Nanopharmaceutics: Part I—Clinical Trials Legislation and Good Manufacturing Practices (GMP) of Nanotherapeutics in the EU

 , , , ,
, , , ,  ,
, 

Abstract
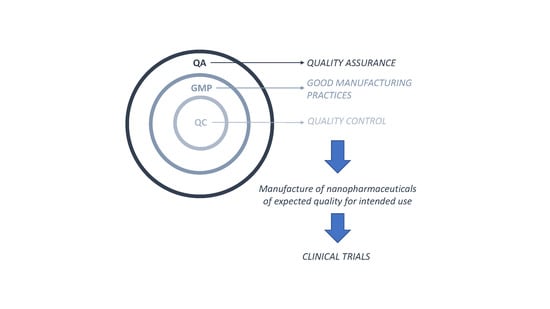
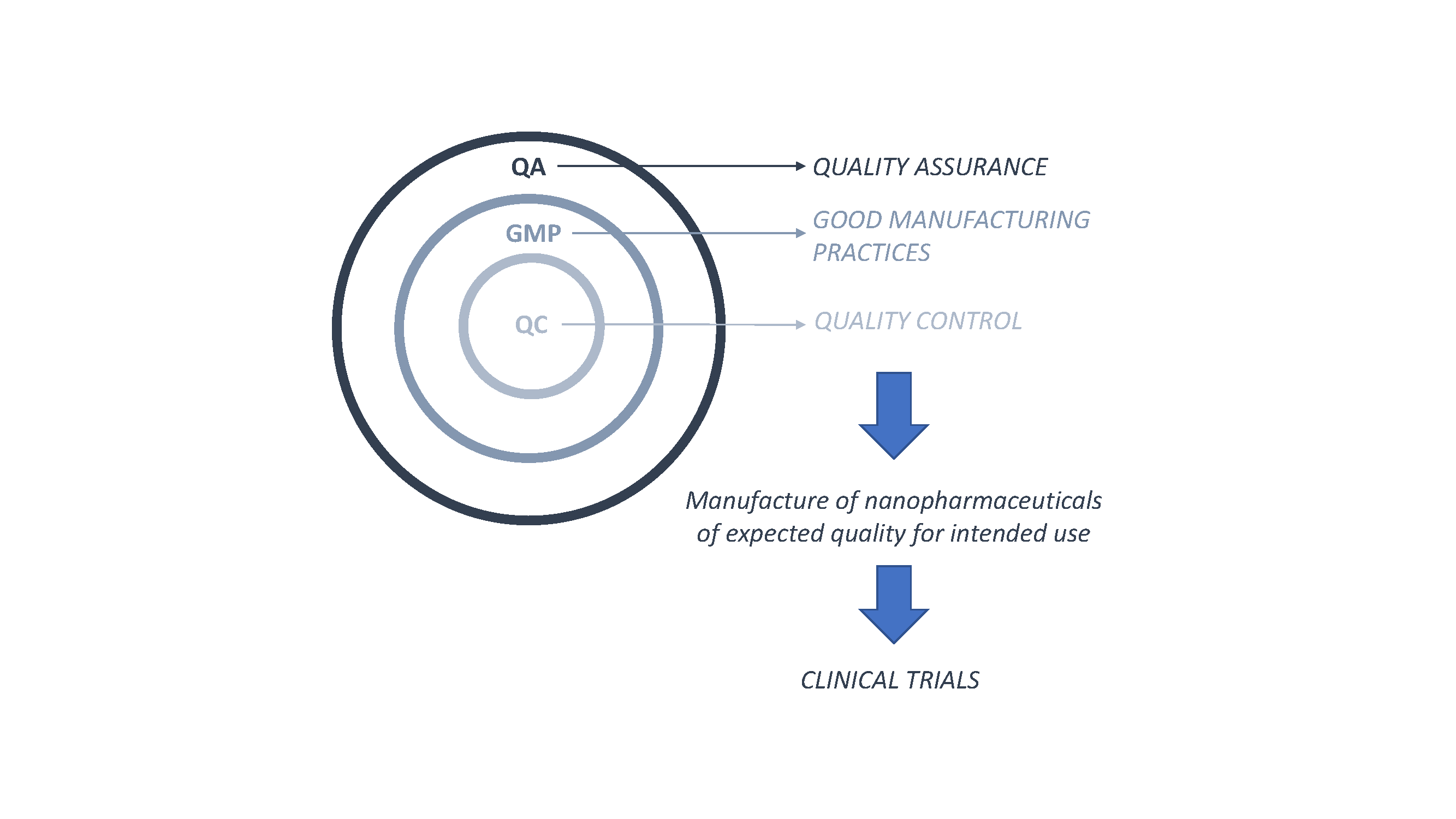{kind=link}
{kind=link}
1. Introduction
2. Regulatory Framework of Clinical Trials
2.1. European Legislation
2.2. Nanopharmaceuticals in Clinical Trials
3. Good Manufacturing Practices Applied to Nanopharmaceuticals
3.1. Intravenous Iron-Based Nanopharmaceuticals
3.2. Liposomal-Based Nanopharmaceuticals
3.3. Block Copolymer Micelle-Based Nanopharmaceuticals
3.4. Surface Coating Requirements
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vieira, R.; Souto, S.B.; Sanchez-Lopez, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; Garcia, M.L.; Silva, A.M.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome-Review of Classical and New Compounds: Part-I. Pharmaceuticals 2019, 12, 152. [Google Scholar] [CrossRef] [PubMed]
- Vieira, R.; Souto, S.B.; Sanchez-Lopez, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Silva, A.M.; Fortuna, A.; Garcia, M.L.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome-Strategies for In Vivo Administration: Part-II. J. Clin. Med. 2019, 8, 1332. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Guerra, M.; Dias-Ferreira, J.; Lopez-Machado, A.; Ettcheto, M.; Cano, A.; Espina, M.; Camins, A.; Garcia, M.L.; Souto, E.B. Current Applications of Nanoemulsions in Cancer Therapeutics. Nanomaterials 2019, 9, 821. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lopez, E.; Egea, M.A.; Davis, B.M.; Guo, L.; Espina, M.; Silva, A.M.; Calpena, A.C.; Souto, E.M.B.; Ravindran, N.; Ettcheto, M.; et al. Memantine-Loaded PEGylated Biodegradable Nanoparticles for the Treatment of Glaucoma. Small 2018, 14. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Egea, M.A.; Cano, A.; Espina, M.; Calpena, A.C.; Ettcheto, M.; Camins, A.; Souto, E.B.; Silva, A.M.; Garcia, M.L. PEGylated PLGA nanospheres optimized by design of experiments for ocular administration of dexibuprofen-in vitro, ex vivo and in vivo characterization. Colloids Surf B Biointerfaces 2016, 145, 241–250. [Google Scholar] [CrossRef]
- Souto, E.B.; Zielinska, A.; Luis, M.; Carbone, C.; Martins-Gomes, C.; Souto, S.B.; Silva, A.M. Uveal melanoma: Physiopathology and new in situ-specific therapies. Cancer Chemother. Pharmacol. 2019, 84, 15–32. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Gomes, D.; Esteruelas, G.; Bonilla, L.; Lopez-Machado, A.L.; Galindo, R.; Cano, A.; Espina, M.; Ettcheto, M.; Camins, A.; et al. Metal-Based Nanoparticles as Antimicrobial Agents: An Overview. Nanomaterials 2020, 10, 292. [Google Scholar] [CrossRef]
- Souto, E.B.; Souto, S.B.; Campos, J.R.; Severino, P.; Pashirova, T.N.; Zakharova, L.Y.; Silva, A.M.; Durazzo, A.; Lucarini, M.; Izzo, A.A.; et al. Nanoparticle Delivery Systems in the Treatment of Diabetes Complications. Molecules 2019, 24, 4209. [Google Scholar] [CrossRef]
- Souto, E.B.; Doktorovova, S.; Campos, J.R.; Martins-Lopes, P.; Silva, A.M. Surface-tailored anti-HER2/neu-solid lipid nanoparticles for site-specific targeting MCF-7 and BT-474 breast cancer cells. Eur. J. Pharm. Sci. 2019, 128, 27–35. [Google Scholar] [CrossRef]
- Jose, S.; Cinu, T.A.; Sebastian, R.; Shoja, M.H.; Aleykutty, N.A.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B. Transferrin-Conjugated Docetaxel-PLGA Nanoparticles for Tumor Targeting: Influence on MCF-7 Cell Cycle. Polymers 2019, 11, 1905. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Ettcheto, M.; Egea, M.A.; Espina, M.; Cano, A.; Calpena, A.C.; Camins, A.; Carmona, N.; Silva, A.M.; Souto, E.B.; et al. Memantine loaded PLGA PEGylated nanoparticles for Alzheimer’s disease: In vitro and in vivo characterization. J. Nanobiotechnol. 2018, 16, 32. [Google Scholar] [CrossRef] [PubMed]
- Hofmann-Amtenbrink, M.; Hofmann, H.; Hool, A.; Roubert, F. Nanotechnology in medicine: European research and its implications. Swiss Med. Wkly. 2014, 144, w14044. [Google Scholar] [CrossRef]
- Ji, X.; Lu, W.; Wu, K.; Cho, W.C. Influencing Factors of the Pharmacokinetic Characters on Nanopharmaceutics. Pharm. Nanotechnol. 2017, 5, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, J.; Ferrari, M. Clinical Cancer Nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef]
- Andrade, L.N.; Oliveira, D.M.L.; Chaud, M.V.; Alves, T.F.R.; Nery, M.; da Silva, C.F.; Gonsalves, J.K.C.; Nunes, R.S.; Correa, C.B.; Amaral, R.G.; et al. Praziquantel-Solid Lipid Nanoparticles Produced by Supercritical Carbon Dioxide Extraction: Physicochemical Characterization, Release Profile, and Cytotoxicity. Molecules 2019, 24, 3881. [Google Scholar] [CrossRef] [PubMed]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Furberg, C.D.; Friedman, L.M. Approaches to data analyses of clinical trials. Prog. Cardiovasc. Dis. 2012, 54, 330–334. [Google Scholar] [CrossRef]
- May, G.S.; DeMets, D.L.; Friedman, L.M.; Furberg, C.; Passamani, E. The randomized clinical trial: Bias in analysis. Circulation 1981, 64, 669–673. [Google Scholar] [CrossRef]
- McNally, G.A.; Hrnicek, A.K.; Barr, H. Clinical Trial Amendments: Development of a Process for Consistent Implementation. Clin. J. Oncol. Nurs. 2017, 21, 627–628. [Google Scholar] [CrossRef]
- Friedman, L.; Passamani, E.; May, G.; Furberg, C.; Friedewald, W. Placebo controlled double blind trials. Lancet 1980, 2, 147–148. [Google Scholar] [CrossRef]
- Friedman, L.M.; Furberg, C.D. Community hospitals in clinical trials. N. Engl. J. Med. 1982, 307, 958. [Google Scholar] [CrossRef] [PubMed]
- European Commission—EudraLex—EU Legislation. Available online: https://ec.europa.eu/health/documents/eudralex_en (accessed on 7 March 2017).
- Official Journal of the European Communities—DIRECTIVE 2001/20/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 4 April 2001. 2001. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32001L0020&from=EN (accessed on 5 March 2017).
- Official Journal of the European Communities—COMMISSION DIRECTIVE 2003/94/EC of 8 October 2003. 2003. Available online: http://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2003_94/dir_2003_94_en.pdf (accessed on 7 March 2017).
- EudraLex The Rules Governing Medicinal Products in the European Union Volume 4—Good Manufacturing Practice Medicinal Products for Human and Veterinary Use Annex 13. 2010. Available online: http://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2009_06_annex13.pdf (accessed on 10 March 2017).
- Official Journal of the European Communities—COMMISSION DIRECTIVE 2005/28/EC of 8 April 2005. 2005. Available online: http://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2005_28/dir_2005_28_en.pdf (accessed on 14 March 2017).
- The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)—Mission. Available online: http://www.ich.org/about/mission.html (accessed on 14 March 2017).
- The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)—INTEGRATED ADDENDUM TO ICH E6(R1): GUIDELINE FOR GOOD CLINICAL PRACTICE E6(R2) (Current Step 4 version dated 9 November 2016). Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2Step_4.pdf (accessed on 14 March 2017).
- Petrini, C. Regulation (EU) No 536/2014 on clinical trials on medicinal products for human use: An overview. Ann. Ist. Super. Sanità 2014, 50, 317–321. [Google Scholar] [PubMed]
- Official Journal of the European Union—REGULATION (EU) No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014. 2014. Available online: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf (accessed on 14 March 2017).
- Assembleia da República—Lei n.º 21/2014, de 16 de abril. 2014. Available online: http://www.infarmed.pt/documents/15786/1068535/036-B1_Lei_21_2014_1alt.pdf (accessed on 18 March 2017).
- Assembleia da República—Lei n.º 73/2015 de 27 de julho. Diário da República. 2015. Available online: http://www.ceic.pt/documents/20727/38721/Lei+n.%C2%BA+73-2015+de+27+de+julho/a04332bb-581e-46a2-b0de-ef714aeb1112 (accessed on 18 March 2017).
- INFARMED—Autoridade Nacional do Medicamento e Produtos de Saúde, I.P.—Medicamentos de Uso Humano—Ensaios Clínicos. Available online: http://www.infarmed.pt/web/infarmed/entidades/medicamentos-uso-humano/ensaios-clinicos (accessed on 18 March 2017).
- Zhang, L.; Gu, F.; Chan, J.; Wang, A.; Langer, R.; Farokhzad, O. Nanoparticles in Medicine: Therapeutic Applications and Developments. Clin. Pharm. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef] [PubMed]
- European Union—Clinical Trials Register website. Available online: https://www.clinicaltrialsregister.eu/ (accessed on 25 March 2017).
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.C.; Carbone, C.; Souto, E.B. Beyond liposomes: Recent advances on lipid based nanostructures for poorly soluble/poorly permeable drug delivery. Prog. Lipid. Res. 2017, 68, 1–11. [Google Scholar] [CrossRef]
- Clares, B.; Calpena, A.C.; Parra, A.; Abrego, G.; Alvarado, H.; Fangueiro, J.F.; Souto, E.B. Nanoemulsions (NEs), liposomes (LPs) and solid lipid nanoparticles (SLNs) for retinyl palmitate: Effect on skin permeation. Int. J. Pharm. 2014, 473, 591–598. [Google Scholar] [CrossRef]
- Goncalves, A.S.; Macedo, A.S.; Souto, E.B. Therapeutic nanosystems for oncology nanomedicine. Clin. Transl. Oncol. 2012, 14, 883–890. [Google Scholar] [CrossRef]
- Andreani, T.; Miziara, L.; Lorenzon, E.N.; de Souza, A.L.; Kiill, C.P.; Fangueiro, J.F.; Garcia, M.L.; Gremiao, P.D.; Silva, A.M.; Souto, E.B. Effect of mucoadhesive polymers on the in vitro performance of insulin-loaded silica nanoparticles: Interactions with mucin and biomembrane models. Eur. J. Pharm. Biopharm. 2015, 93, 118–126. [Google Scholar] [CrossRef]
- Andreani, T.; de Souza, A.L.; Kiill, C.P.; Lorenzon, E.N.; Fangueiro, J.F.; Calpena, A.C.; Chaud, M.V.; Garcia, M.L.; Gremiao, M.P.; Silva, A.M.; et al. Preparation and characterization of PEG-coated silica nanoparticles for oral insulin delivery. Int. J. Pharm. 2014, 473, 627–635. [Google Scholar] [CrossRef]
- Severino, P.; Souto, E.B.; Pinho, S.C.; Santana, M.H. Hydrophilic coating of mitotane-loaded lipid nanoparticles: Preliminary studies for mucosal adhesion. Pharm. Dev. Technol. 2013, 18, 577–581. [Google Scholar] [CrossRef]
- Cano, A.; Ettcheto, M.; Chang, J.H.; Barroso, E.; Espina, M.; Kuhne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s disease mice model. J. Control. Release 2019, 301, 62–75. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA’s Approach to Regulation of Nanotechnology Products. USA: Last Updated 8 May 2015. Available online: https://www.fda.gov/scienceresearch/specialtopics/nanotechnology/ucm301114.htm (accessed on 3 September 2019).
- The European Medicines Agency Scientific Guidelines on Nanomedicines Help Medicine Developers Prepare Marketing Authorisation Applications for Human Medicines. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000564.jsp&mid=WC0b01ac05806403e0 (accessed on 3 September 2019).
- Havel, H.; Finch, G.; Strode, P.; Wolfgang, M.; Zale, S.; Bobe, I.; Youssoufian, H.; Peterson, M.; Liu, M. Nanomedicines: From Bench to Bedside and Beyond. AAPS J. 2016, 18, 1373–1378. [Google Scholar] [CrossRef]
- Guo, J.-W.; Lee, Y.-H.; Huang, H.-W.; Tzou, M.-C.; Wang, Y.-J.; Tsai, J.-C. Development of Taiwan’s strategies for regulating nanotechnology-based pharmaceuticals harmonized with international considerations. Int. J. Nanomed. 2014, 9, 4773–4783. [Google Scholar] [CrossRef]
- EMA Issues Draft Reflection Paper for Iron-Based Nano-Colloidal Products. Belgium: 11 February 2014. Available online: http://gabi-journal.net/news/ema-issues-draft-reflection-paper-for-iron-based-nano-colloidal-products (accessed on 3 September 2019).
- Committee for Medicinal Products for Human Use (CHMP). Reflection Paper on the Data Requirements for Intravenous Iron-Based Nano-Colloidal Products Developed with Reference to an Innovator Medicinal Product. EMA, 2015. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/03/WC500184922.pdf (accessed on 3 September 2019).
- Committee for Medicinal Products for Human Use (CHMP). Reflection Paper on the Data Requirements for Intravenous Liposomal Products Developed with Reference to an Innovator Liposomal Product. EMA, 2013. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/03/WC500140351.pdf (accessed on 3 September 2019).
- Gaucher, G.; Dufresne, M.-H.; Sant, V.P.; Kang, N.; Maysinger, D.; Leroux, J.-C. Block copolymer micelles: Preparation, characterization and application in drug delivery. J. Control. Release 2005, 109, 169–188. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP). Joint MHLW/EMA Reflection Paper on the Development of Block Copolymer Micelle Medicinal Products. EMA, 2013. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/01/WC500159411.pdf (accessed on 10 September 2019).
- Doktorovova, S.; Shegokar, R.; Martins-Lopes, P.; Silva, A.M.; Lopes, C.M.; Muller, R.H.; Souto, E.B. Modified Rose Bengal assay for surface hydrophobicity evaluation of cationic solid lipid nanoparticles (cSLN). Eur. J. Pharm. Sci. 2012, 45, 606–612. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP). Reflection Paper on Surface Coatings: General Issues for Consideration Regarding Parenteral Administration of Coated Nanomedicine Products. EMA, 2013. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/08/WC500147874.pdf (accessed on 10 September 2019).
- Daliu, P.; Santini, A.; Novellino, E. A decade of nutraceutical patents: Where are we now in 2018? Expert Opin. Ther. Pat. 2018, 28, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.; Tenore, G.C.; Novellino, E. Nutraceuticals: A paradigm of proactive medicine. Eur. J. Pharm. Sci. 2017, 96, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, A.; D’Addezio, L.; Camilli, E.; Piccinelli, R.; Turrini, A.; Marletta, L.; Marconi, S.; Lucarini, M.; Lisciani, S.; Gabrielli, P.; et al. From Plant Compounds to Botanicals and Back: A Current Snapshot. Molecules 2018, 23, 1844. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, A.; Lucarini, M. Extractable and Non-Extractable Antioxidants. Molecules 2019, 24, 1933. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M. A current shot and re-thinking of antioxidant research strategy. Braz. J. Anal. Chem. 2018, 5, 9–11. [Google Scholar] [CrossRef]
- Daliu, P.; Santini, A.; Novellino, E. From pharmaceuticals to nutraceuticals: Bridging disease prevention and management. Expert Rev. Clin. Pharmacol. 2019, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, A.; Lucarini, M.; Souto, E.B.; Cicala, C.; Caiazzo, E.; Izzo, A.A.; Novellino, E.; Santini, A. Polyphenols: A concise overview on the chemistry, occurrence, and human health. Phytother. Res. 2019, 33, 2221–2243. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.; Cammarata, S.M.; Capone, G.; Ianaro, A.; Tenore, G.C.; Pani, L.; Novellino, E. Nutraceuticals: Opening the debate for a regulatory framework. Br. J. Clin. Pharmacol. 2018, 84, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.; Novellino, E. Nutraceuticals—Shedding light on the grey area between pharmaceuticals and food. Expert Rev. Clin. Pharmacol. 2018, 11, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Pimentel-Moral, S.; Teixeira, M.C.; Fernandes, A.R.; Arráez-Román, D.; Martínez-Férez, A.; Segura-Carretero, A.; Souto, E.B. Lipid nanocarriers for the loading of polyphenols—A comprehensive review. Adv. Colloid Interface Sci. 2018, 260, 85–94. [Google Scholar] [CrossRef]
- Pimentel-Moral, S.; Teixeira, M.C.; Fernandes, A.R.; Borrás-Linares, I.; Arráez-Román, D.; Martínez-Férez, A.; Segura-Carretero, A.; Souto, E.B. Polyphenols-enriched Hibiscus sabdariffa extract-loaded nanostructured lipid carriers (NLC): Optimization by multi-response surface methodology. J. Drug Deliv. Sci. Technol. 2019, 49, 660–667. [Google Scholar] [CrossRef]
- Santos, I.S.; Ponte, B.M.; Boonme, P.; Silva, A.M.; Souto, E.B. Nanoencapsulation of polyphenols for protective effect against colon–rectal cancer. Biotechnol. Adv. 2013, 31, 514–523. [Google Scholar] [CrossRef]
- Pereira, I.; Zielinska, A.; Ferreira, N.R.; Silva, A.M.; Souto, E.B. Optimization of linalool-loaded solid lipid nanoparticles using experimental factorial design and long-term stability studies with a new centrifugal sedimentation method. Int. J. Pharm. 2018, 549, 261–270. [Google Scholar] [CrossRef]
- Zielinska, A.; Martins-Gomes, C.; Ferreira, N.R.; Silva, A.M.; Nowak, I.; Souto, E.B. Anti-inflammatory and anti-cancer activity of citral: Optimization of citral-loaded solid lipid nanoparticles (SLN) using experimental factorial design and LUMiSizer(R). Int. J. Pharm. 2018, 553, 428–440. [Google Scholar] [CrossRef]
- Zielinska, A.; Ferreira, N.R.; Durazzo, A.; Lucarini, M.; Cicero, N.; Mamouni, S.E.; Silva, A.M.; Nowak, I.; Santini, A.; Souto, E.B. Development and Optimization of Alpha-Pinene-Loaded Solid Lipid Nanoparticles (SLN) Using Experimental Factorial Design and Dispersion Analysis. Molecules 2019, 24, 2683. [Google Scholar] [CrossRef]
- Carbone, C.; Martins-Gomes, C.; Caddeo, C.; Silva, A.M.; Musumeci, T.; Pignatello, R.; Puglisi, G.; Souto, E.B. Mediterranean essential oils as precious matrix components and active ingredients of lipid nanoparticles. Int. J. Pharm. 2018, 548, 217–226. [Google Scholar] [CrossRef]
- Carbone, C.; Teixeira, M.D.C.; Sousa, M.D.C.; Martins-Gomes, C.; Silva, A.M.; Souto, E.M.B.; Musumeci, T. Clotrimazole-Loaded Mediterranean Essential Oils NLC: A Synergic Treatment of Candida Skin Infections. Pharmaceutics 2019, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Izzo, A.A.; Milić, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Santini, A.; Novellino, E. Nutraceuticals in hypercholesterolaemia: An overview. Br. J. Pharmacol. 2017, 174, 1450–1463. [Google Scholar] [CrossRef] [PubMed]
- Putignano, D.; Bruzzese, D.; Orlando, V.; Fiorentino, D.; Tettamanti, A.; Menditto, E. Differences in drug use between men and women: An Italian cross sectional study. BMC Womens Health 2017, 17, 73. [Google Scholar] [CrossRef]
- Menditto, E.; Cahir, C.; Aza-Pascual-Salcedo, M.; Bruzzese, D.; Poblador-Plou, B.; Malo, S.; Costa, E.; González-Rubio, F.; Gimeno-Miguel, A.; Orlando, V.; et al. Adherence to chronic medication in older populations: Application of a common protocol among three European cohorts. Patient Prefer. Adherence 2018, 12, 1975–1987. [Google Scholar] [CrossRef]
- Iolascon, G.; Gimigliano, F.; Moretti, A.; Riccio, I.; Di Gennaro, M.; Illario, M.; Monetti, V.M.; Orlando, V.; Menditto, E. Rates and reasons for lack of persistence with anti-osteoporotic drugs: Analysis of the Campania region database. Clin. Cases Miner. Bone Metab. 2016, 13, 127–130. [Google Scholar] [CrossRef]
- Scala, D.; Menditto, E.; Armellino, M.F.; Manguso, F.; Monetti, V.M.; Orlando, V.; Antonino, A.; Makoul, G.; De Palma, M. Italian translation and cultural adaptation of the communication assessment tool in an outpatient surgical clinic. BMC Health Serv. Res. 2016, 16, 163. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souto, E.B.; Silva, G.F.; Dias-Ferreira, J.; Zielinska, A.; Ventura, F.; Durazzo, A.; Lucarini, M.; Novellino, E.; Santini, A. Nanopharmaceutics: Part I—Clinical Trials Legislation and Good Manufacturing Practices (GMP) of Nanotherapeutics in the EU. Pharmaceutics 2020, 12, 146. https://doi.org/10.3390/pharmaceutics12020146
Souto EB, Silva GF, Dias-Ferreira J, Zielinska A, Ventura F, Durazzo A, Lucarini M, Novellino E, Santini A. Nanopharmaceutics: Part I—Clinical Trials Legislation and Good Manufacturing Practices (GMP) of Nanotherapeutics in the EU. Pharmaceutics. 2020; 12(2):146. https://doi.org/10.3390/pharmaceutics12020146
Chicago/Turabian StyleSouto, Eliana B., Gabriela F. Silva, João Dias-Ferreira, Aleksandra Zielinska, Fátima Ventura, Alessandra Durazzo, Massimo Lucarini, Ettore Novellino, and Antonello Santini. 2020. "Nanopharmaceutics: Part I—Clinical Trials Legislation and Good Manufacturing Practices (GMP) of Nanotherapeutics in the EU" Pharmaceutics 12, no. 2: 146. https://doi.org/10.3390/pharmaceutics12020146
APA StyleSouto, E. B., Silva, G. F., Dias-Ferreira, J., Zielinska, A., Ventura, F., Durazzo, A., Lucarini, M., Novellino, E., & Santini, A. (2020). Nanopharmaceutics: Part I—Clinical Trials Legislation and Good Manufacturing Practices (GMP) of Nanotherapeutics in the EU. Pharmaceutics, 12(2), 146. https://doi.org/10.3390/pharmaceutics12020146