The Importance of Poly(ethylene glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles

 , and
, and 
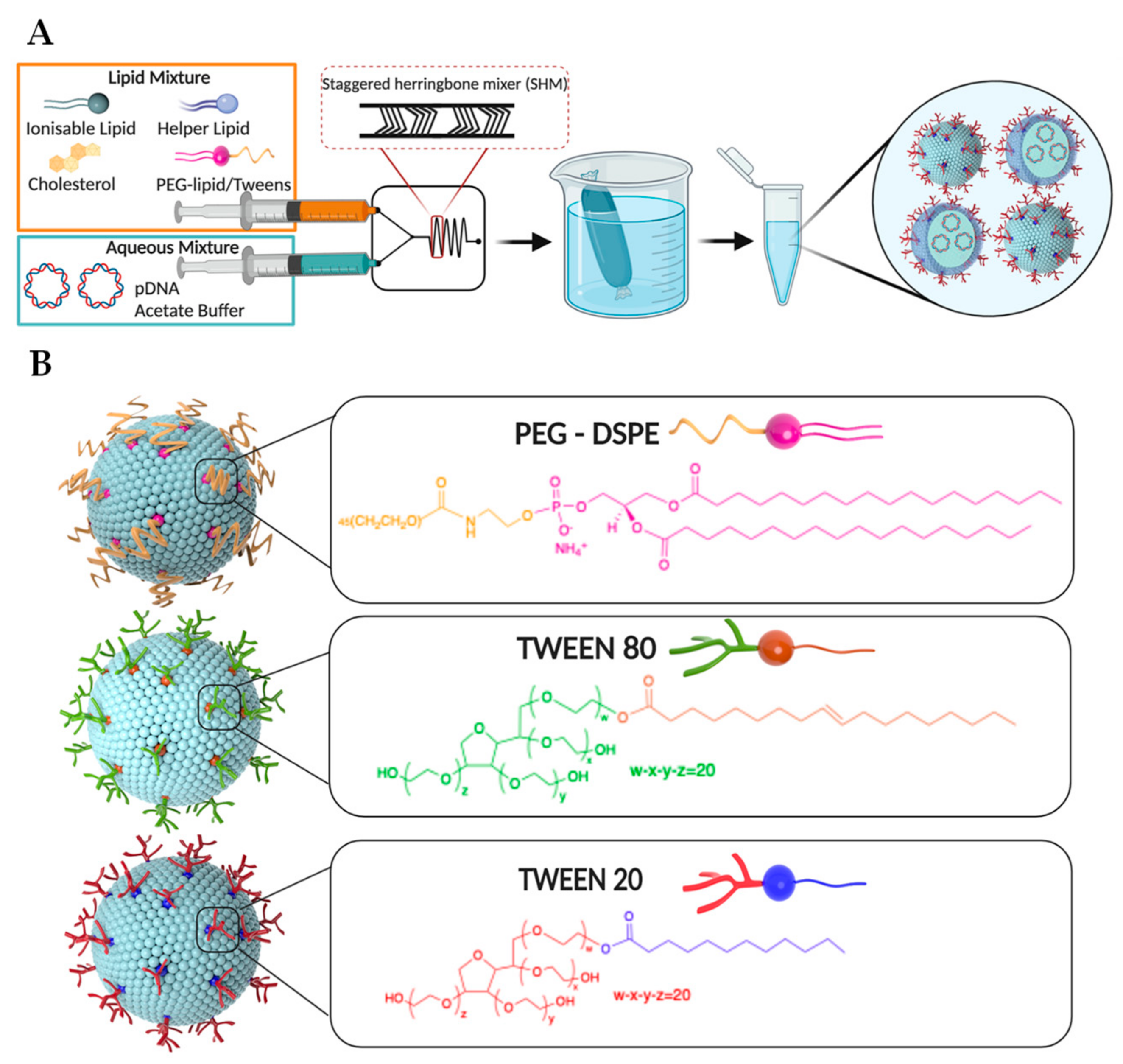{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Formulation of LNPs Containing pDNA
2.2.2. LNP Characterization
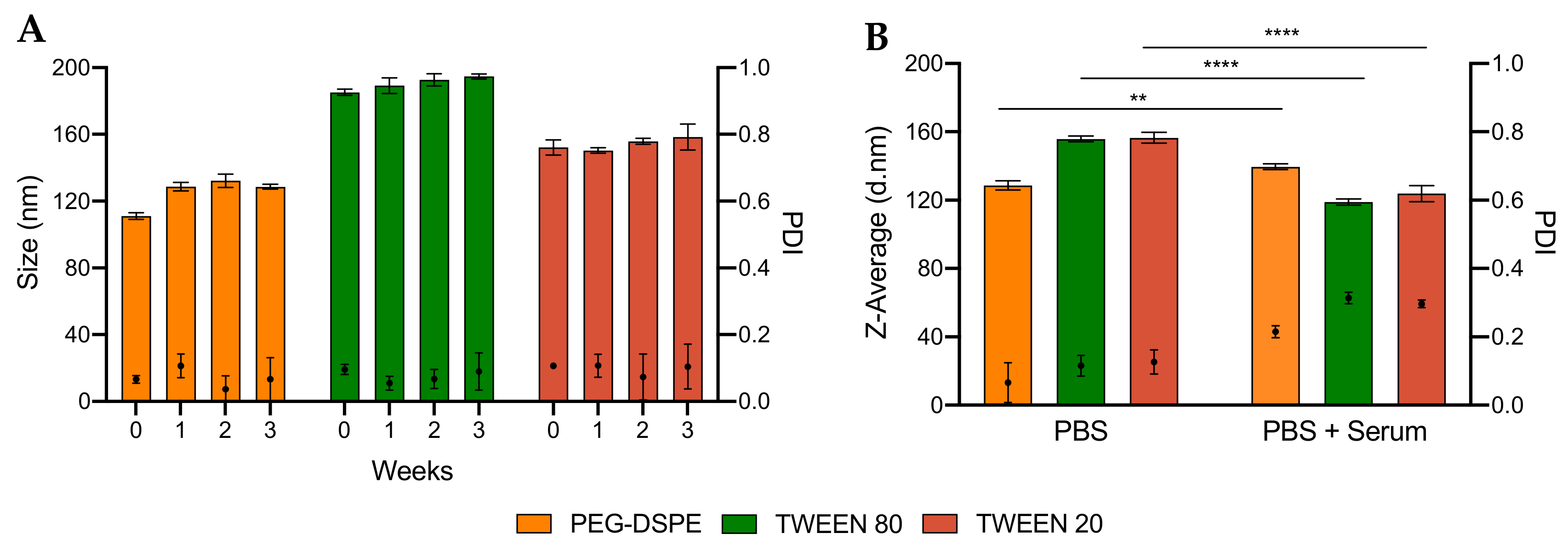
2.2.3. Analysis of Particle Stability
2.2.4. In vivo Gene Expression nLuc by LNPs
2.2.5. Ex vivo Whole-Organ Bioluminescence Imaging
2.2.6. NanoLuciferase (nLuc) Assay
2.2.7. Statistical Analysis
3. Results and Discussion
3.1. Synthesis of LNPs Encapsulating Plasmid DNA
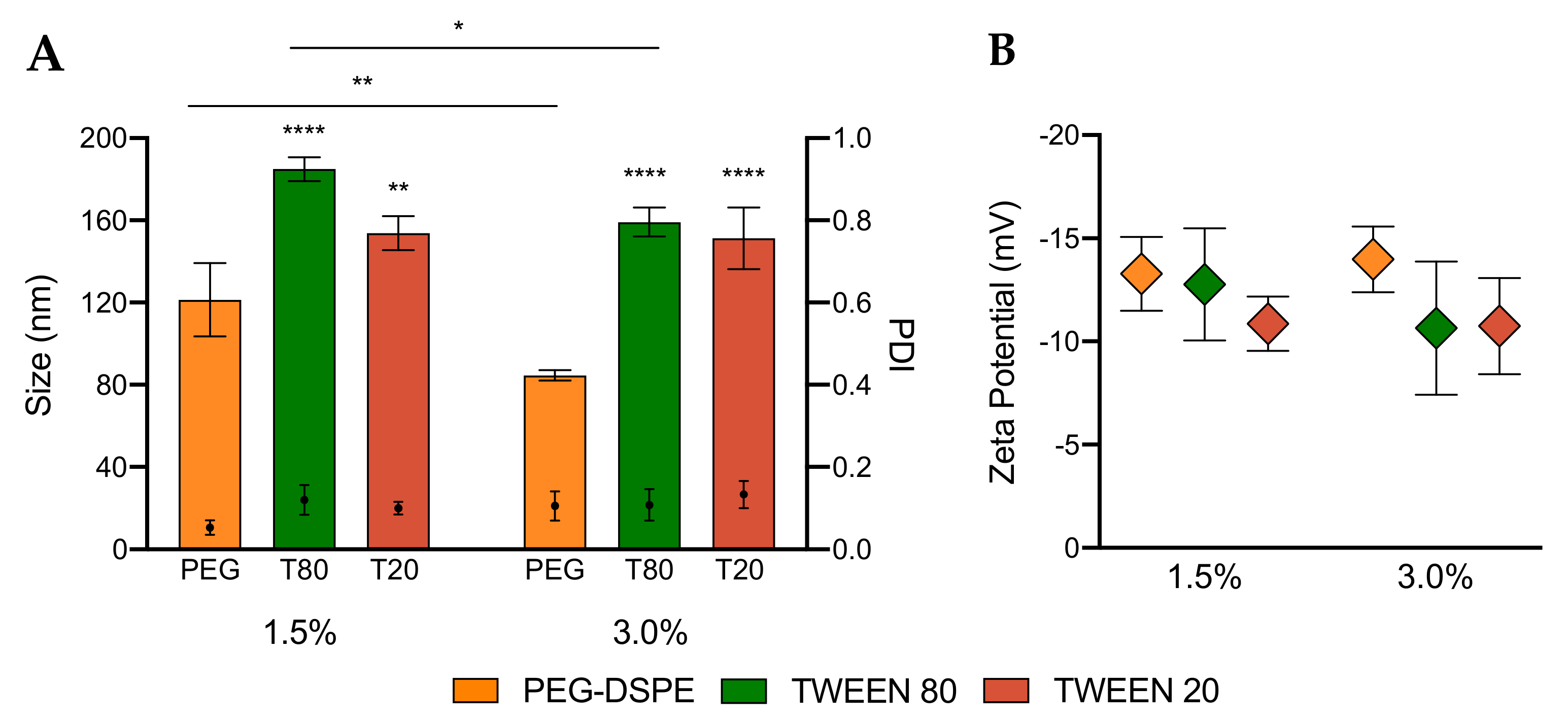
3.2. Characterization of LNPs
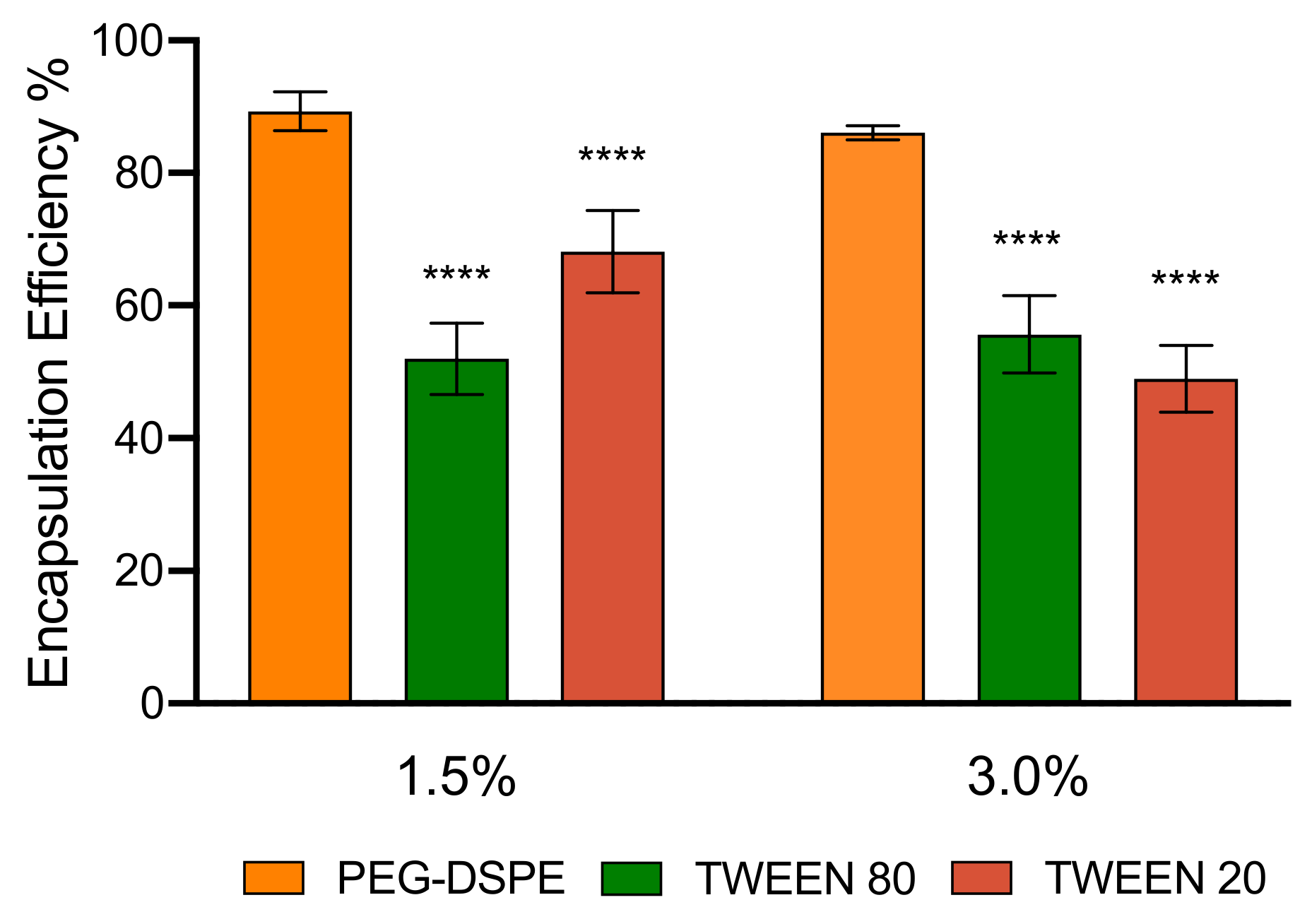
3.3. Encapsulation Efficiency and Stability of LNPs
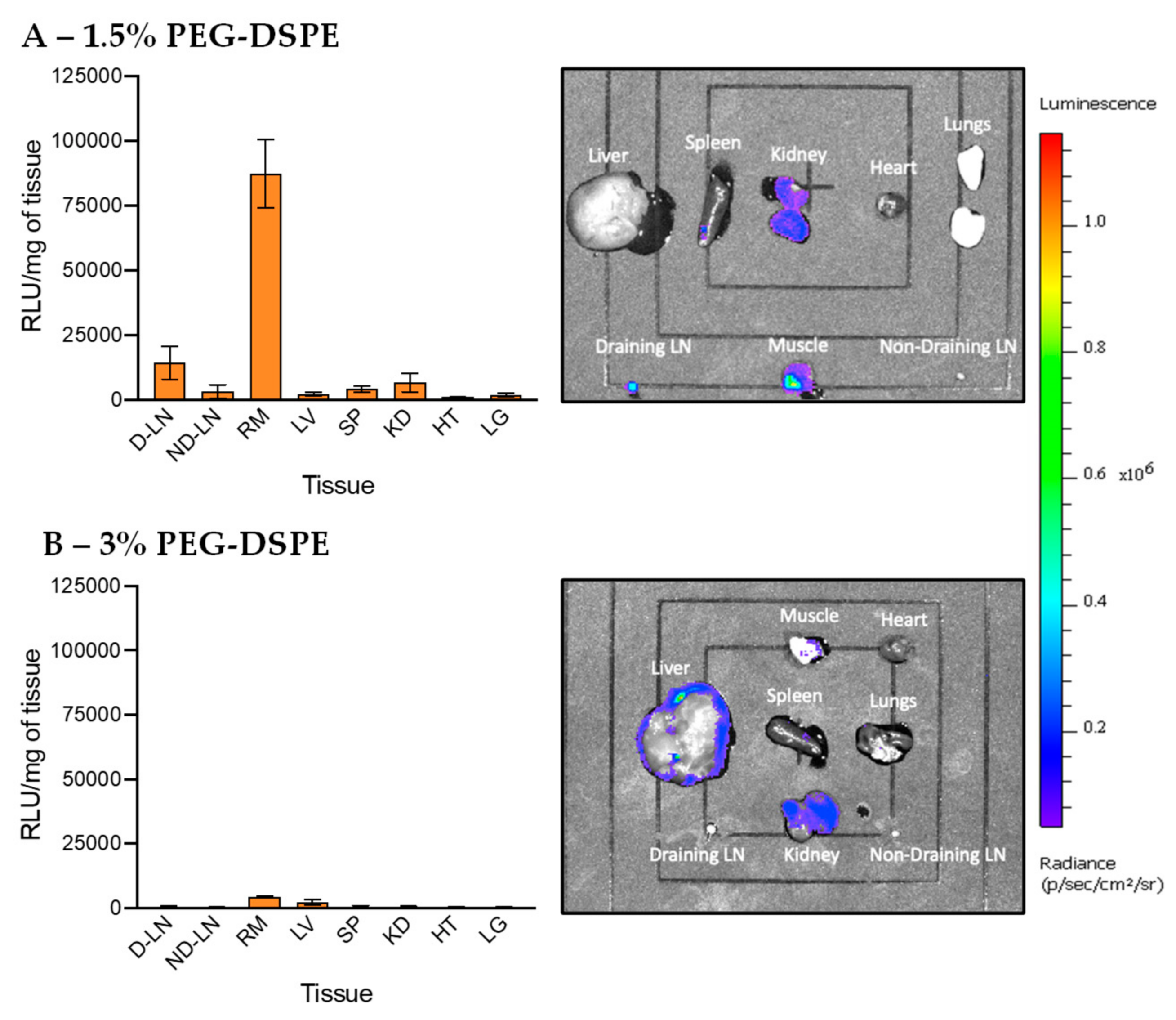
3.4. In Vivo Transfection of LNPs Stabilized by PEG-DSPE
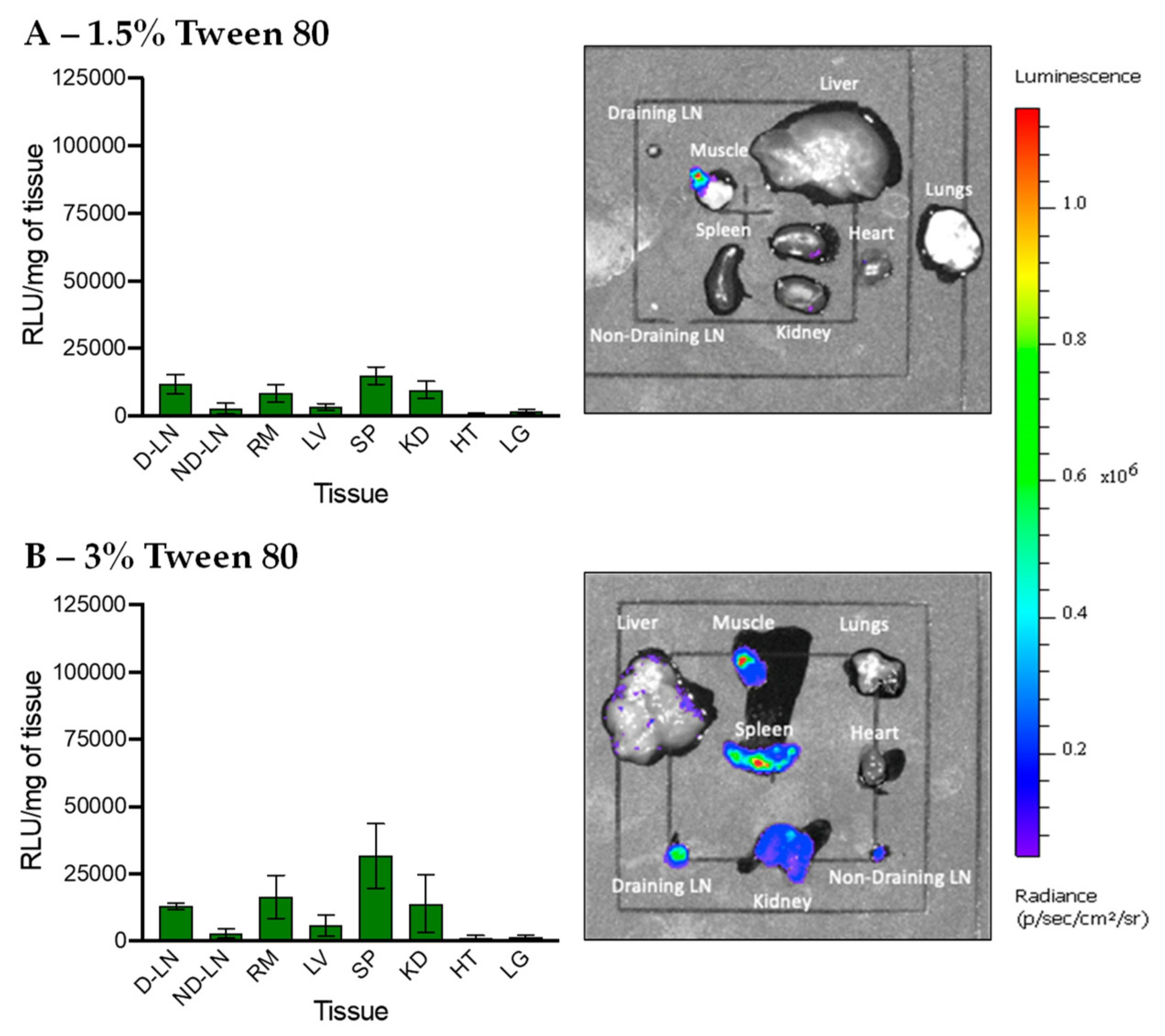
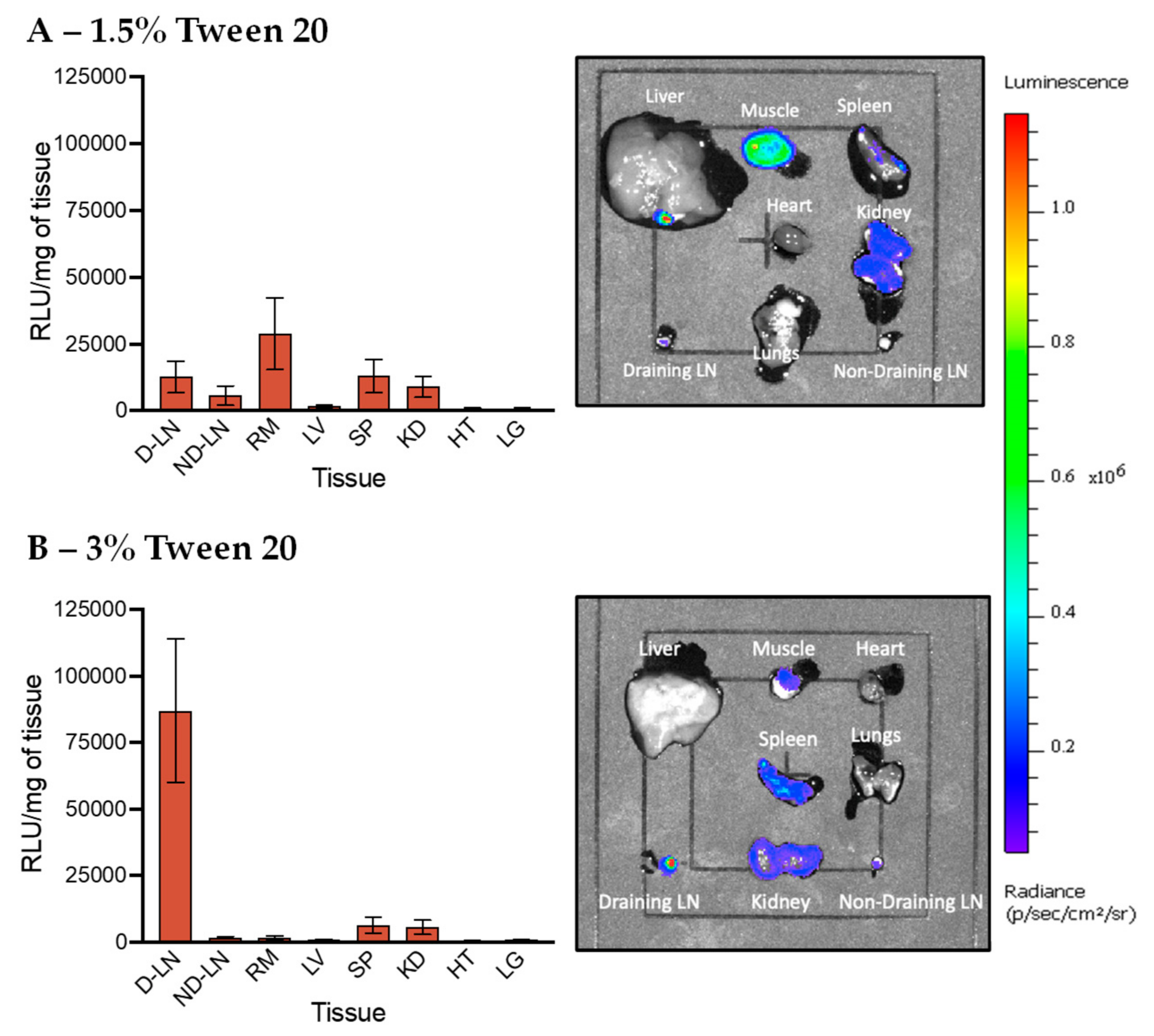
3.5. In Vivo Transfection of LNPs Stabilized by Tween 80 and Tween 20
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.P.; Gu, W.Y.; Prasadam, I.; Jia, Z.F.; Crawford, R.; Xiao, Y.; Monteiro, M.J. An influenza virus-inspired polymer system for the timed release of siRNA. Nat. Commun. 2013, 4, 1902. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.A.; Ko, S.Y.; Morabito, K.M.; Yang, E.S.; Pelc, R.S.; DeMaso, C.R.; Castilho, L.R.; Abbink, P.; Boyd, M.; Nityanandam, R.; et al. Rapid development of a DNA vaccine for Zika virus. Science 2016, 354, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Rivas-Garcia, L.; Baptista, P.V.; Fernandes, A.R. Gene Therapy in Cancer Treatment: Why Go Nano? Pharmaceutics 2020, 12, 233. [Google Scholar] [CrossRef]
- Cohen, J. New coronavirus threat galvanizes scientists. Science 2020, 367, 492–493. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.Q.; Li, Y.Z.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Lostale-Seijo, I.; Montenegro, J. Synthetic materials at the forefront of gene delivery. Nat. Rev. Chem. 2018, 2, 258–277. [Google Scholar] [CrossRef]
- Mastrobattista, E.; Hennink, W.E.; Schiffelers, R.M. Delivery of nucleic acids. Pharm. Res. 2007, 24, 1561–1563. [Google Scholar] [CrossRef]
- Carvalho, A.M.; Cordeiro, R.A.; Faneca, H. Silica-Based Gene Delivery Systems: From Design to Therapeutic Applications. Pharmaceutics 2020, 12, 649. [Google Scholar] [CrossRef]
- Buck, J.; Grossen, P.; Cullis, P.R.; Huwyler, J.; Witzigmann, D. Lipid-Based DNA Therapeutics: Hallmarks of Non-Viral Gene Delivery. ACS Nano 2019, 13, 3754–3782. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Negro, M.; Sanchez-Arribas, N.; Guerrero-Martinez, A.; Moya, M.L.; de Ilarduya, C.T.; Mendicuti, F.; Aicart, E.; Junquera, E. A Non-Viral Plasmid DNA Delivery System Consisting on a Lysine-Derived Cationic Lipid Mixed with a Fusogenic Lipid. Pharmaceutics 2019, 11, 632. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.Y.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Vu, M.N.; Kelly, H.G.; Wheatley, A.K.; Peng, S.; Pilkington, E.H.; Veldhuis, N.A.; Davis, T.P.; Kent, S.J.; Truong, N.P. Cellular Interactions of Liposomes and PISA Nanoparticles during Human Blood Flow in a Microvascular Network. Small 2020, 16, 2002861. [Google Scholar] [CrossRef]
- Moss, K.H.; Popova, P.; Hadrup, S.R.; Astakhova, K.; Taskova, M. Lipid Nanoparticles for Delivery of Therapeutic RNA Oligonucleotides. Mol. Pharmaceut. 2019, 16, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug. Deliv. Rev. 2020. [Google Scholar] [CrossRef]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Trevaskis, N.L. Targeting immune cells within lymph nodes. Nat. Nanotechnol. 2020, 15, 423–425. [Google Scholar] [CrossRef]
- Najibi, A.J.; Mooney, D.J. Cell and tissue engineering in lymph nodes for cancer immunotherapy. Adv. Drug. Deliv. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schudel, A.; Francis, D.M.; Thomas, S.N. Material design for lymph node drug delivery. Nat. Rev. Mater. 2019, 4, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Schudel, A.; Chapman, A.P.; Yau, M.K.; Higginson, C.J.; Francis, D.M.; Manspeaker, M.P.; Avecilla, A.R.C.; Rohner, N.A.; Finn, M.G.; Thomas, S.N. Programmable multistage drug delivery to lymph nodes. Nat. Nanotechnol. 2020, 15, 491–499. [Google Scholar] [CrossRef]
- Jiang, H.; Wang, Q.; Sun, X. Lymph node targeting strategies to improve vaccination efficacy. J. Control. Release 2017, 267, 47–56. [Google Scholar] [CrossRef]
- Cai, S.; Yang, Q.H.; Bagby, T.R.; Forrest, M.L. Lymphatic drug delivery using engineered liposomes and solid lipid nanoparticles. Adv. Drug. Deliv. Rev. 2011, 63, 901–908. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar]
- Truong, N.P.; Quinn, J.F.; Anastasaki, A.; Rolland, M.; Vu, M.; Haddleton, D.; Whittaker, M.R.; Davis, T.P. Surfactant-free RAFT emulsion polymerization using a novel biocompatible thermoresponsive polymer. Polym. Chem. 2017, 8, 1353–1363. [Google Scholar] [CrossRef]
- Truong, N.P.; Dussert, M.V.; Whittaker, M.R.; Quinn, J.F.; Davis, T.P. Rapid synthesis of ultrahigh molecular weight and low polydispersity polystyrene diblock copolymers by RAFT-mediated emulsion polymerization. Polym. Chem. 2015, 6, 3865–3874. [Google Scholar] [CrossRef]
- Mui, B.L.; Tam, Y.K.; Jayaraman, M.; Ansell, S.M.; Du, X.Y.; Tam, Y.Y.C.; Lin, P.J.C.; Chen, S.; Narayanannair, J.K.; Rajeev, K.G.; et al. Influence of Polyethylene Glycol Lipid Desorption Rates on Pharmacokinetics and Pharmacodynamics of siRNA Lipid Nanoparticles. Mol. Ther-Nucl. Acids 2013, 2, e139. [Google Scholar] [CrossRef]
- Sainz-Ramos, M.; Villate-Beitia, I.; Gallego, I.; Na, L.Q.; Lopez-Mendez, T.B.; Eritja, R.; Grijalvo, S.; Puras, G.; Pedraz, J.L. Non-viral mediated gene therapy in human cystic fibrosis airway epithelial cells recovers chloride channel functionality. Int. J. Pharm. 2020, 588, 119757. [Google Scholar] [CrossRef]
- Attia, N.; Mashal, M.; Soto-Sanchez, C.; Martinez-Navarrete, G.; Fernandez, E.; Grijalvo, S.; Eritja, R.; Puras, G.; Pedraz, J.L. Gene transfer to rat cerebral cortex mediated by polysorbate 80 and poloxamer 188 nonionic surfactant vesicles. Drug. Des. Dev. Ther. 2018, 12, 3937–3949. [Google Scholar] [CrossRef] [PubMed]
- Gallego, I.; Villate-Beitia, I.; Martinez-Navarrete, G.; Menendez, M.; Lopez-Mendez, T.; Soto-Sanchez, C.; Zarate, J.; Puras, G.; Fernandez, E.; Pedraz, J.L. Non-viral vectors based on cationic niosomes and minicircle DNA technology enhance gene delivery efficiency for biomedical applications in retinal disorders. Nanomed-Nanotechnol. 2019, 17, 308–318. [Google Scholar] [CrossRef]
- Carafa, M.; Di Marzio, L.; Marianecci, C.; Cinque, B.; Lucania, G.; Kajiwara, K.; Cifone, M.G.; Santucci, E. Designing novel pH-sensitive non-phospholipid vesicle: Characterization and cell interaction. Eur. J. Pharm. Sci. 2006, 28, 385–393. [Google Scholar] [CrossRef]
- Huang, Y.Z.; Rao, Y.F.; Chen, J.L.; Yang, V.C.; Liang, W.Q. Polysorbate cationic synthetic vesicle for gene delivery. J. Biomed. Mater. Res. A 2011, 96, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Puras, G.; Mashal, M.; Zarate, J.; Agirre, M.; Ojeda, E.; Grijalvo, S.; Eritja, R.; Diaz-Tahoces, A.; Navarrete, G.M.; Aviles-Trigueros, M.; et al. A novel cationic niosome formulation for gene delivery to the retina. J. Control. Release 2014, 174, 27–36. [Google Scholar] [CrossRef]
- Villate-Beitia, I.; Gallego, I.; Martinez-Navarrete, G.; Zarate, J.; Lopez-Mendez, T.; Soto-Sanchez, C.; Santos-Vizcaino, E.; Puras, G.; Fernandez, E.; Pedraz, J.L. Polysorbate 20 non-ionic surfactant enhances retinal gene delivery efficiency of cationic niosomes after intravitreal and subretinal administration. Int. J. Pharm. 2018, 550, 388–397. [Google Scholar] [CrossRef]
- Belliveau, N.M.; Huft, J.; Lin, P.J.C.; Chen, S.; Leung, A.K.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther-Nucl. Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Chen, S.; Tam, Y.Y.C.; Lin, P.J.C.; Leung, A.K.K.; Tam, Y.K.; Cullis, P.R. Development of lipid nanoparticle formulations of siRNA for hepatocyte gene silencing following subcutaneous administration. J. Control. Release 2014, 196, 106–112. [Google Scholar] [CrossRef]
- Silberstein, E.; Serna, C.; Fragoso, S.P.; Nagarkatti, R.; Debrabant, A. A novel nanoluciferase-based system to monitor Trypanosoma cruzi infection in mice by bioluminescence imaging. PLoS ONE 2018, 13, e0195879. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.J.W.; Kulkarni, J.A.; van der Meel, R.; Cullis, P.R.; Vader, P.; Schiffelers, R.M. State-of-the-Art Design and Rapid-Mixing Production Techniques of Lipid Nanoparticles for Nucleic Acid Delivery. Small Methods 2018, 2, 1700375. [Google Scholar] [CrossRef]
- Sato, Y.; Note, Y.; Maeki, M.; Kaji, N.; Baba, Y.; Tokeshi, M.; Harashima, H. Elucidation of the physicochemical properties and potency of siRNA-loaded small-sized lipid nanoparticles for siRNA delivery. J. Control. Release 2016, 229, 48–57. [Google Scholar] [CrossRef]
- Kaurav, M.; Madan, J.; Sudheesh, M.S.; Pandey, R.S. Combined adjuvant-delivery system for new generation vaccine antigens: Alliance has its own advantage. Artif. Cell Nanomed. B 2018, 46, S818–S831. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Lou, G.; Anderluzzi, G.; Schmidt, S.T.; Woods, S.; Gallorini, S.; Brazzoli, M.; Giusti, F.; Ferlenghi, I.; Johnson, R.N.; Roberts, C.W.; et al. Delivery of self-amplifying mRNA vaccines by cationic lipid nanoparticles: The impact of cationic lipid selection. J. Control. Release 2020, 325, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; Xia, Y.; Mihai, C.; Griffith, J.P.; Hou, S.G.; Esposito, A.A.; Ketova, T.; Welsher, K.; et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 2020, 11, 983. [Google Scholar] [CrossRef]
- Cheng, X.W.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug. Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc. Chem. Res. 2019, 52, 2435–2444. [Google Scholar] [CrossRef]
- Khor, S.Y.; Vu, M.N.; Pilkington, E.H.; Johnston, A.P.R.; Whittaker, M.R.; Quinn, J.F.; Truong, N.P.; Davis, T.P. Elucidating the Influences of Size, Surface Chemistry, and Dynamic Flow on Cellular Association of Nanoparticles Made by Polymerization-Induced Self-Assembly. Small 2018, 14, 1801702. [Google Scholar] [CrossRef]
- Karimi, F.; McKenzie, T.G.; O’Connor, A.J.; Qiao, G.G.; Heath, D.E. Nano-scale clustering of integrin-binding ligands regulates endothelial cell adhesion, migration, and endothelialization rate: Novel materials for small diameter vascular graft applications. J. Mater. Chem. B 2017, 5, 5942–5953. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.X.; Du, X.Y.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo. Angew. Chem. Int. Edit. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Jin, J.F.; Zhu, L.L.; Chen, M.; Xu, H.M.; Wang, H.F.; Feng, X.Q.; Zhu, X.P.; Zhou, Q. The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient Prefer. Adher. 2015, 9, 923–942. [Google Scholar]
- Truong, N.P.; Zhang, C.; Nguyen, T.A.H.; Anastasaki, A.; Schulze, M.W.; Quinn, J.F.; Whittaker, A.K.; Hawker, C.J.; Whittaker, M.R.; Davis, T.P. Overcoming Surfactant-Induced Morphology Instability of Noncrosslinked Diblock Copolymer Nano-Objects Obtained by RAFT Emulsion Polymerization. ACS Macro. Lett. 2018, 7, 159–165. [Google Scholar] [CrossRef]
- Jia, Z.F.; Truong, N.P.; Monteiro, M.J. Reversible polymer nanostructures by regulating SDS/PNIPAM binding. Polym. Chem. 2013, 4, 233–236. [Google Scholar] [CrossRef]
- Zayas, H.A.; Truong, N.P.; Valade, D.; Jia, Z.F.; Monteiro, M.J. Narrow molecular weight and particle size distributions of polystyrene 4-arm stars synthesized by RAFT-mediated miniemulsions. Polym. Chem. 2013, 4, 592–599. [Google Scholar] [CrossRef]
- Cao, X.; Ashfaq, R.; Cheng, F.; Maharjan, S.; Li, J.; Ying, G.L.; Hassan, S.; Xiao, H.Y.; Yue, K.; Zhang, Y.S. A Tumor-on-a-Chip System with Bioprinted Blood and Lymphatic Vessel Pair. Adv. Funct. Mater. 2019, 29, 1807173. [Google Scholar] [CrossRef]
- Shanti, A.; Samara, B.; Abdullah, A.; Hallfors, N.; Accoto, D.; Sapudom, J.; Alatoom, A.; Teo, J.; Danti, S.; Stefanini, C. Multi-Compartment 3D-Cultured Organ-on-a-Chip: Towards a Biomimetic Lymph Node for Drug Development. Pharmaceutics 2020, 12, 464. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zukancic, D.; Suys, E.J.A.; Pilkington, E.H.; Algarni, A.; Al-Wassiti, H.; Truong, N.P. The Importance of Poly(ethylene glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles. Pharmaceutics 2020, 12, 1068. https://doi.org/10.3390/pharmaceutics12111068
Zukancic D, Suys EJA, Pilkington EH, Algarni A, Al-Wassiti H, Truong NP. The Importance of Poly(ethylene glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles. Pharmaceutics. 2020; 12(11):1068. https://doi.org/10.3390/pharmaceutics12111068
Chicago/Turabian StyleZukancic, Danijela, Estelle J. A. Suys, Emily H. Pilkington, Azizah Algarni, Hareth Al-Wassiti, and Nghia P. Truong. 2020. "The Importance of Poly(ethylene glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles" Pharmaceutics 12, no. 11: 1068. https://doi.org/10.3390/pharmaceutics12111068
APA StyleZukancic, D., Suys, E. J. A., Pilkington, E. H., Algarni, A., Al-Wassiti, H., & Truong, N. P. (2020). The Importance of Poly(ethylene glycol) and Lipid Structure in Targeted Gene Delivery to Lymph Nodes by Lipid Nanoparticles. Pharmaceutics, 12(11), 1068. https://doi.org/10.3390/pharmaceutics12111068