1. Introduction
Topical/transdermal drug delivery refers to the delivery of drugs via skin and is an attractive alternative to conventional methods such as oral and parenteral routes. Advantages associated with topical/transdermal delivery include non-invasive delivery, bypass of first pass metabolism, prolonged duration of action, reduced dosing frequency, constant levels of drug in the plasma, reduced drug toxicity/adverse events, improved patient compliance, and others [
1,
2,
3]. However, skin acts as a major barrier for the entry of drugs and foreign compounds due to the presence of stratum corneum, a thin keratin-rich layer (15 µm) of dead cells embedded in an intricate lipid environment made of cholesterol, ceramides, and free fatty acids [
4,
5,
6]. In addition, several other factors such as physicochemical properties of the drug (lipophilicity, solubility, molecular weight or size, and hydrogen bonding) and characteristics of a formulation/vehicle or a drug delivery system influence the permeation [
7]. To overcome these challenges, several physical and chemical methods have been employed to enhance the transport of drugs through the skin. Physical methods include approaches such as microneedles, thermal ablation, radiofrequency, iontophoresis, ballistic liquid jets, laser, and others [
4,
8,
9,
10,
11]. However, these methods are known to cause irritation to the skin due to mechanical, thermal, magnetic, and electrical energy [
8]. Chemical methods include the use of penetration enhancers such as propylene glycol, ethanol, transcutol, and others to enhance the drug transport through the skin. They increase the diffusion of drugs through the skin by interacting and altering the complex structure of skin and thus enhancing the partition of drug into different layers [
12,
13]. Several penetration enhancers have been approved in the market, but their application in topical and transdermal formulations is limited as there is no clear understanding on how these agents enhance drug transport [
14]. In addition to penetration enhancers, several other excipients/additives such as solvents, co-solvents, surfactants, humectants, thickening agents, and others are used in the development of topical/transdermal formulation. These agents act as inactive ingredients and control the extent of absorption (thermodynamic activity and partition coefficient), maintain the viscosity and pH, improve the stability as well as organoleptic properties, and increase the bulk of the formulation [
15,
16]. Similar to every other dosage form, topical formulation development program also involves pre-formulation development, formulation development, performance (in vitro and in vivo), and stability. A well-designed Quality Target Product Profile (QTPP) provides a structure to ensure that a formulation scientist embarks on a product development program that is efficient and yet defines a listing of all relevant medical, technical, and scientific information required to reach the desired commercial development outcome [
17]. However, formulation scientists face several challenges while developing a drug product with desirable QTPP. In case of topical product development, achieving the target flux is a challenge as it is dependent of several factors.
Percutaneous drug absorption is a process which involves steps such as (i) dissolution and release of drug from the vehicle/formulation, (ii) partition of drug into the stratum corneum, (iii) diffusion of the solubilized drug across the stratum corneum, and (iv) penetration of drug into the layers of the skin [
18]. The goal in the development of any topical/transdermal drug formulation is to achieve maximum flux across the skin without any drug build-up. Critical factors which influence the flux across the skin include concentration of drug in the vehicle/formulation, physical state of drug in the formulation, and other formulation properties. Concentration of drug in the formulation is important as a proportional increase in the flux can be achieved by increasing the concentration of the dissolved drug. According to Fick’s law of diffusion (Equation (1)), at a higher concentration above the solubility, the excess drug in the formulation acts as a reservoir and helps in maintaining constant flux for a prolonged period and thus increases the permeation [
19]. Physical state of the drug in the formulation (solubilized drug vs. dispersed/suspended drug) can also significantly affect the permeation. It is known that greater flux is achieved when the drug is in solubilized form compared to suspended form. Enhanced permeation is attributed to increase in thermodynamic activity and partition with solubilized drug. Thus, the solubilized systems have advantages such as increased efficacy at lower concentrations, low drug irritation potential and cost efficient [
16]. In addition to the above, formulation properties such as type of formulation (monophasic vs. multiphasic systems), viscosity, pH, and other organoleptic properties significantly affect the transport of drug across the skin. Therefore, pharmaceutical formulation scientists must consider all the above factors in the development of any new topical drug product. Since the inception of topical dosage forms, numerous excipients have been investigated for their application in conventional dosage forms, such as creams, gels, and ointments. Although several excipients have been approved by the United States Food and Drug Administration (USFDA) for topical use, formulation scientists face challenges in the development of topical drug product with desired permeation profile. In addition, efforts have also been made in development of novel formulations, such as microemulsions, nanoparticulate drug delivery systems, eutectic mixtures, patches, and others to enhance the permeation of drugs across the skin [
20,
21]. In general, there is a lack of scientific evaluation on how the formulation properties, such as concentration of drug, physical state of the drug, and formulation type, influence the bioavailability of conventional topical dosage forms, such as creams and gels. Although several studies have evaluated the effect of formulation properties on transdermal permeation of drugs, the scientific data show that the research was limited to only one factor, such as concentration of drug [
22], concentration of excipients [
7,
23], and formulation type [
24,
25]. Therefore, the current study was designed to provide ready information to the formulators who design and develop topical semisolid formulations on the impact of formulation properties (concentration of drug, physical state of the drug, mucoadhesive agents, and formulation type) on transdermal permeation.
Equation (1): Fick’s law of diffusion, where J is the steady-state flux of the drug molecule through the skin (µg/cm2·h), K is the permeability coefficient (cm/s), ΔC is the difference in concentration (µg/cm3), D is the diffusion coefficient (cm2/s), Kp is the apparent partition coefficient, and h is the thickness of the layer of skin (cm).
Non-steroidal anti-inflammatory drugs (NSAIDs) are used to treat local pain and inflammation associated with injuries, rheumatoid arthritis, osteoarthritis, and other musculoskeletal problems [
7,
26]. Although, NSAIDs are very effective, their oral absorption is associated with severe gastric irritation leading to gastric bleeding and ulcers. Therefore, topical/transdermal delivery of NSAIDs is preferred as it bypasses hepatic first pass metabolism and also results in targeted effect at the site of inflammation/pain [
24]. Majority of the NSAIDs (salicylates, acetic acid derivatives, enol acid derivatives, and propionic acid derivatives) approved by USFDA have similar physicochemical properties (molecular mass, log
P, and p
Ka) [
27,
28,
29,
30]. Hence, it can be assumed that there may be similarities in transdermal permeation for these compounds [
29]. Among these agents, ibuprofen is the most commonly used NSAID. Ibuprofen (α-methyl-4-(2-methylpropyl) benzeneacetic acid) is a weak acid (p
Ka 4.5–4.6), thus the pH of the skin (~4.8) favors passive diffusion as majority of the molecules will be in unionized form. However, poor aqueous solubility (0.084 and 0.685 mg/L at pH 4.5 and 5.54, respectively) limits the skin permeation of ibuprofen. Ibuprofen is considered as an attractive candidate for topical/percutaneous delivery due to the physicochemical properties (low molecular weight (MW: 206.29 g·mol
−1), suitable partition coefficient (log
P: 3.68), and short elimination half-life (
t1/2 2–4 h), [
7]. Currently, topical formulations of ibuprofen are not approved in the United States. Therefore, taking into account all the above factors and availability of drug for research purposes, ibuprofen was chosen as the model drug for our study. The main goal of the study was to prepare semisolid formulations and investigate the effect of concentration of drug, formulation type and physical state of drug on transdermal permeation of ibuprofen. All the excipients (except Sepineo SE 68) used were approved by the USFDA for topical use and were within the limits listed in the inactive ingredient database. In the present study, we have developed ibuprofen topical creams at two concentrations (3% and 5%
w/
w)—emulgel (3%
w/
w) and clear non-aqueous gel (3%
w/
w). Further, in-vitro permeation studies were performed across the Strat-M
® membrane to study the effect of various formulation parameters on the permeation of ibuprofen.
2. Materials
Ibuprofen, hydroxy propyl methyl cellulose (HPMC) (MW: 86,000, viscosity 4000 cps at 2% solution), and hydroxy propyl cellulose were purchased from Acros Organics (Fair Lawn, NJ, USA). Absolute ethanol, sorbitan monolaurate (Span 20), sodium chloride, glacial acetic acid, acetonitrile (HPLC grade) were procured from Fisher Chemicals (Fair Lawn, NJ, USA). Deionized water used in all the experiments was obtained from in-house Milli-Q® IQ 7000 Ultrapure Water System (EMD Millipore, Bedford, MA, USA). Mineral oil NF and white petrolatum were purchased from PCCA (Houston, TX, USA). Tefose® 63 (mixture of PEG-6 stearate NF/JPE and Ethylene glycol palmitostearate EP/NF/JPE) and Transcutol® (Diethylene glycol monoethyl ether EP/NF) were gift samples from Gattefossé (Paramus, NJ, USA). Kollicream® IPM (isopropyl myristate), Kollicream® OA (oleyl alcohol), Kollisolv® MCT 70 (medium-chain triglycerides), Kollisolv® PEG 400 (polyethylene glycol 400), Kollisolv® PG (propylene glycol), Kolliphor® CS 20 (macrogol cetostearyl ether 20/polyoxyl 20 cetostearyl ether), Kolliphor® PS 80 (polysorbate 80), Kolliphor® CS A (cetostearyl alcohol (type A)), Kolliwax® CA (cetyl alcohol), and Kolliwax® SA (stearyl alcohol) were generous samples from BASF (Tarrytown, NY, USA). Glycerol monostearate was obtained from Alfa Aesar (Ward Hill, MA, USA). Strat-M® membrane and glycerol were procured from Sigma-Aldrich (St. Louis, MO, USA). Carbopol 974P (Carbomer Homopolymer Type B) was a sample from Lubrizol Life Sciences (Cleveland, OH, USA). Sepineo™ P600 (acrylamide/sodium acryloyldimethyl taurate copolymer/isohexadecane and Polysorbate 80) and Sepineo™ SE 68 (cetearyl alcohol, cetearyl glucoside) were gift samples from Seppic Inc (Fairfield, NJ, USA).
3. Methods
3.1. Solubility of Ibuprofen in Solvents
The solubility of ibuprofen in liquid excipients was determined using visual solubility protocol. In this method, the excipients were accurately weighed (2.5 g) in individually-labelled 20 mL scintillation vials. To these vials, accurately weighed aliquots of ibuprofen (~5 mg for glycerol due to poor solubility and ~25 mg for other excipients) was added and tightly closed. Further, the vials were placed in a shaking water bath (Fisher Scientific, Waltham, MA, USA) maintained at 25 °C for at least 15 min to allow proper mixing. After 15 min, the vials were visually inspected, and additional aliquots of ibuprofen were added periodically (every 15 min) until saturation was achieved. Following this, the vials were placed in the shaking water bath for 24 h and visually inspected the following day. The final weight of the vials was measured to determine the approximate solubility of ibuprofen in each excipient and reported as mg/g and percentage (%).
3.2. Formulation of Ibuprofen Creams and Gels
3.2.1. Optimization of Formulations
Optimization of all the formulations was performed by evaluating the effect of different concentrations of excipients on the stability, excipient instability, viscosity, and any visual changes in the formulations (
Appendix A). After optimization, stable formulations which provided acceptable appearance and viscosity were chosen for further evaluation. Compositions of all the optimized creams, emulgel and clear gel are provided in
Table 1,
Table 2, and
Table 3, respectively. All the formulations had differences in composition since the main aim of this study was to evaluate on how formulation parameters such as concentration and physical state of the drug, formulation type and mucoadhesive agents influence the permeation of ibuprofen.
3.2.2. Formulation of Creams
Compositions of the creams developed is provided in
Table 1. Ibuprofen creams were developed at two different strengths (3%
w/
w (F1A, F1B, F2A, F2B) and 5%
w/
w (F3, F4)) using a water-in-oil (w/o) emulsion method. Briefly, ibuprofen was accurately weighed and transferred into a 250 mL beaker containing all the required oil phase components for each formulation. In another 100 mL beaker, accurately weighed water soluble components were dissolved in water. Both the beakers were placed in a water bath and heated to 65 ± 2 °C. Once both the phases reached approximately similar temperature, the aqueous phase was added to the oil phase and homogenized at 5000 rpm using a high shear homogenizer (Fisherbrand™ 850, Waltham, MA, USA) for 10 min to form an emulsion. After homogenization, the emulsion was allowed to cool down to room temperature by mixing it using an overhead stirrer (IKA RW20 Digital, Wilmington, NC, USA) at 300 rpm for 2 h until a smooth cream was obtained. Sepineo P600 was added to the mixture during the process of homogenization for formulations F3 and F5.
3.2.3. Formulation of Emulgel
Composition of ibuprofen emulgel (F5) (3%
w/
w) is provided in
Table 2. Ibuprofen (3 g) was dissolved in ethanol (30 mL) and water was added to ibuprofen solution. To this mixture, accurately weighed Sepineo P600 was added immediately and vigorously mixed using a glass rod until a smooth emulgel was formed.
3.2.4. Formulation of Clear Non-Aqueous Gel
Composition of ibuprofen clear non aqueous gel (F6) (3%
w/
w) is provided in
Table 3. Ibuprofen was weighed and added to a mixture of propylene glycol, ethanol, transcutol, and glycerin to obtain a clear solution. To this clear solution, PEG 400 was added and mixed on a magnetic stirrer. Accurately weighed HPC (4 g) was dispersed in the mixture and allowed to thicken at room temperature using an overhead mixer (IKA RW20 Digital, Wilmington, NC, USA) at 500 rpm for 2 h.
3.3. Polarized Light Microscopy
Polarized light microscopy was used to study the microscopic features of the optimized creams and gels. All the formulations were applied on a microscopic glass slide and evenly spread with a coverslip. The cover slipped slides were observed under Amscope® PZ300 series polarized light microscope (Amscope, Irvine, CA, USA) in the transmission mode at 180× magnification and photomicrographs were captured on a laboratory PC.
3.4. HPLC Analysis of Ibuprofen
The amount of ibuprofen in the samples was quantified using Waters Alliance e2695 HPLC equipped with 2998 photodiode array detector and Empower 3.0 software. The analysis was carried out on a reverse phase Phenomenex® C18 column (250 × 4.6 mm; 5 µm particle size) at 25 °C. The mobile phase was a mixture (60:40) of acetonitrile and water (adjusted to pH 3.8 with acetic acid) at a constant flow rate of 1.5 mL/min. Samples (60 µL) were injected into the column using autosampler and monitored at 220 nm. Retention time of ibuprofen was 6.5 min. All the samples injected were filtered through 0.45 µm membrane filter.
3.5. Measurement of Viscosity and pH
Rheological experiments were conducted to measure the viscosity of the optimized formulations. Measurements were performed at room temperature using a Viscolead-one digital viscometer (Fungilab Inc. New York, NY, USA) equipped with a spindle rotor (R6) set at 20 rpm. The method was validated by using 2% HPMC gel (4000 cps) as a control. The pH of the formulations was evaluated on day 0 and day 60 using a calibrated Mettler Toledo InLab® pH meter equipped with LE422 micro pH electrode (Mettler Toledo, Columbus, OH, USA).
3.6. In-Vitro Permeation Studies
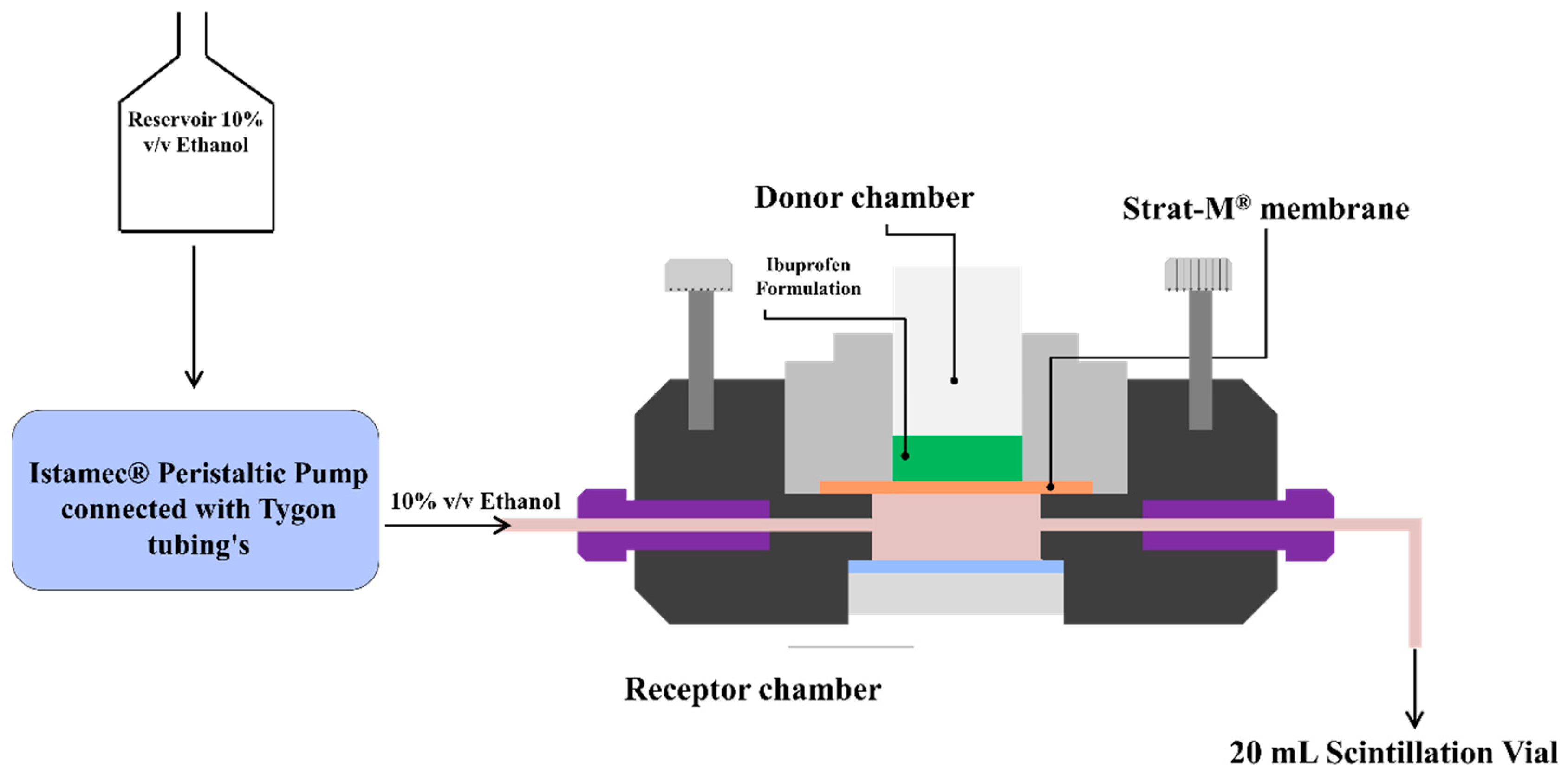
In-vitro permeation studies were conducted using a PermeGear® ILC-07 automated system (PermeGear, Riegelsville, PA, USA) equipped with seven in-line flow-through diffusion cells, made of Kel-F. Each diffusion cell had a donor and receptor chamber clamped with threaded rods and adjustable locking nuts. Receptor chambers (volume: 254 µL receptor) had inlet and outlet ports connected to the Tygon tubings having 1/4-28 HPLC fittings. Temperature of the cells was maintained at 32 °C using Julabo BC4 circulating water bath (Seelbach, Germany). Diameter of the diffusional area was 1 cm (total diffusional area: 0.785 cm
2) and the cells were connected to a 7-channel peristaltic pump® IPC (Ismatec, Zurich, Switzerland) which draws receptor solution from a reservoir (
Figure 1). Strat-M
® was used as a diffusion membrane, which was mounted on the cells and sandwiched between the donor and receptor chambers using the adjustable locking nuts. Formulations (~10 mg ibuprofen) were placed on the diffusion membrane and the receptor fluid (10%
v/
v ethanol) was allowed to flow at a rate of 4 mL/h for 24 h. At pre-determined time intervals, the receptor fluid was collected in 20 mL scintillation vials and analyzed using HPLC to determine the amount of ibuprofen permeated through the Strat-M
® membrane.
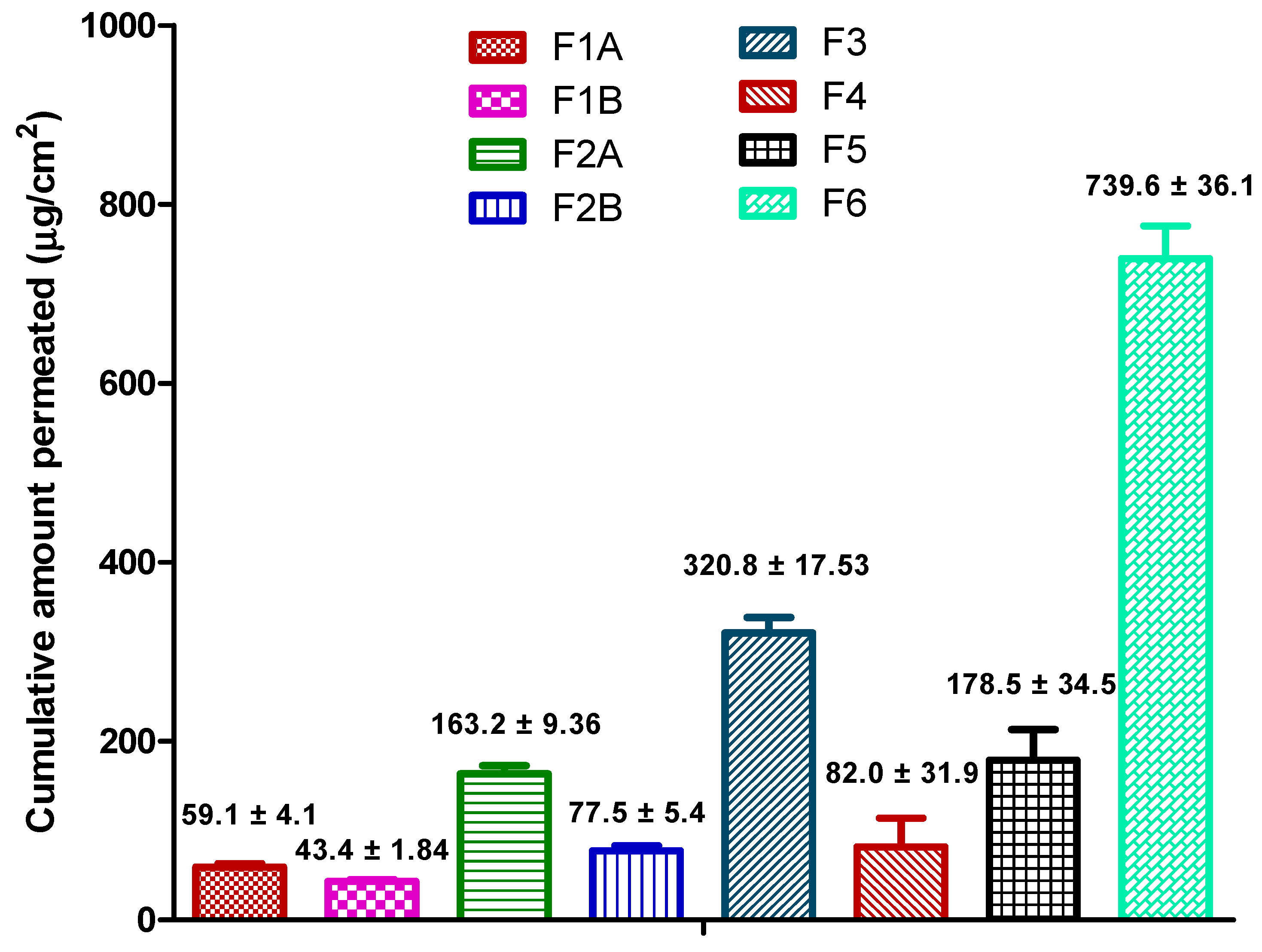
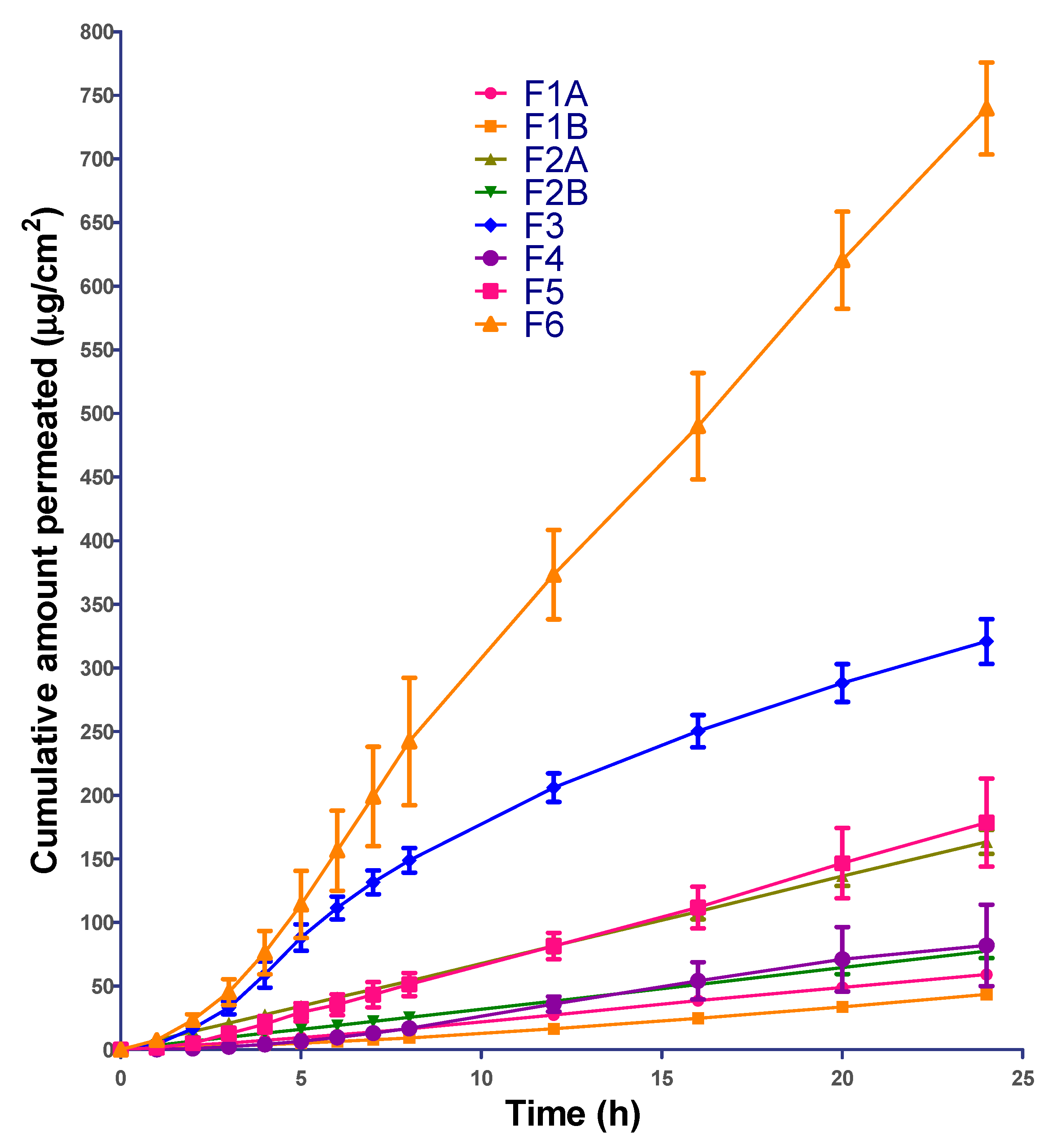
3.7. Permeation Data Analysis
The permeation profile from the formulations was plotted as cumulative amount of ibuprofen permeated vs. time. The flux (µg/cm
2/h) and lag-time (h) estimates were generated using Skin and Membrane Permeation Data Analysis (SAMPA) software, version 1.04, a free software tool used for skin and membrane permeation data analysis [
31].
3.8. Physical Stability
Physical stability studies were conducted for all the formulations at 25 ± 2 °C and at 40 ± 2 °C. All the samples were transferred to glass scintillation vials, closed tightly and stored at 25 ± 2 °C and 40 ± 2 °C. Samples were evaluated for stability, changes in color, and any other physical instability for 90 days.
3.9. Statistical Analysis
All the data was statistically analyzed using GraphPad Prism software (Version 5.0, San Diego, CA, USA). Permeation data analysis was performed using SAMPA software, version 1.04. A p-value of <0.05 was considered as statistically significant.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




