Model-Based Optimisation and Control Strategy for the Primary Drying Phase of a Lyophilisation Process



Abstract

1. Introduction
2. Materials and Methods
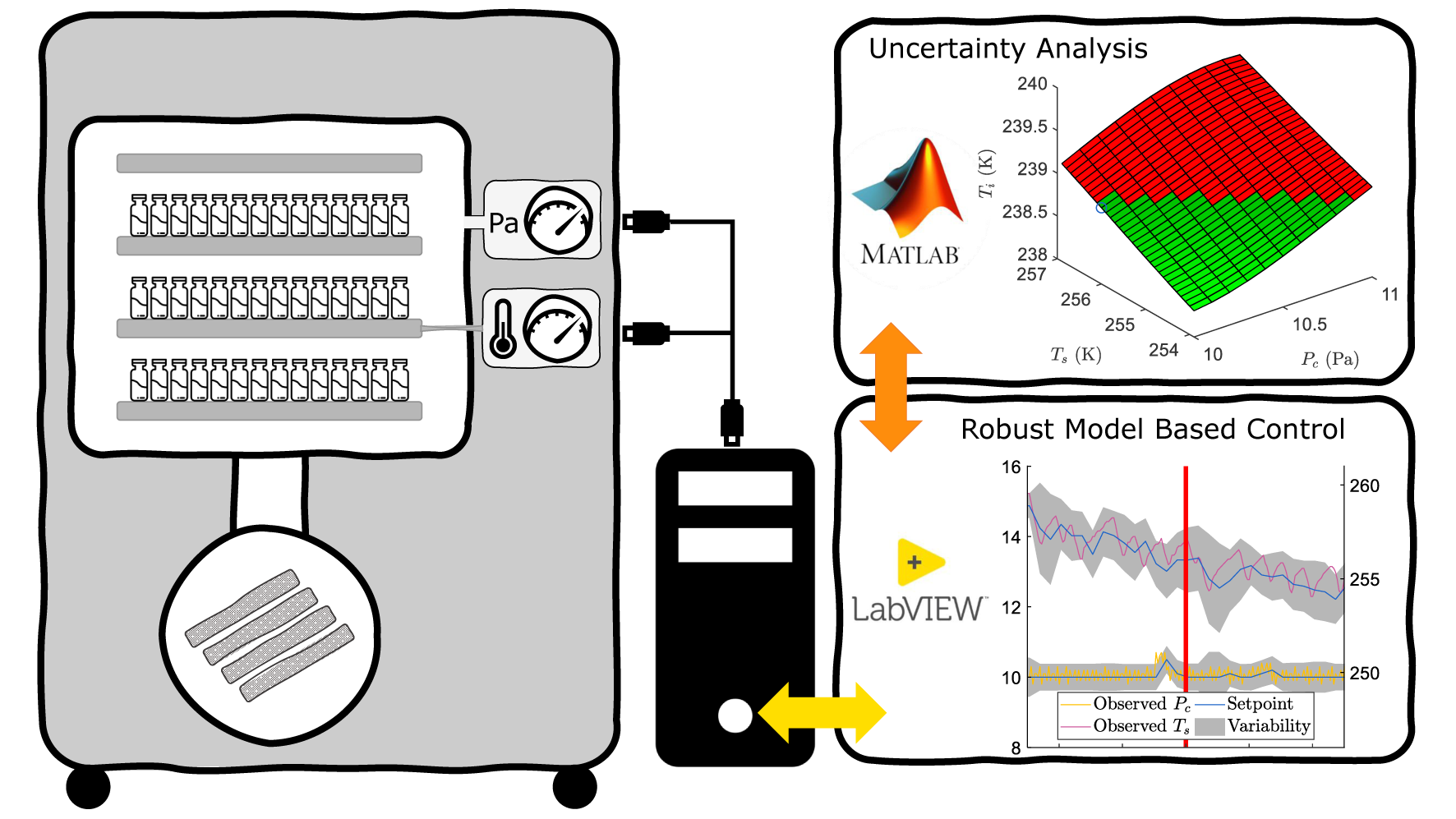
2.1. Supervisory Control of the Freeze-Dryer
2.2. Primary Drying Model
2.3. Input Parameters and Variability Estimation
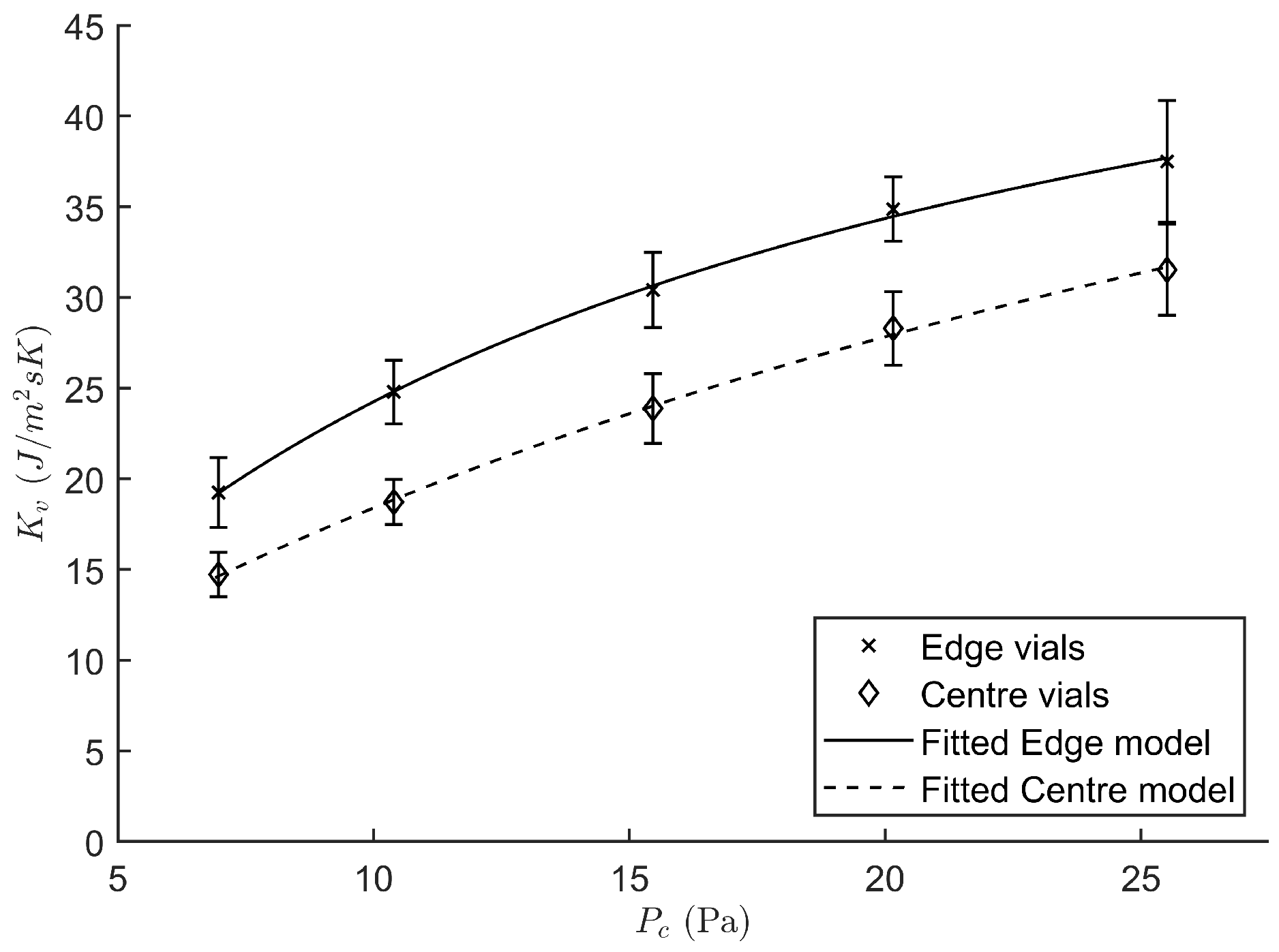
2.3.1. Determination of the Heat Transfer Coefficient
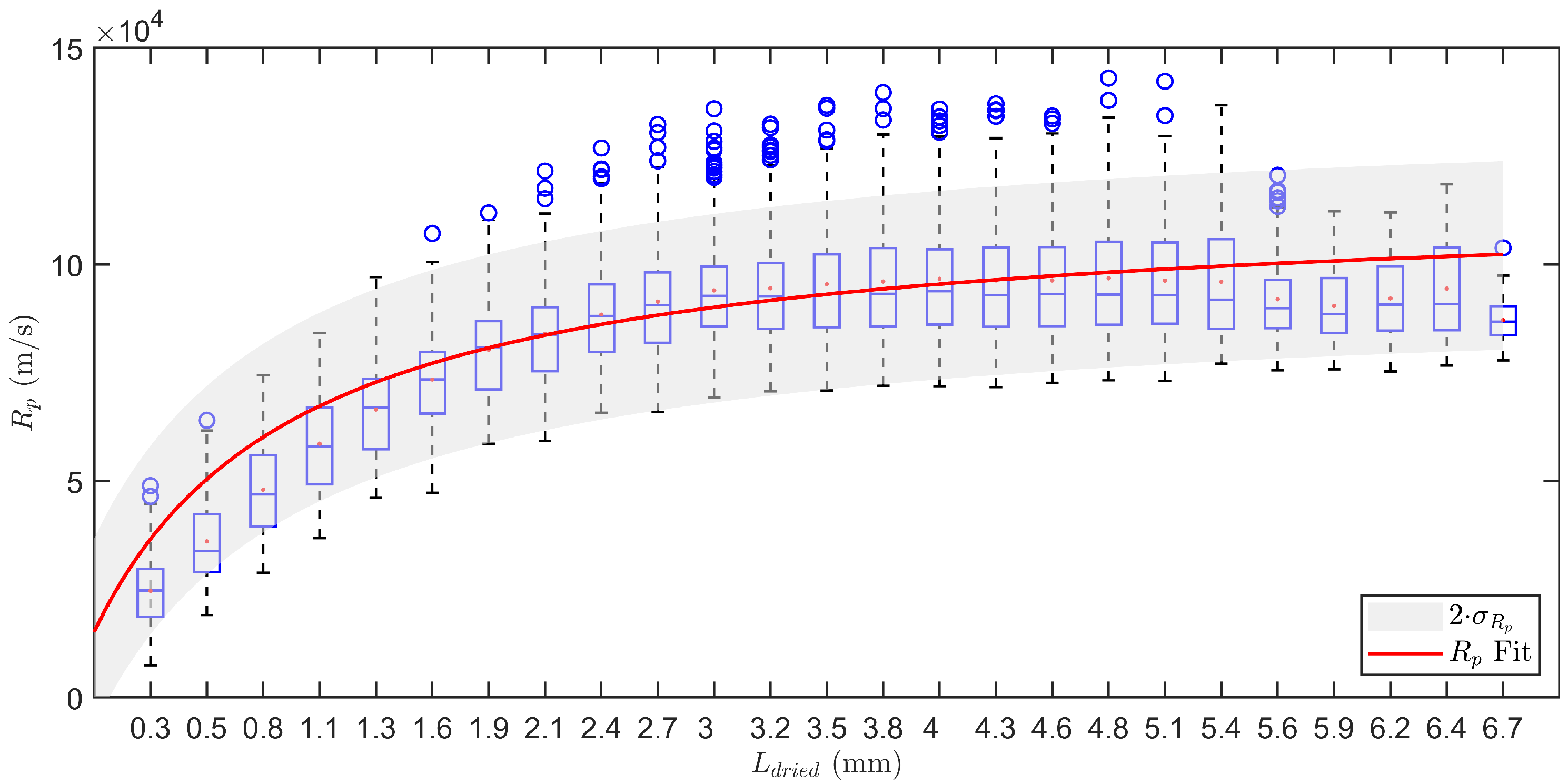
2.3.2. Determination of the Dry Layer Resistance
2.3.3. Determination of Filling Volume Variability
2.3.4. Determination of Vial Radii Variability
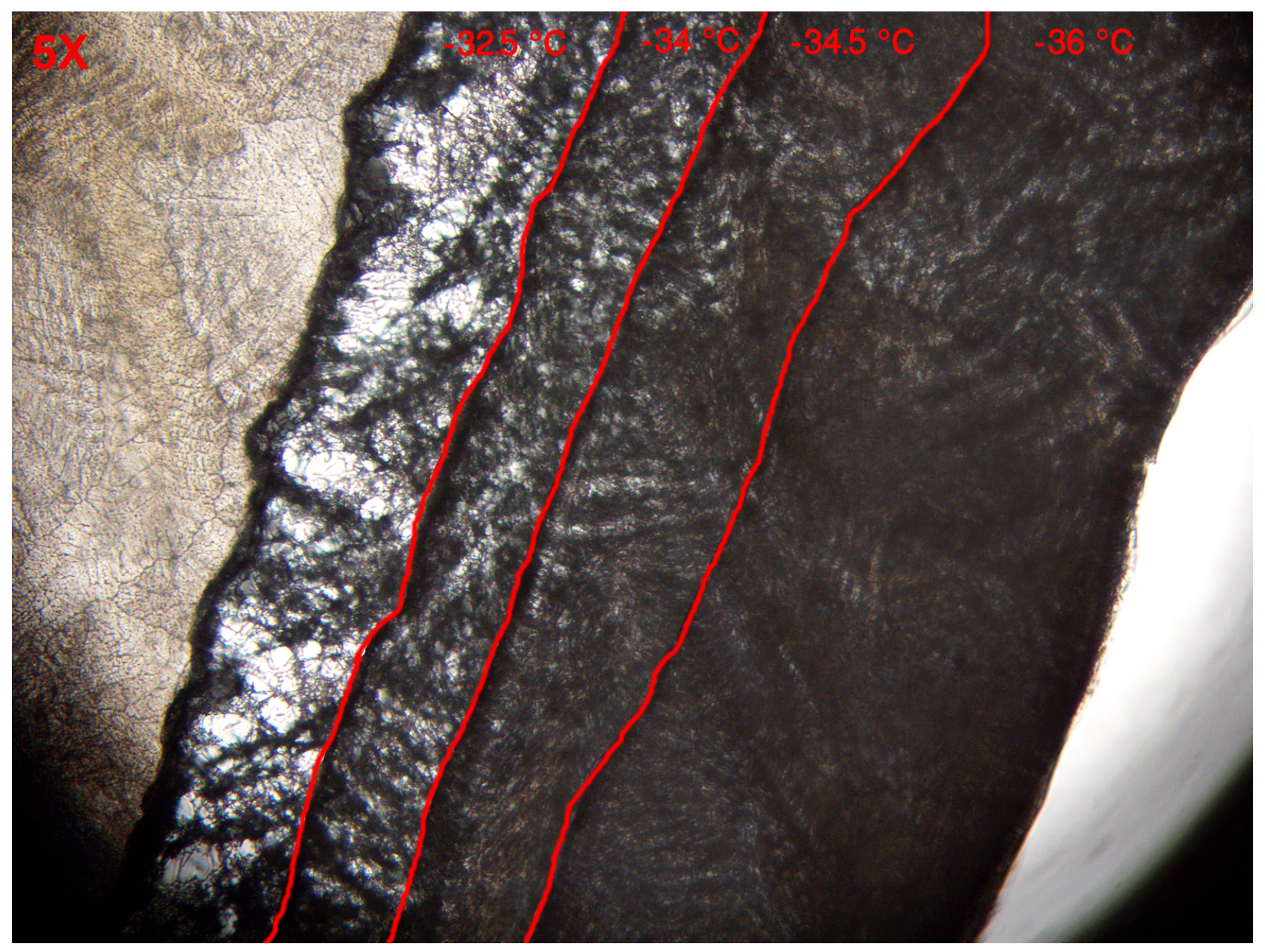
2.3.5. Determination of the Critical Temperature
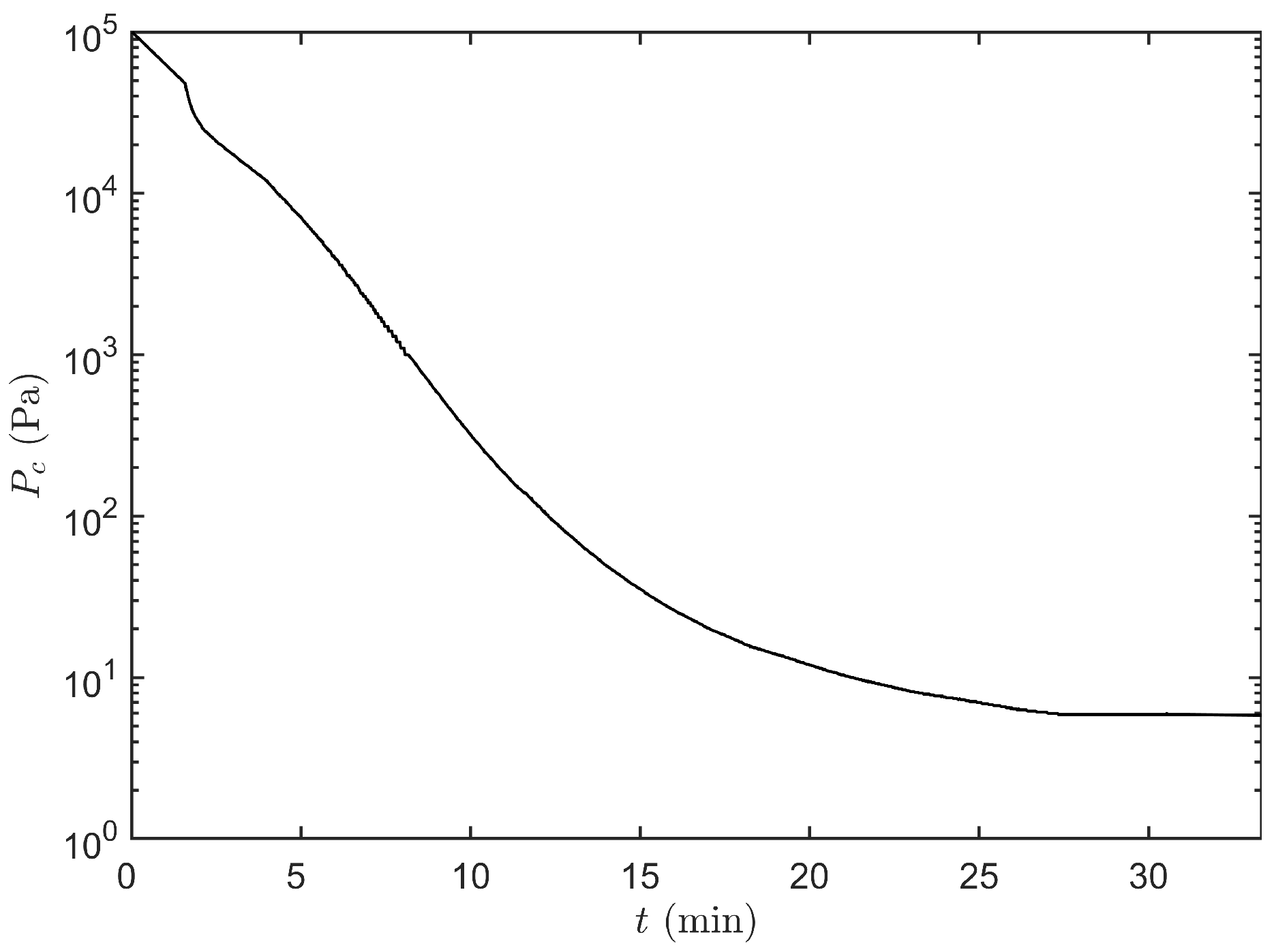
2.3.6. Determination of the Pressure Decrease Curve
2.4. Freezing and Primary Drying Initialization Phase
2.5. Primary Drying
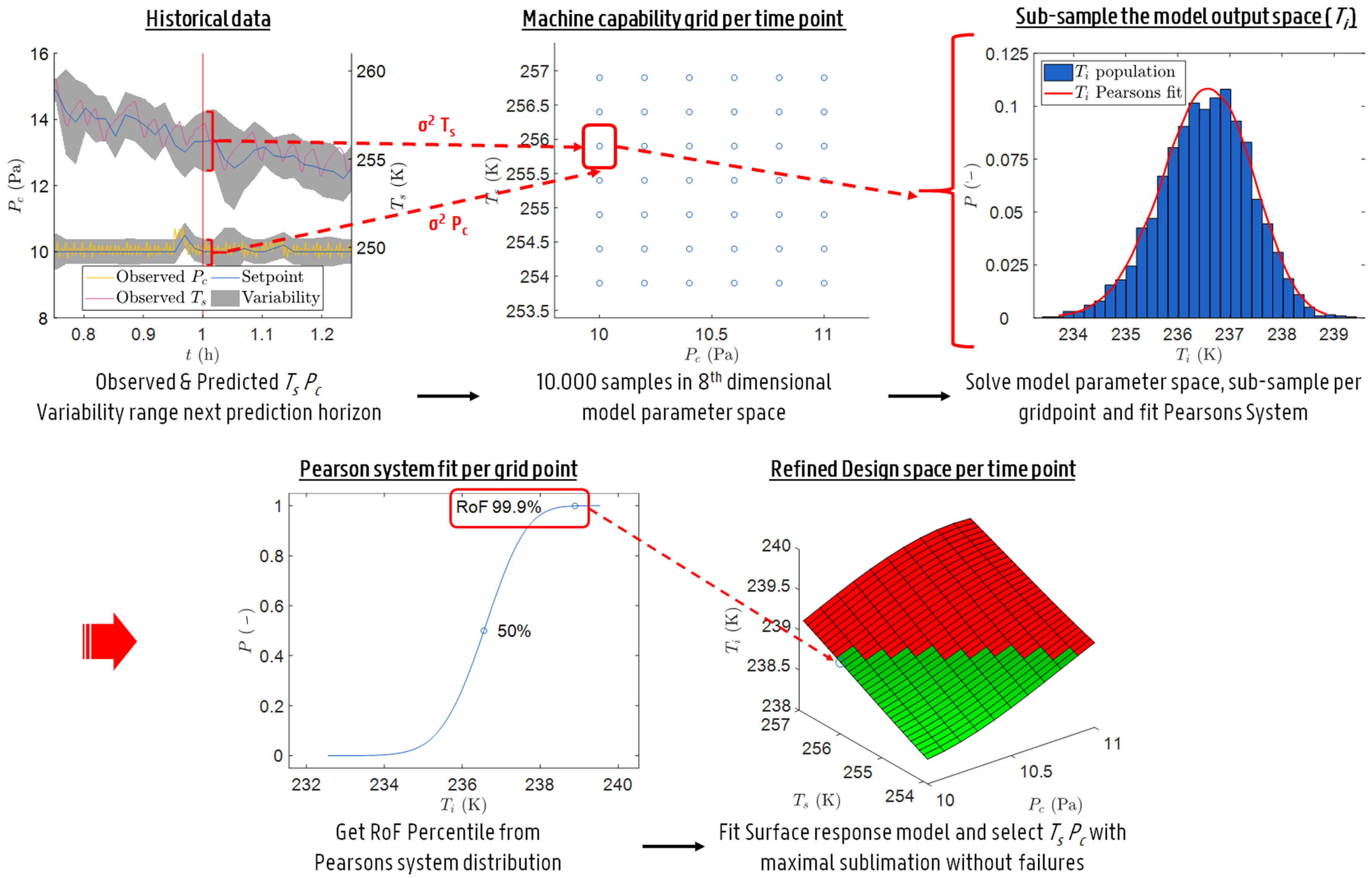
2.5.1. Uncertainty Analysis
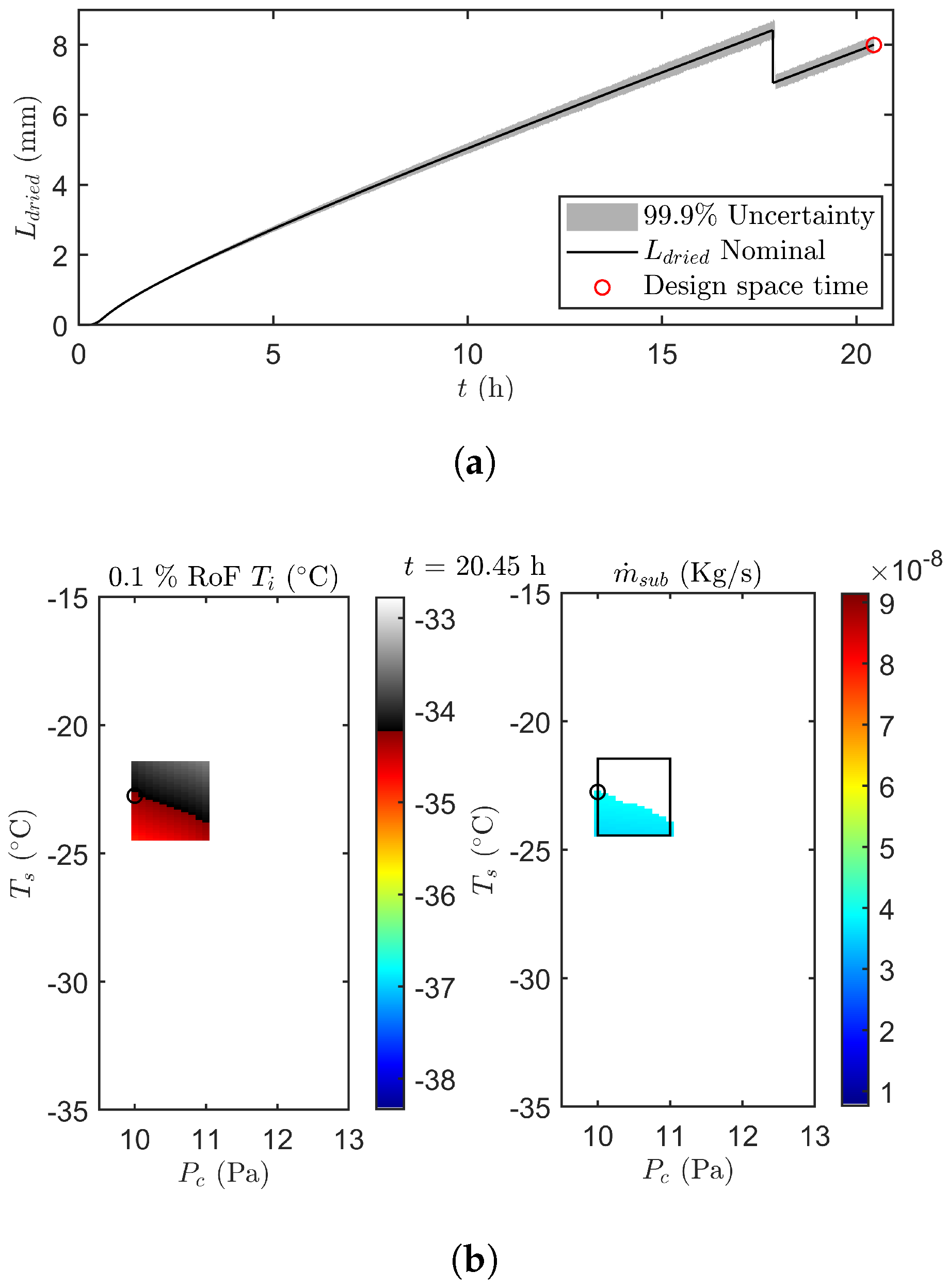
2.5.2. Optimisation and Control of the Primary Drying Phase
2.6. Primary Drying Endpoint
2.7. Secondary Drying Phase
2.8. Experimental Verification
3. Results
3.1. Input Parameters and Variability
3.1.1. Determination of the Heat Transfer Coefficient
3.1.2. Determination of the Dry Layer Resistance
3.1.3. Variation of Vial Radii and Filling Volume
3.1.4. Determination of the Critical Temperature
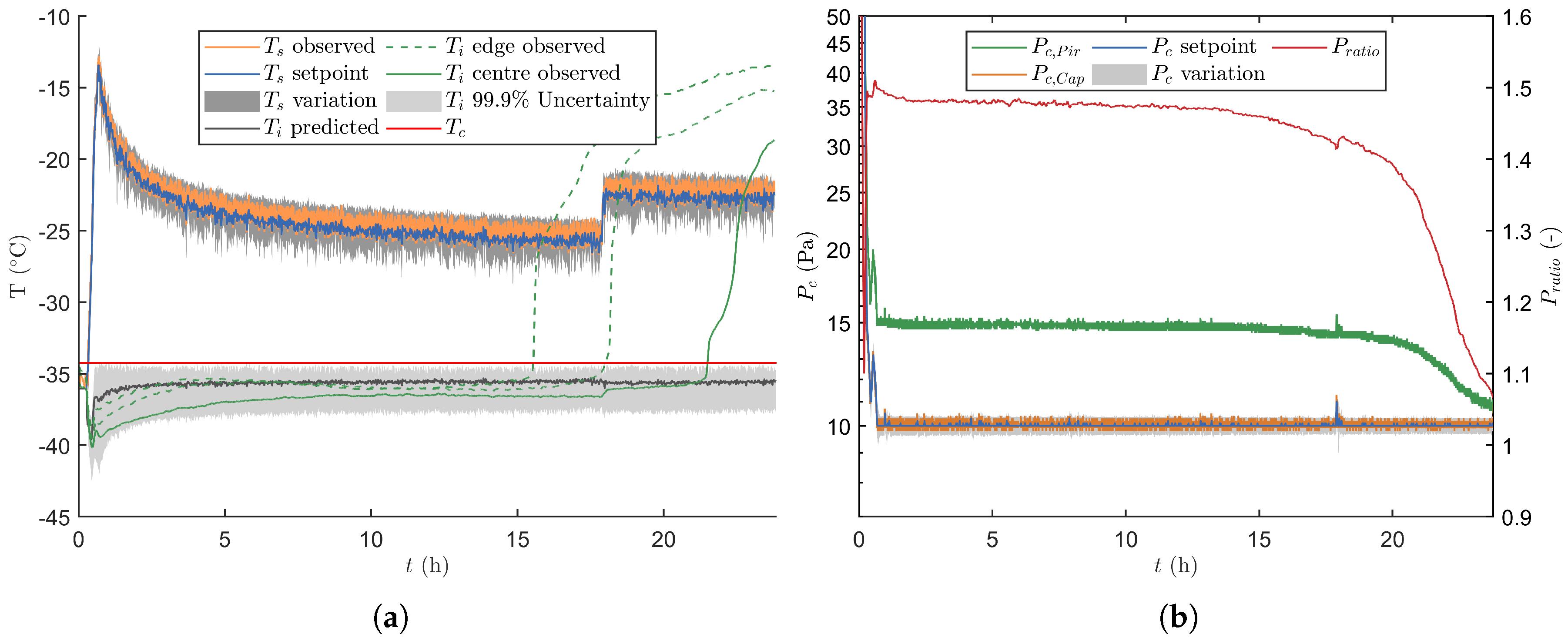
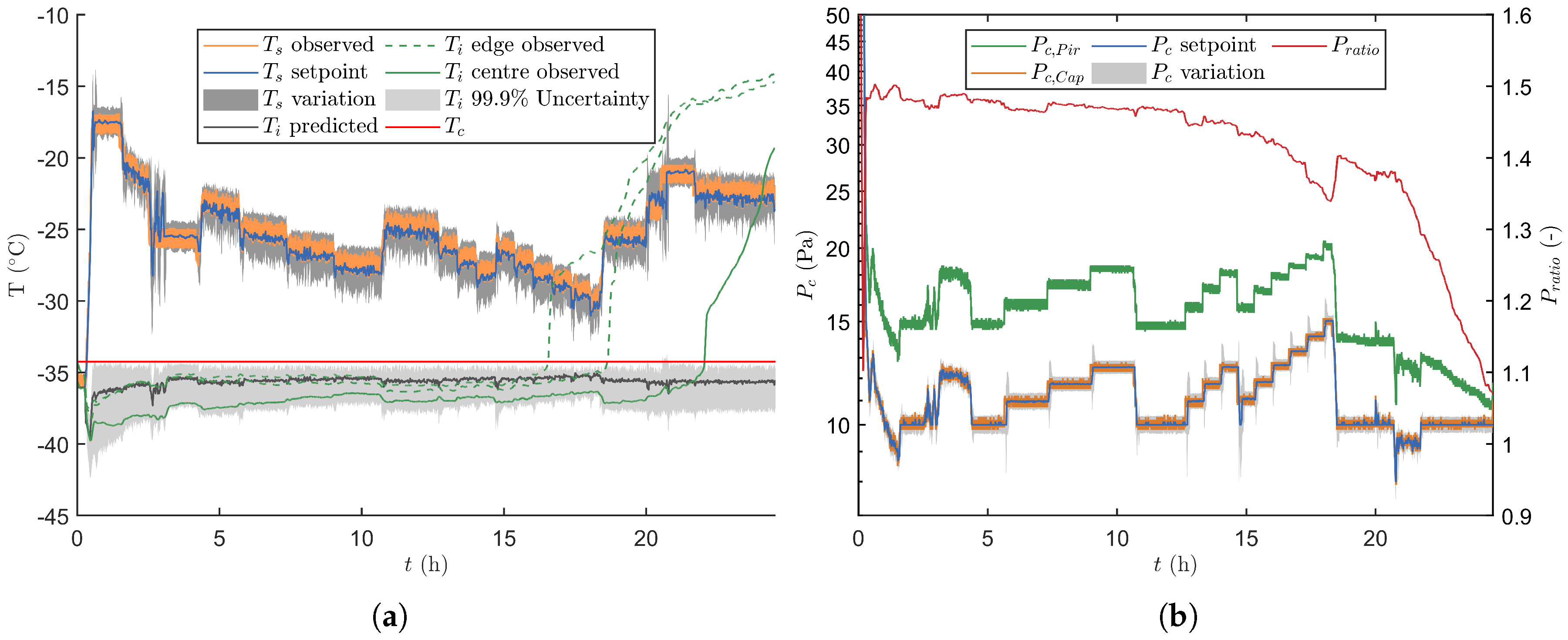
3.2. Experimental Verification of the Real-Time Optimisation Strategy
3.2.1. Verification Runs without Process Disturbances
3.2.2. Verification Runs with Process Disturbances
3.2.3. End of Primary Drying
4. Discussion and Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Fissore, D.; McCoy, T. Editorial: Freeze-drying and process analytical technology for pharmaceuticals. Front. Chem. 2018, 6, 622. [Google Scholar] [CrossRef]
- Renteria Gamiz, A.G.; Van Bockstal, P.J.; De Meester, S.; De Beer, T.; Corver, J.; Dewulf, J. Analysis of a pharmaceutical batch freeze dryer: resource consumption, hotspots, and factors for potential improvement. Dry. Technol. 2019, 37, 1563–1582. [Google Scholar] [CrossRef]
- Overcashier, D.; Patapoff, T.; Hsu, C. Lyophilization of protein formulations in vials: Investigation of the relationship between resistance to vapor flow during primary drying and small-scale product collapse. J. Pharm. Sci. 1999, 88, 688–695. [Google Scholar] [CrossRef]
- Fissore, D.; Pisano, R.; Barresi, A.A. Advanced Approach to Build the Design Space for the Primary Drying of a Pharmaceutical Freeze-Drying Process. J. Pharm. Sci. 2011, 100, 4922–4933. [Google Scholar] [CrossRef]
- Patel, S.M.; Chaudhuri, S.; Pikal, M.J. Choked flow and importance of Mach I in freeze-drying process design. Chem. Eng. Sci. 2010, 65, 5716–5727. [Google Scholar] [CrossRef]
- Schneid, S.C.; Gieseler, H.; Kessler, W.J.; Luthra, S.A.; Pikal, M.J. Optimization of the secondary drying step in freeze drying using tdlas technology. Aaps Pharmscitech 2011, 12, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Pisano, R.; Fissore, D.; Barresi, A.A. Quality by Design in the Secondary Drying Step of a Freeze-Drying Process. Dry. Technol. 2012, 30, 1307–1316. [Google Scholar] [CrossRef]
- Mortier, S.T.F.; Van Bockstal, P.J.; Corver, J.; Nopens, I.; Gernaey, K.V.; De Beer, T. Uncertainty analysis as essential step in the establishment of the dynamic Design Space of primary drying during freeze-drying. Eur. Pharm. Biopharm. 2016, 103, 71–83. [Google Scholar] [CrossRef]
- Tang, X.; Pikal, M. Design of freeze-drying processes for pharmaceuticals: Practical advice. Pharm. Res. 2004, 21, 191–200. [Google Scholar] [CrossRef]
- Pikal, M.J. Freeze-drying of proteins. part i: Process design. BioPharm 1990, 3, 18–27. [Google Scholar]
- Pikal, M.; Roy, M.; Shah, S. Mass and heat transfer in vial freeze-drying of pharmaceuticals: Role of the vial. J. Pharm. Sci. 1984, 73, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Nail, S.L.; Jiang, S.; Chongprasert, S.; Knopp, S.A. Fundamentals of freeze-drying. In Development and Manufacture of Protein Pharmaceuticals; Springer: New York, NY, USA, 2002; pp. 281–360. [Google Scholar]
- Giordano, A.; Barresi, A.A.; Fissore, D. On the use of mathematical models to build the design space for the primary drying phase of a pharmaceutical lyophilization process. J. Pharm. Sci. 2011, 100, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Pisano, R.; Fissore, D.; Barresi, A.A. Freeze-drying cycle optimization using model predictive control techniques. Ind. Eng. Chem. 2011, 50, 7363–7379. [Google Scholar] [CrossRef]
- McKay, M.D.; Morrison, J.D.; Upton, S.C. Evaluating prediction uncertainty in simulation models. Comput. Phys. Commun. 1999, 117, 44–51. [Google Scholar] [CrossRef]
- Nail, S.; Tchessalov, S.; Shalaev, E.; Ganguly, A.; Renzi, E.; Dimarco, F.; Wegiel, L.; Ferris, S.; Kessler, W.; Pikal, M.; et al. Recommended best practices for process monitoring instrumentation in pharmaceutical freeze drying—2017. AAPS PharmSciTech 2017, 18, 2379–2393. [Google Scholar] [CrossRef]
- Patel, S.M.; Doen, T.; Pikal, M.J. Determination of End Point of Primary Drying in Freeze-Drying Process Control. AAPS PharmSciTech 2010, 11, 73–84. [Google Scholar] [CrossRef]
- Tang, X.C.; Nail, S.L.; Pikal, M.J. Freeze-drying process design by manometric temperature measurement: Design of a smart freeze-dryer. Pharm. Res. 2005, 22, 685–700. [Google Scholar] [CrossRef]
- Murphy, D.M.; Koop, T. Review of the vapour pressures of ice and supercooled water for atmospheric applications. Quaterly J. R. Soc. 2005, 131, 1539–1565. [Google Scholar] [CrossRef]
- Van Bockstal, P.J.; Mortier, S.T.F.; Corver, J.; Nopens, I.; Gernaey, K.V.; De Beer, T. Quantitative risk assessment via uncertainty analysis in combination with error propagation for the determination of the dynamic Design Space of the primary drying step during freeze-drying. Eur. Pharm. Biopharm. 2017, 121, 32–41. [Google Scholar] [CrossRef]
- Johnson, N.; Kotz, S.; Balakrishnan, N. Continuous Univariate Distributions; Wiley-Interscience: New York, NY, USA, 1995; Volume 2. [Google Scholar]
- Kritzer, P.; Niederreiter, H.; Pillichshammer, F.; Niederreiter, H.; Winterhof, A. Uniform Distribution and Quasi-Monte Carlo Methods. Discrepancy, Integration and Applications; De Gruyter: Boston, MA, USA, 2014. [Google Scholar]
- Rambhatla, S.; Obert, J.; Luthra, S.; Bhugra, C.; Pikal, M.J. Cake shrinkage during freeze drying: A combined experimental and theoretical study. Pharm. Dev. Technol. 2005, 10, 33–40. [Google Scholar] [CrossRef]
- Rambhatla, S.; Pikal, M.J. Heat and mass transfer scale-up issues during freeze-drying, i: Atypical radiation and the edge vial effect. AAPS PharmSciTech 2003, 4, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Pisano, R.; Fissore, D.; Barresi, A. Developments in Heat Transfer; Ch. Heat Transfer in Freeze-Drying Apparatus; IntechOpen: Rijeka, Croatia, 2011; pp. 91–114. [Google Scholar]
- Kuu, W.Y.; O’Bryan, K.R.; Hardwick, L.M.; Paul, T.W. Product mass transfer resistance directly determined during freeze-drying cycle runs using tunable diode laser absorption spectroscopy (tdlas) and pore diffusion model. Pharm. Dev. Technol. 2011, 16, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Kuu, W.Y.; Hardwick, L.M.; Akers, M.J. Rapid determination of dry layer mass transfer resistance for various pharmaceutical formulations during primary drying using product temperature profiles. Int. J. Pharm. 2006, 313, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Kasraian, K.; Spitznagel, T.M.; Juneau, J.A.; Yim, K. Characterization of the sucrose/glycine/water system by differential scanning calorimetry and freeze-drying microscopy. Pharm. Dev. Technol. 1998, 3, 233–239. [Google Scholar] [CrossRef]
- Pikal, M.J.; Shah, S. The collapse temperature in freeze drying: Dependence on measurement methodology and rate of water removal from the glassy phase. Int. J. Pharm. 1990, 62, 165–186. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Symbol | Unit | Variation Term |
|---|---|---|---|
| Parameters | |||
| Inner vial radius | m | standard deviation | |
| Outer vial radius | m | standard deviation | |
| Heat transfer coefficient | J/ms·K | relative standard deviation | |
| Dry layer resistance | m/s | standard deviation | |
| Filling volume | m | standard deviation | |
| Variables | |||
| Shelf temperature | K | iterative determination | |
| Chamber pressure | Pa | iterative determination | |
| Dried layer thickness | m | error propagation |
| Description | Symbol | Numerical Value | Unit |
|---|---|---|---|
| Conversion factor a | a | 88,9200 | - |
| Conversion factor b | b | 1.02 | - |
| Inner vial radius | 0.0110 | m | |
| Inner vial radius standard variation | 5.44 × 10−5 | m | |
| Outer vial radius | 0.0120 | m | |
| Outer vial radius standard variation | 5.44 × 10−5 | m | |
| Vial neck radius | 0.00630 | m | |
| Condenser duct radius | 0.0800 | m | |
| coefficient | 9.550426 | Pa | |
| coefficient | 5723.2658 | K | |
| coefficient | 3.53068 | 1/K | |
| coefficient | 0.00728332 | Pa | |
| coefficient | 4.68 × 104 | J/mol | |
| coefficient | 35.9 | J/mol·K | |
| coefficient | 0.0741 | J/mol·K | |
| coefficient | 542 | J/mol | |
| coefficient | 124 | K | |
| coefficient for edge vials | −1.14 | J/ms·K | |
| coefficient for centre vials | 3.46 | J/ms·K | |
| coefficient for edge vials | 4.46 | J/ms·KPa | |
| coefficient for centre vials | 1.93 | J/ms·KPa | |
| coefficient for edge vials | 0.0757 | 1/Pa | |
| coefficient for centre vials | 0.0292 | 1/Pa | |
| relative standard deviation | 0.0761 | - | |
| coefficient | 1.51 × 104 | m/s | |
| coefficient | 9.68 × 107 | 1/s | |
| coefficient | 968 | 1/m | |
| standard deviation | 1.10 × 104 | m/s | |
| Critical temperature | 238.9 | K | |
| Filling volume | 3.00 × 10-6 | m | |
| Filling volume standard deviation | 1.29 × 10-8 | m | |
| Density of ice | 919.4 | kg/m | |
| Density of solution | 1000 | kg/m | |
| Porosity of dried layer | 0.97 | - | |
| Molecular weight of water | M | 0.018015 | kg/mol |
| Latent heat of sublimation | 51,136.82 | J/mol | |
| Thermal conductivity of ice | 2.45 | W/mK | |
| Number of samples | 10,000 | - | |
| Time resolution primary drying prediction | 60 | s | |
| Width of the prediction horizon | 120 | s | |
| Width of the machine capability space | 1 | Pa | |
| Risk of Failure | 0.1 | % |
| Verification Experiment | (h) | UA end (h) | Relative Difference Midpoint and UA (%) | |
|---|---|---|---|---|
| Midpoint | Offset | |||
| Normal operation 1 | 20.78 | 22.73 | 21.55 | 3.57 |
| Normal operation 2 | 20.96 | 23.99 | 21.48 | 2.42 |
| Normal operation 3 | 21.68 | 23.77 | 21.49 | 0.88 |
| Disturbances 1 | 22.22 | 23.87 | 22.35 | 0.58 |
| Disturbances 2 | 21.38 | 23.32 | 22.37 | 4.43 |
| Disturbances 3 | 22.08 | 24.52 | 23.45 | 5.64 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanbillemont, B.; Nicolaï, N.; Leys, L.; De Beer, T. Model-Based Optimisation and Control Strategy for the Primary Drying Phase of a Lyophilisation Process. Pharmaceutics 2020, 12, 181. https://doi.org/10.3390/pharmaceutics12020181
Vanbillemont B, Nicolaï N, Leys L, De Beer T. Model-Based Optimisation and Control Strategy for the Primary Drying Phase of a Lyophilisation Process. Pharmaceutics. 2020; 12(2):181. https://doi.org/10.3390/pharmaceutics12020181
Chicago/Turabian StyleVanbillemont, Brecht, Niels Nicolaï, Laurens Leys, and Thomas De Beer. 2020. "Model-Based Optimisation and Control Strategy for the Primary Drying Phase of a Lyophilisation Process" Pharmaceutics 12, no. 2: 181. https://doi.org/10.3390/pharmaceutics12020181
APA StyleVanbillemont, B., Nicolaï, N., Leys, L., & De Beer, T. (2020). Model-Based Optimisation and Control Strategy for the Primary Drying Phase of a Lyophilisation Process. Pharmaceutics, 12(2), 181. https://doi.org/10.3390/pharmaceutics12020181