M., de Jong, I. J., Hillebrands, J.-L., van Goor, H., Boersema, M., Bank, R. A., & Olinga, P. (2020). Predictive Value of Precision-Cut Kidney Slices as an Ex Vivo Screening Platform for Therapeutics in Human Renal Fibrosis. Pharmaceutics, 12(5), 459. https://doi.org/10.3390/pharmaceutics12050459