Inhibitory Effect of AB-PINACA, Indazole Carboxamide Synthetic Cannabinoid, on Human Major Drug-Metabolizing Enzymes and Transporters


 , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Inhibitory Effects of AB-PINACA on Eight Major CYP Activities in Human Liver Microsomes
2.3. Inhibitory Effects of AB-PINACA on Six Major UGT Activities in Human Liver Microsomes
2.4. Inhibitory Effects of AB-PINACA on the Transport Activities of OCTs, OATs, OATPs, P-gp, and BCRP
2.5. Molecular Docking of AB-PINACA
2.6. Data Analysis
3. Results
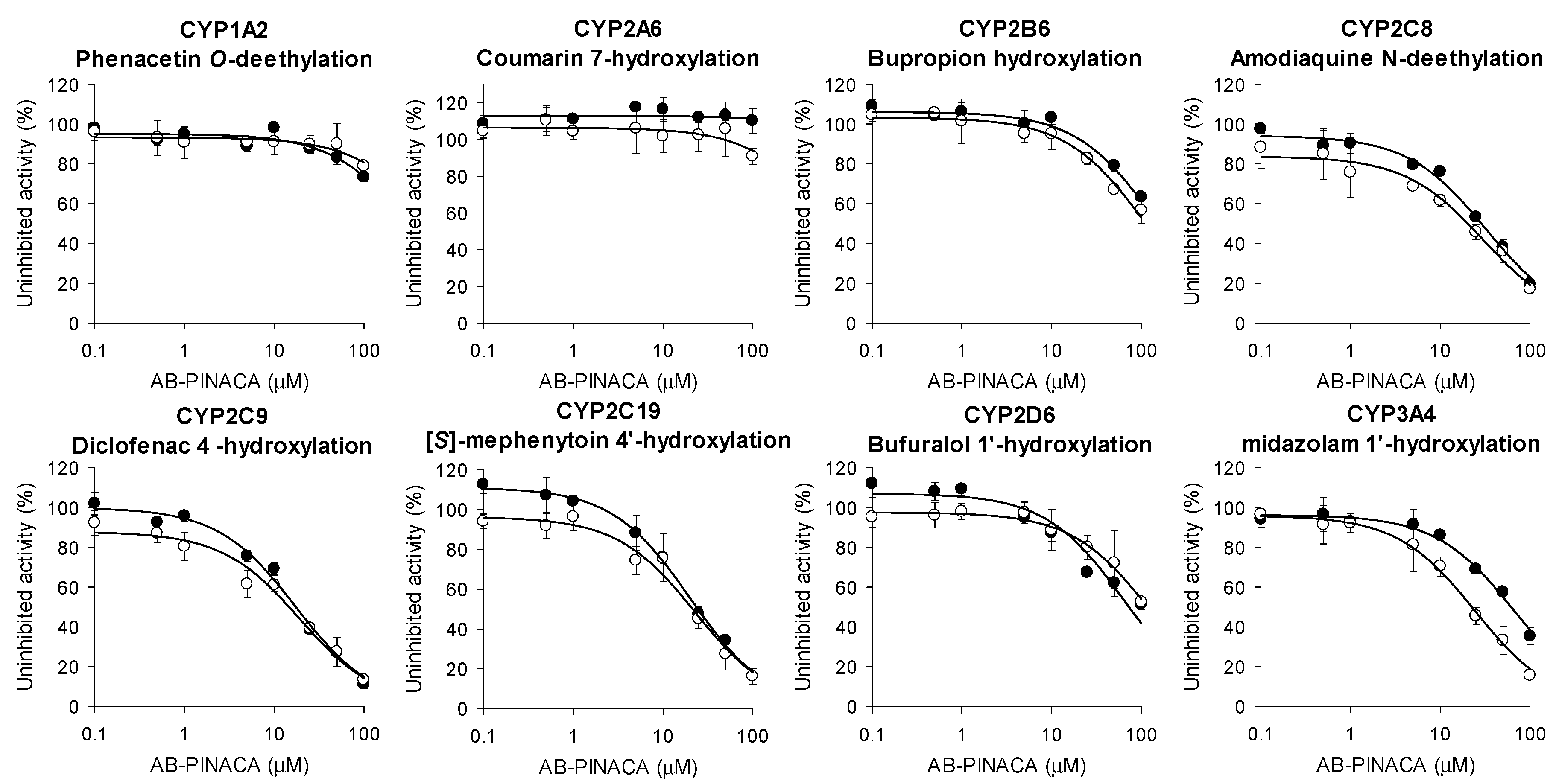
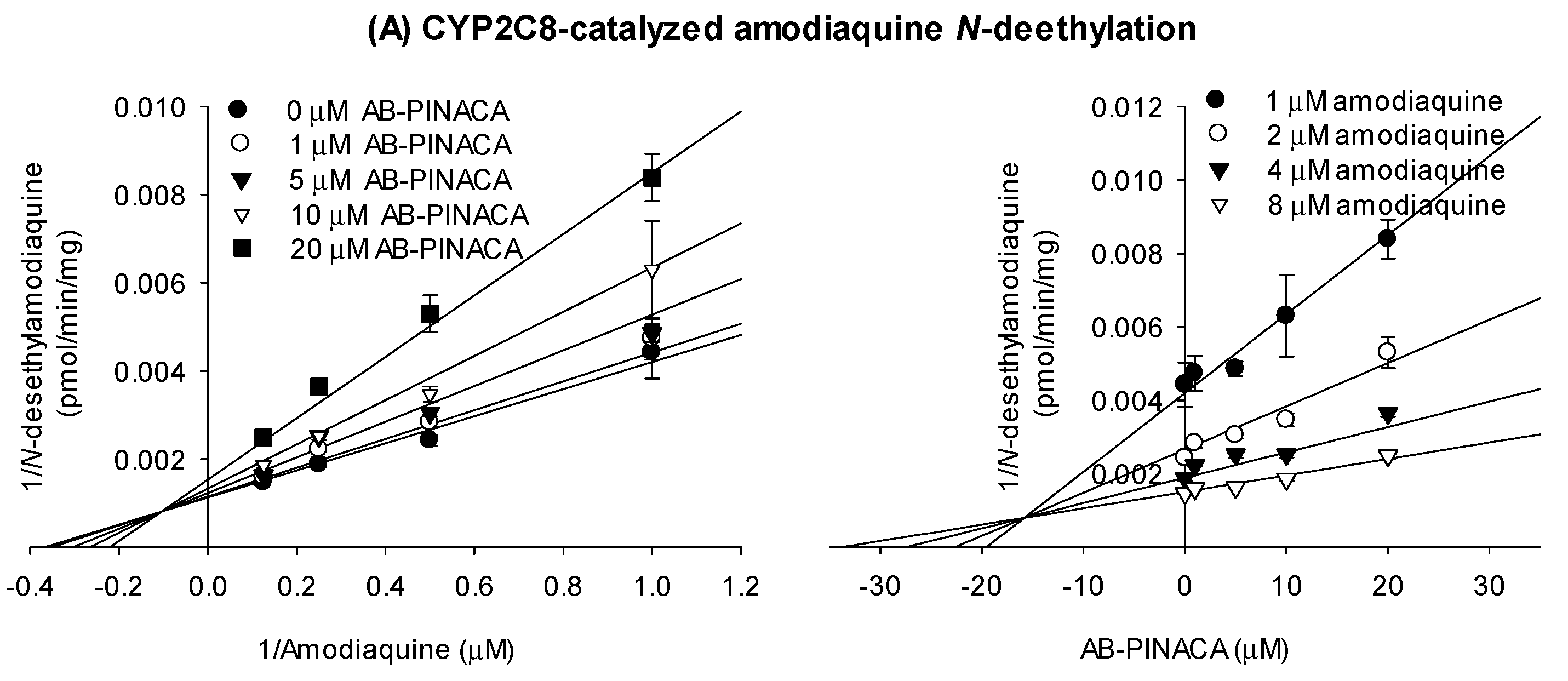
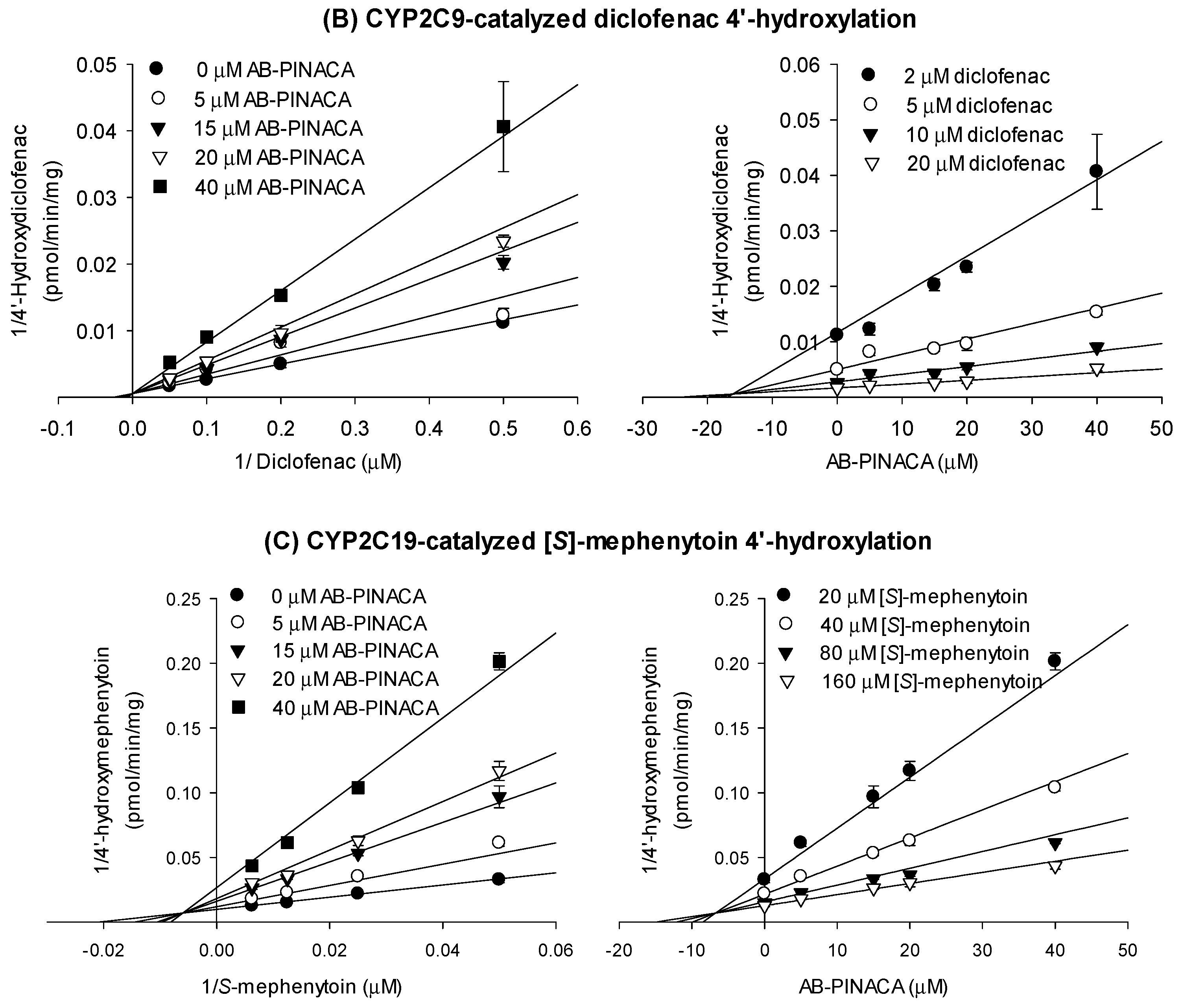
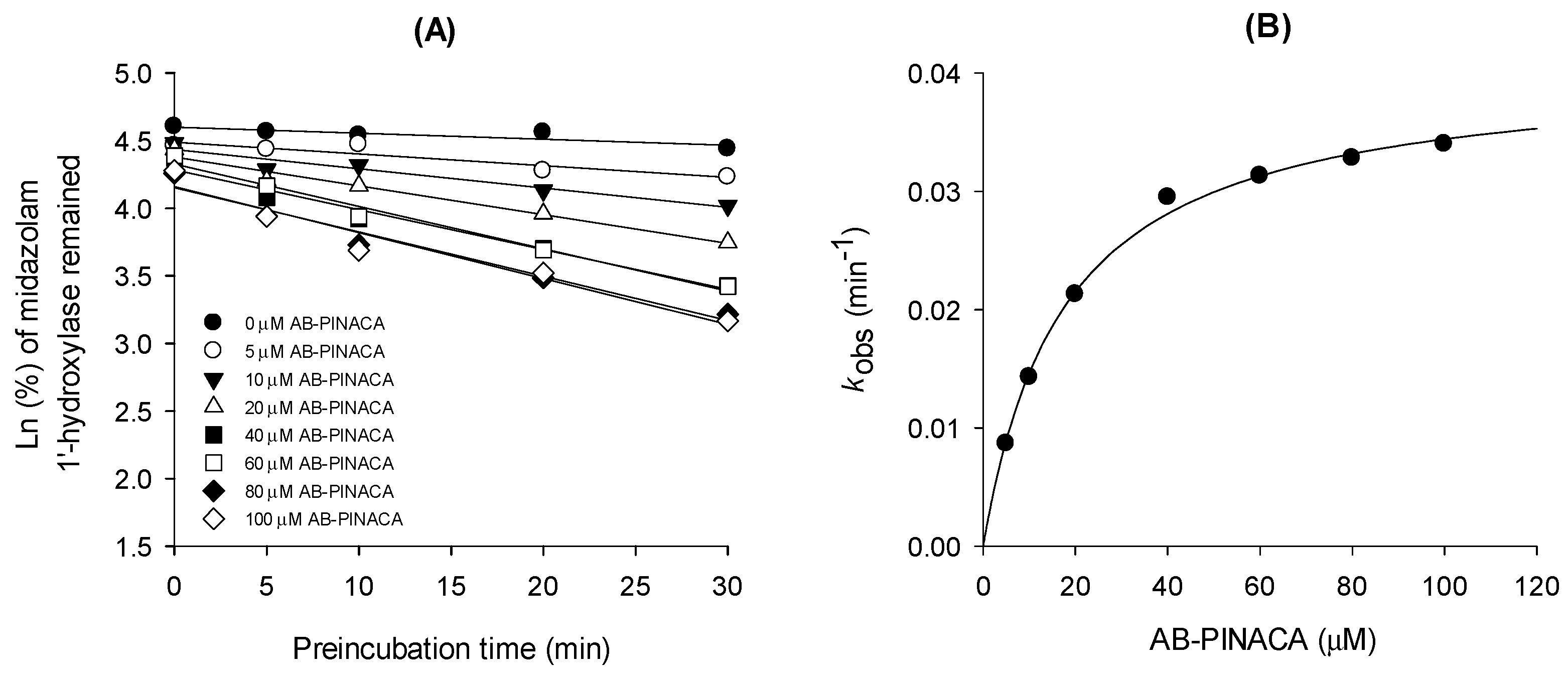
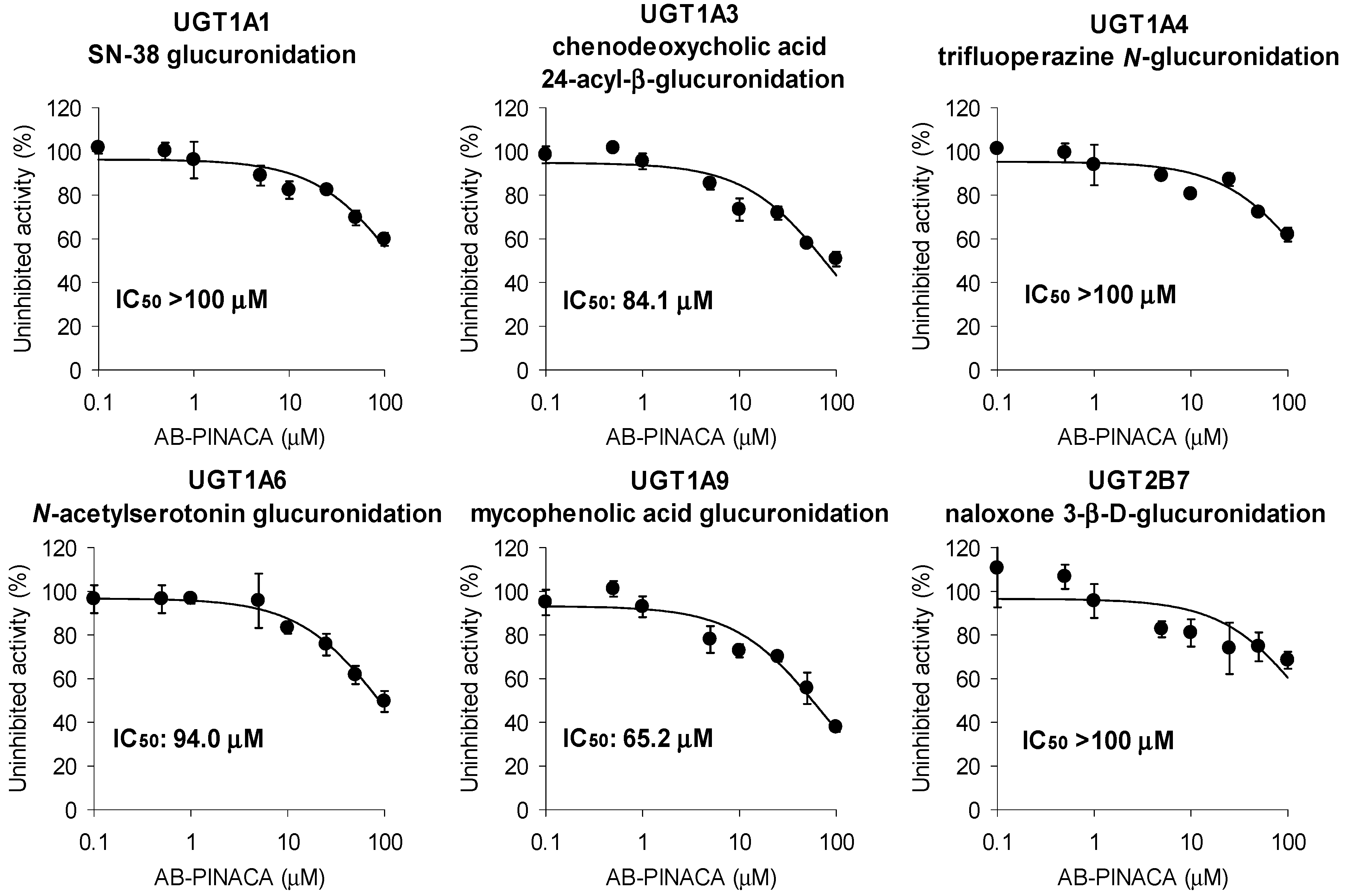
3.1. Inhibitory Effects of AB-PINACA on CYP and UGT Activities in Human Liver Microsomes
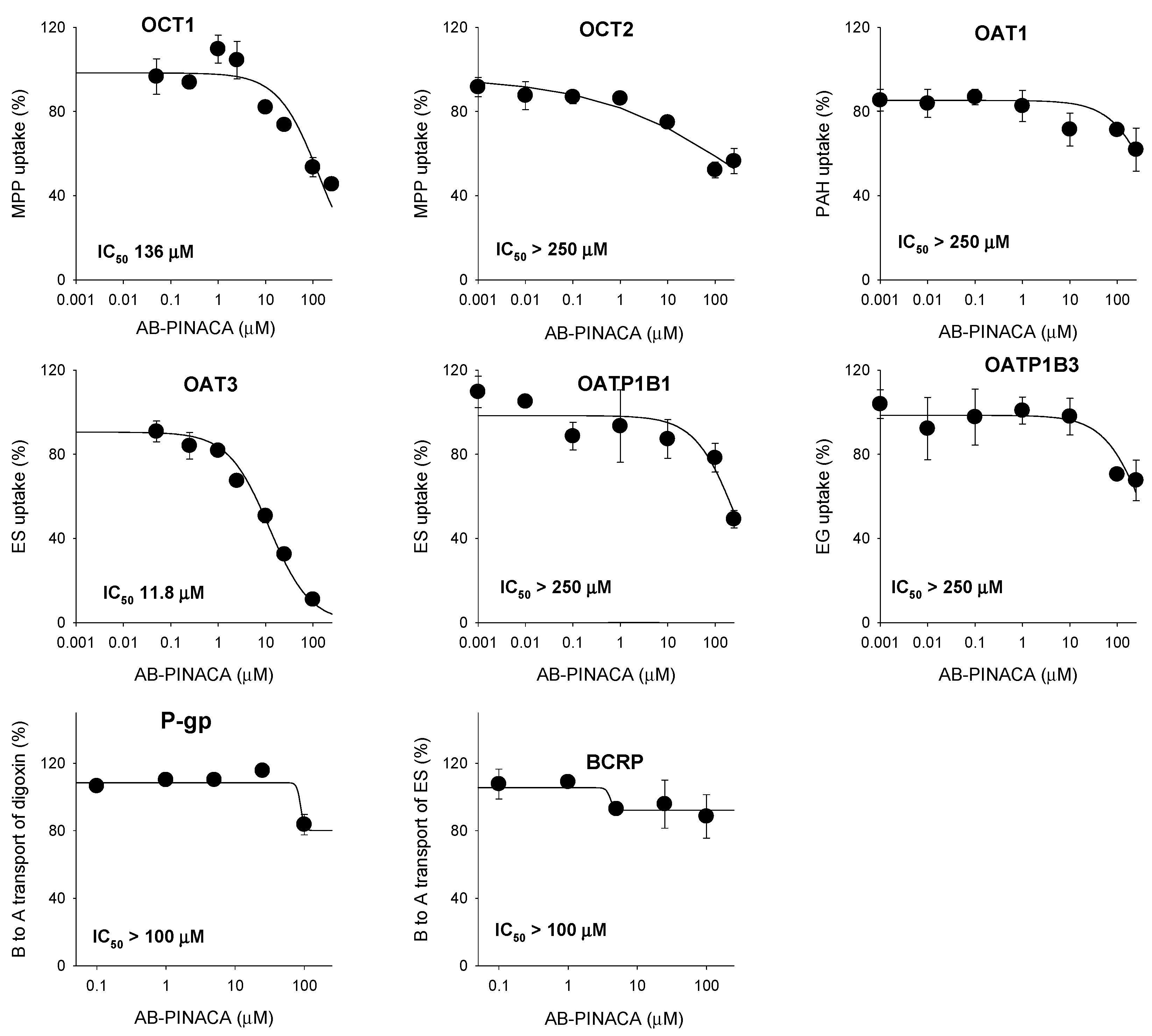
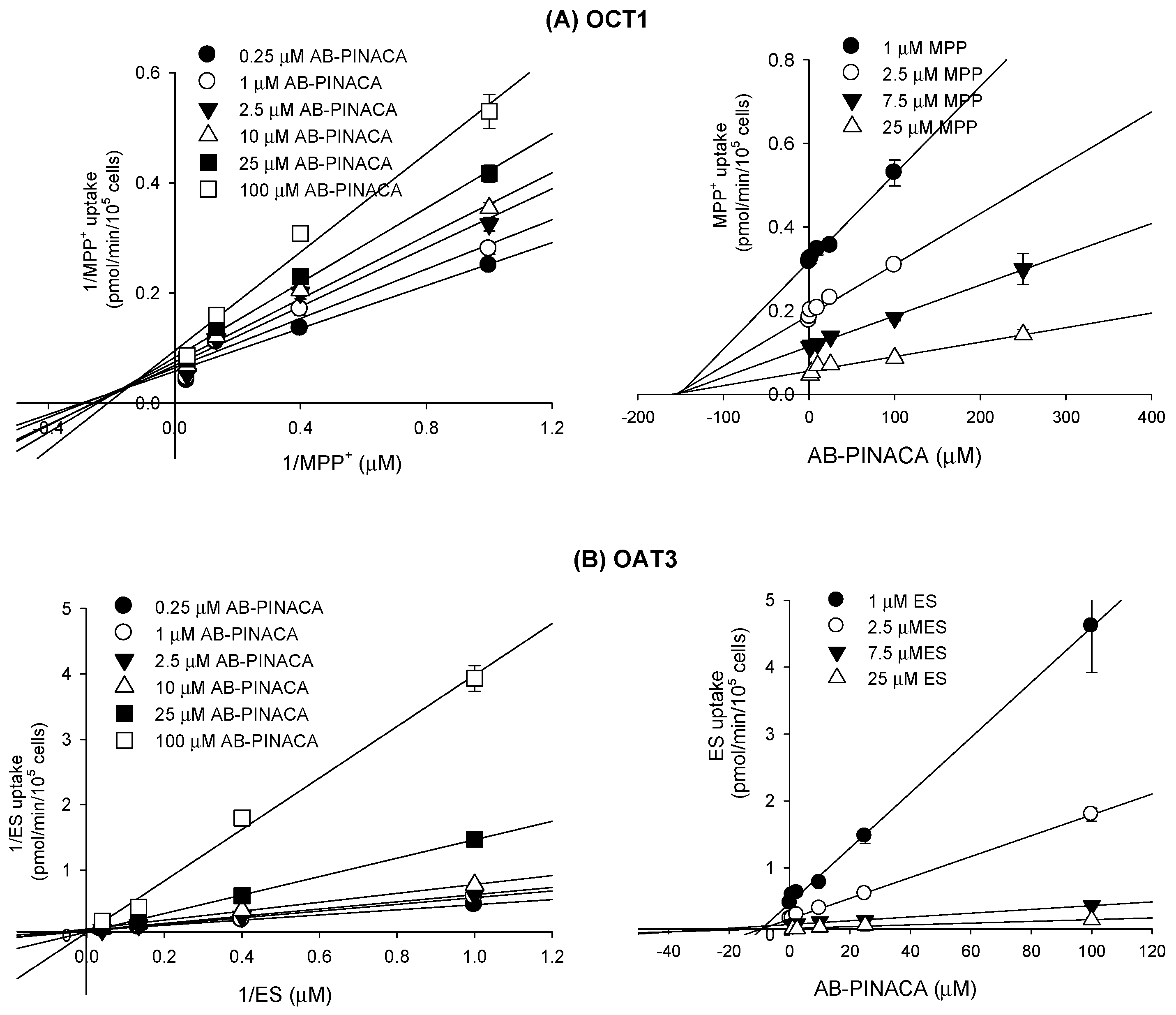
3.2. Inhibitory Effect of AB-PINACA on Drug Transporters
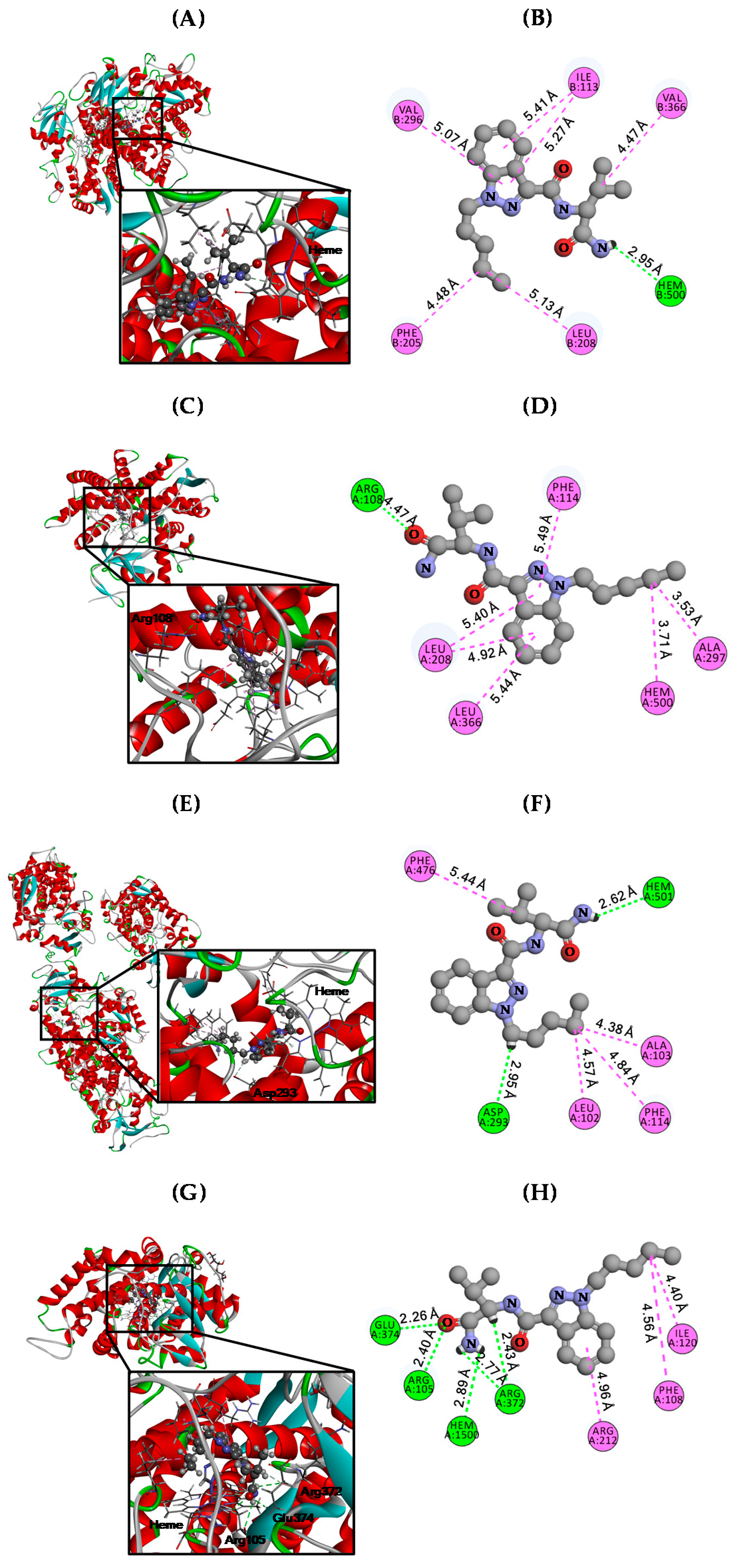
3.3. Molecular Docking Analysis
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Expert Committee on Drug Dependence. Ab-pinaca critical review report agenda item 4.4. In Expert Committee on Drug Dependence Thirty-Ninth Meeting; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Wiley, J.L.; Marusich, J.A.; Lefever, T.W.; Antonazzo, K.R.; Wallgren, M.T.; Cortes, R.A.; Patel, P.R.; Grabenauer, M.; Moore, K.N.; Thomas, B.F. Ab-chminaca, ab-pinaca, and fubimina: Affinity and potency of novel synthetic cannabinoids in producing delta(9)-tetrahydrocannabinol-like effects in mice. J. Pharmacol. Exp. Ther. 2015, 354, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.L.; Marusich, J.A.; Lefever, T.W.; Grabenauer, M.; Moore, K.N.; Thomas, B.F. Cannabinoids in disguise: Delta(9)-tetrahydrocannabinol-like effects of tetramethylcyclopropyl ketone indoles. Neuropharmacology 2013, 75, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.Y.; Kim, J.H.; Kim, D.K.; Lee, H.S. Synthetic cannabinoids are substrates and inhibitors of multiple drug-metabolizing enzymes. Arch. Pharm. Res. 2018, 41, 691–710. [Google Scholar] [CrossRef]
- Wohlfarth, A.; Castaneto, M.S.; Zhu, M.S.; Pang, S.K.; Scheidweiler, K.B.; Kronstrand, R.; Huestis, M.A. Pentylindole/pentylindazole synthetic cannabinoids and their 5-fluoro analogs produce different primary metabolites: Metabolite profiling for ab-pinaca and 5f-ab-pinaca. AAPS J. 2015, 17, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Suzuki, M.; Todoroki, K.; Inoue, K.; Min, J.Z.; Kikura-Hanajiri, R.; Goda, Y.; Toyo’oka, T. Uplc/esi-ms/ms-based determination of metabolism of several new illicit drugs, adb-fubinaca, ab-fubinaca, ab-pinaca, qupic, 5f-qupic and alpha-pvt, by human liver microsome. Biomed. Chromatogr. 2014, 28, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Nielsen, L.M.; Holm, N.B.; Rasmussen, H.B.; Linnet, K.; Consortium, I. Synthetic cannabimimetic agents metabolized by carboxylesterases. Drug Test. Anal. 2015, 7, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Richter, L.H.J.; Maurer, H.H.; Meyer, M.R. New psychoactive substances: Studies on the metabolism of xlr-11, ab-pinaca, fub-pb-22, 4-methoxy-alpha-pvp, 25-i-nbome, and meclonazepam using human liver preparations in comparison to primary human hepatocytes, and human urine. Toxicol. Lett. 2017, 280, 142–150. [Google Scholar] [CrossRef]
- Uchiyama, N.; Matsuda, S.; Wakana, D.; Kikura-Hanajiri, R.; Goda, Y. New cannabimimetic indazole derivatives, n-(1-amino-3-methyl-1-oxobutan-2-yl)-1-pentyl-1h-indazole-3-carboxamide (ab-pinaca) and n-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(4-fluorobenzyl)-1h-indazole-3-carboxamide (ab-fubinaca) identified as designer drugs in illegal products. Forensic Toxicol. 2013, 31, 93–100. [Google Scholar]
- Peterson, B.L.; Couper, F.J. Concentrations of ab-chminaca and ab-pinaca and driving behavior in suspected impaired driving cases. J. Anal. Toxicol. 2015, 39, 642–647. [Google Scholar] [CrossRef]
- Thornton, S.L.; Akpunonu, P.; Glauner, K.; Hoehn, K.S.; Gerona, R. Unintentional pediatric exposure to a synthetic cannabinoid (ab-pinaca) resulting in coma and intubation. Ann. Emerg. Med. 2015, 66, 343–344. [Google Scholar] [CrossRef]
- Stage, C.; Jurgens, G.; Guski, L.S.; Thomsen, R.; Bjerre, D.; Ferrero-Miliani, L.; Lyauk, Y.K.; Rasmussen, H.B.; Dalhoff, K.; Consortium, I. The pharmacokinetics of enalapril in relation to ces1 genotype in healthy danish volunteers. Basic Clin. Pharmacol. Toxicol. 2017, 121, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Stage, C.; Jurgens, G.; Guski, L.S.; Thomsen, R.; Bjerre, D.; Ferrero-Miliani, L.; Lyauk, Y.K.; Rasmussen, H.B.; Dalhoff, K.; Consortium, T.I. The impact of ces1 genotypes on the pharmacokinetics of methylphenidate in healthy danish subjects. Br. J. Clin. Pharmacol. 2017, 83, 1506–1514. [Google Scholar] [CrossRef] [PubMed]
- Suzaki, Y.; Uemura, N.; Takada, M.; Ohyama, T.; Itohda, A.; Morimoto, T.; Imai, H.; Hamasaki, H.; Inano, A.; Hosokawa, M.; et al. The effect of carboxylesterase 1 (ces1) polymorphisms on the pharmacokinetics of oseltamivir in humans. Eur. J. Clin. Pharmacol. 2013, 69, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kwon, S.S.; Kong, T.Y.; Cheong, J.C.; Kim, H.S.; In, M.K.; Lee, H.S. AM-2201 inhibits multiple cytochrome p450 and uridine 5-diphospho-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2017, 22, 443. [Google Scholar] [CrossRef]
- Kong, T.Y.; Kim, J.H.; Kwon, S.S.; Cheong, J.C.; Kim, H.S.; In, M.K.; Lee, H.S. Inhibition of cytochrome p450 and uridine 5′-diphospho-glucuronosyltransferases by MAM-2201 in human liver microsomes. Arch. Pharm. Res. 2017, 40, 727–735. [Google Scholar] [CrossRef]
- Kong, T.Y.; Kwon, S.S.; Cheong, J.C.; Kim, H.S.; Kim, J.Y.; Lee, H.S. In vitro inhibitory effects of synthetic cannabinoid EAM-2201 on cytochrome p450 and udp-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules 2018, 23, 920. [Google Scholar] [CrossRef]
- Kim, S.; Choi, W.G.; Kwon, M.; Lee, S.; Cho, Y.Y.; Lee, J.Y.; Kang, H.C.; Song, I.S.; Lee, H.S. In vitro inhibitory effects of APINACA on human major cytochrome p450, udp-glucuronosyltransferase enzymes, and drug transporters. Molecules 2019, 24, 3000. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.K.; Shin, Y.; Jeon, J.H.; Song, I.S.; Lee, H.S. In vitro interaction of AB-FUBINACA with human Cytochrome P450, UDP-glucuronosyltransferase enzymes and drug transporters. Molecules 2020, 25, 4589. [Google Scholar] [CrossRef]
- Shi, S.J.; Li, Y.Q. Interplay of drug-metabolizing enzymes and transporters in drug absorption and disposition. Curr. Drug Metab. 2014, 15, 915–941. [Google Scholar] [CrossRef]
- Choi, M.K.; Song, I.S. Interactions of ginseng with therapeutic drugs. Arch. Pharm. Res. 2019, 42, 862–878. [Google Scholar] [CrossRef]
- Huang, S.M.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; Al Habet, S.; Baweja, R.K.; Burckart, G.J.; et al. New era in drug interaction evaluation: Us food and drug administration update on cyp enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Taub, M.E.; Chothe, P.P.; Chu, X.Y.; Giacomini, K.M.; Kim, R.B.; Ray, A.S.; Stocker, S.L.; Unadkat, J.D.; Wittwer, M.B.; et al. Transporters in drug development: 2018 itc recommendations for transporters of emerging clinical importance. Clin. Pharmacol. Ther. 2018, 104, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Lee, S.; Lee, W.; Jin, S.; Kwon, M.; Shin, C.H.; Choi, M.K.; Song, I.S. Herb-drug interaction of red ginseng extract and ginsenoside rc with valsartan in rats. Molecules 2020, 25, 622. [Google Scholar] [CrossRef]
- Jeong, H.U.; Kwon, M.; Lee, Y.; Yoo, J.S.; Shin, D.H.; Song, I.S.; Lee, H.S. Organic anion transporter 3- and organic anion transporting polypeptides 1b1- and 1b3-mediated transport of catalposide. Drug Des. Devel. Ther. 2015, 9, 643–653. [Google Scholar] [PubMed]
- Saeed, A.; Saddique, G.; Channar, P.A.; Larik, F.A.; Abbas, Q.; Hassan, M.; Raza, H.; Fattah, T.A.; Seo, S.Y. Synthesis of sulfadiazinyl acyl/aryl thiourea derivatives as calf intestinal alkaline phosphatase inhibitors, pharmacokinetic properties, lead optimization, lineweaver-burk plot evaluation and binding analysis. Bioorg. Med. Chem. 2018, 26, 3707–3715. [Google Scholar]
- Schoch, G.A.; Yano, J.K.; Sansen, S.; Dansette, P.M.; Stout, C.D.; Johnson, E.F. Determinants of cytochrome p450 2c8 substrate binding: Structures of complexes with montelukast, troglitazone, felodipine, and 9-cis-retinoic acid. J. Biol. Chem. 2008, 283, 17227–17237. [Google Scholar] [CrossRef] [PubMed]
- Wester, M.R.; Yano, J.K.; Schoch, G.A.; Yang, C.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. The structure of human cytochrome p450 2c9 complexed with flurbiprofen at 2.0-a resolution. J. Biol. Chem. 2004, 279, 35630–35637. [Google Scholar] [CrossRef]
- Reynald, R.L.; Sansen, S.; Stout, C.D.; Johnson, E.F. Structural characterization of human cytochrome p450 2c19: Active site differences between p450s 2c8, 2c9, and 2c19. J. Biol. Chem. 2012, 287, 44581–44591. [Google Scholar] [CrossRef]
- Kaur, P.; Chamberlin, A.R.; Poulos, T.L.; Sevrioukova, I.F. Structure-based inhibitor design for evaluation of a cyp3a4 pharmacophore model. J. Med. Chem. 2016, 59, 4210–4220. [Google Scholar] [CrossRef]
- Albaugh, D.R.; Fullenwider, C.L.; Fisher, M.B.; Hutzler, J.M. Time-dependent inhibition and estimation of cyp3a clinical pharmacokinetic drug-drug interactions using plated human cell systems. Drug Metab. Dispos. 2012, 40, 1336–1344. [Google Scholar] [CrossRef]
- Zhou, S.F. Drugs behave as substrates, inhibitors and inducers of human cytochrome p450 3a4. Curr. Drug Metab. 2008, 9, 310–322. [Google Scholar] [CrossRef]
- Michael, R.; Wester, E.F.J. Cristina Marques-Soares, Patrick, M.; Dansette, Daniel Mansuy, C. David Stout. Structure of a substrate complex of mammalian cytochrome p450 2c5 at 2.3 resolution_evidence for multiple substrate binding modes. Biochemistry 2003, 42, 6370–6379. [Google Scholar]
- Wester, M.R.; Johnson, E.F.; Marques-Soares, C.; Dijols, S.; Dansette, P.M.; Mansuy, D.; Stout, C.D. Structure of mammalian cytochrome p450 2c5 complexed with diclofenac at 2.1 a resolution: Evidence for an induced fit model of substrate binding. Biochemistry 2003, 42, 9335–9345. [Google Scholar] [CrossRef]
- Tatrai, P.; Schweigler, P.; Poller, B.; Domange, N.; de Wilde, R.; Hanna, I.; Gaborik, Z.; Huth, F. A systematic in vitro investigation of the inhibitor preincubation effect on multiple classes of clinically relevant transporters. Drug Metab. Dispos. 2019, 47, 768–778. [Google Scholar] [CrossRef]
- Jung, N.; Lehmann, C.; Rubbert, A.; Knispel, M.; Hartmann, P.; van Lunzen, J.; Stellbrink, H.J.; Faetkenheuer, G.; Taubert, D. Relevance of the organic cation transporters 1 and 2 for antiretroviral drug therapy in human immunodeficiency virus infection. Drug Metab. Dispos. 2008, 36, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Panfen, E.; Chen, W.Q.; Zhang, Y.P.; Sinz, M.; Marathe, P.; Gan, J.P.; Shen, H. Enhanced and persistent inhibition of organic cation transporter 1 activity by preincubation of cyclosporine A. Drug Metab. Dispos. 2019, 47, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Eraly, S.A.; Tsigelny, I.; Nigam, S.K. Interaction of organic cations with organic anion transporters. J. Biol. Chem. 2009, 284, 31422–31430. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.K.; Jin, Q.R.; Ahn, S.H.; Bae, M.A.; Song, I.S. Sitagliptin attenuates metformin-mediated ampk phosphorylation through inhibition of organic cation transporters. Xenobiotica 2010, 40, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Khamdang, S.; Takeda, M.; Noshiro, R.; Narikawa, S.; Enomoto, A.; Anzai, N.; Piyachaturawat, P.; Endou, H. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2002, 303, 534–539. [Google Scholar] [CrossRef]
- Tachampa, K.; Takeda, M.; Khamdang, S.; Noshiro-Kofuji, R.; Tsuda, M.; Jariyawat, S.; Fukutomi, T.; Sophasan, S.; Anzai, N.; Endou, H. Interactions of organic anion transporters and organic cation transporters with mycotoxins. J. Pharmacol. Sci. 2008, 106, 435–443. [Google Scholar] [CrossRef]
- Liu, H.C.; Goldenberg, A.; Chen, Y.C.; Lun, C.; Wu, W.; Bush, K.T.; Balac, N.; Rodriguez, P.; Abagyan, R.; Nigam, S.K. Molecular properties of drugs interacting with slc22 transporters oat1, oat3, oct1, and oct2: A machine-learning approachs. J. Pharmacol. Exp. Ther. 2016, 359, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Khamdang, S.; Takeda, M.; Shimoda, M.; Noshiro, R.; Narikawa, S.; Huang, X.L.; Enomoto, A.; Piyachaturawat, P.; Endou, H. Interactions of human- and rat-organic anion transporters with pravastatin and cimetidine. J. Pharmacol. Sci. 2004, 94, 197–202. [Google Scholar] [CrossRef]
- Jin, S.; Lee, S.; Jeon, J.H.; Kim, H.; Choi, M.K.; Song, I.S. Enhanced intestinal permeability and plasma concentration of metformin in rats by the repeated administration of red ginseng extract. Pharmaceutics 2019, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, A.N.; Burckhardt, G. Organic anion transporters of the slc22 family: Biopharmaceutical, physiological, and pathological roles. Pharm. Res. 2007, 24, 450–470. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYPs | Enzyme Activities | IC50 (μM) | ||
|---|---|---|---|---|
| Without NADPH Preincubation | With NADPH Preincubation | IC50 Shift | ||
| CYP1A2 | Phenacetin O-deethylase | >100 | >100 | - |
| CYP2A6 | Coumarin 7-hydroxylase | >100 | >100 | - |
| CYP2B6 | Bupropion hydroxylase | >100 | >100 | - |
| CYP2C8 | Amodiaquine N-deethylase | 32.5 | 30.1 | 1.1 |
| CYP2C9 | Diclofenac 4’-hydroxylase | 17.4 | 19.3 | 0.9 |
| CYP2C19 | [S]-Mephenytoin 4’-hydroxylase | 20.0 | 23.0 | 0.9 |
| CYP2D6 | Bufuralol 1’-hydroxylase | >100 | >100 | - |
| CYP3A4 | Midazolam 1’-hydroxylase | 66.4 | 24.4 | 2.7 |
| CYPs and Transporters | Ki (μM) | Mode of Inhibition |
|---|---|---|
| CYP2C8 | 16.9 | mixed inhibition |
| CYP2C9 | 16.1 | competitive inhibition |
| CYP2C19 | 6.7 | mixed inhibition |
| CYP3A4 | 17.6 (kinact 0.04047 min−1) | time-dependent inhibition |
| OCT1 | 145.7 | mixed inhibition |
| OAT3 | 8.3 | competitive inhibition |
| CYPs | CDOCKER Energy (kcal/mol) | Hydrogen Bond (Å) | Hydrophobic (Å) |
|---|---|---|---|
| CYP2C8 | −29.36 | Heme (4.47) | Ile113 (5.27 & 5.41) |
| Phe205 (4.48) | |||
| Leu208 (5.13) | |||
| Val296 (5.07) | |||
| Val366 (4.47) | |||
| CYP2C9 | −26.99 | Arg108 (4.47) | Leu208 (5.40 & 4.92) |
| Phe114 (5.49) | |||
| Ala297 (3.53) | |||
| Leu366 (5.44) | |||
| Heme (3.71) | |||
| CYP2C19 | −33.15 | Asp239 (2.95) Heme (2.62) | Leu102 (4.57) |
| Aal103 (4.38) | |||
| Phe114 (4.84) | |||
| Phe476 (5.44) | |||
| CYP3A4 | −32.34 | Arg105 (2.40) | Phe108 (4.56) Ile120 (4.40) Arg212 (4.96) |
| Arg372 (2.77 & 2.43) | |||
| Glu374 (2.26) | |||
| Heme (2.89) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, E.J.; Park, R.; Jeon, J.-H.; Cho, Y.-Y.; Lee, J.Y.; Kang, H.C.; Song, I.-S.; Lee, H.S. Inhibitory Effect of AB-PINACA, Indazole Carboxamide Synthetic Cannabinoid, on Human Major Drug-Metabolizing Enzymes and Transporters. Pharmaceutics 2020, 12, 1036. https://doi.org/10.3390/pharmaceutics12111036
Park EJ, Park R, Jeon J-H, Cho Y-Y, Lee JY, Kang HC, Song I-S, Lee HS. Inhibitory Effect of AB-PINACA, Indazole Carboxamide Synthetic Cannabinoid, on Human Major Drug-Metabolizing Enzymes and Transporters. Pharmaceutics. 2020; 12(11):1036. https://doi.org/10.3390/pharmaceutics12111036
Chicago/Turabian StylePark, Eun Jeong, Ria Park, Ji-Hyeon Jeon, Yong-Yeon Cho, Joo Young Lee, Han Chang Kang, Im-Sook Song, and Hye Suk Lee. 2020. "Inhibitory Effect of AB-PINACA, Indazole Carboxamide Synthetic Cannabinoid, on Human Major Drug-Metabolizing Enzymes and Transporters" Pharmaceutics 12, no. 11: 1036. https://doi.org/10.3390/pharmaceutics12111036
APA StylePark, E. J., Park, R., Jeon, J.-H., Cho, Y.-Y., Lee, J. Y., Kang, H. C., Song, I.-S., & Lee, H. S. (2020). Inhibitory Effect of AB-PINACA, Indazole Carboxamide Synthetic Cannabinoid, on Human Major Drug-Metabolizing Enzymes and Transporters. Pharmaceutics, 12(11), 1036. https://doi.org/10.3390/pharmaceutics12111036