Induction of WNT16 via Peptide-mRNA Nanoparticle-Based Delivery Maintains Cartilage Homeostasis

 ,
,
Abstract
1. Introduction
2. Results
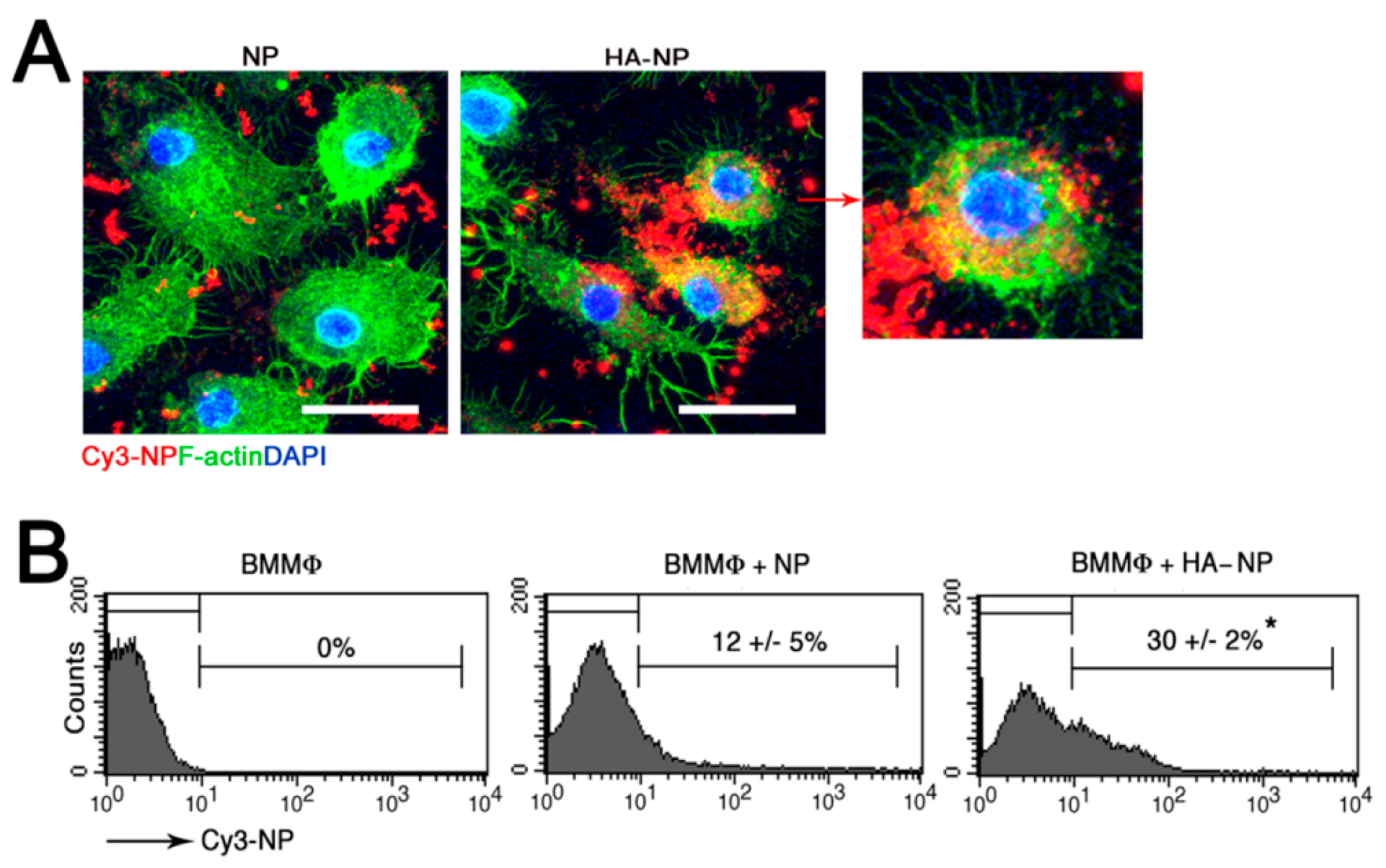
2.1. Hyaluronic Acid (HA) Coating Enhances Cellular Uptake of the Nanoparticle (NP)
2.2. p5RHH-mRNA NP Preparation and Characterization
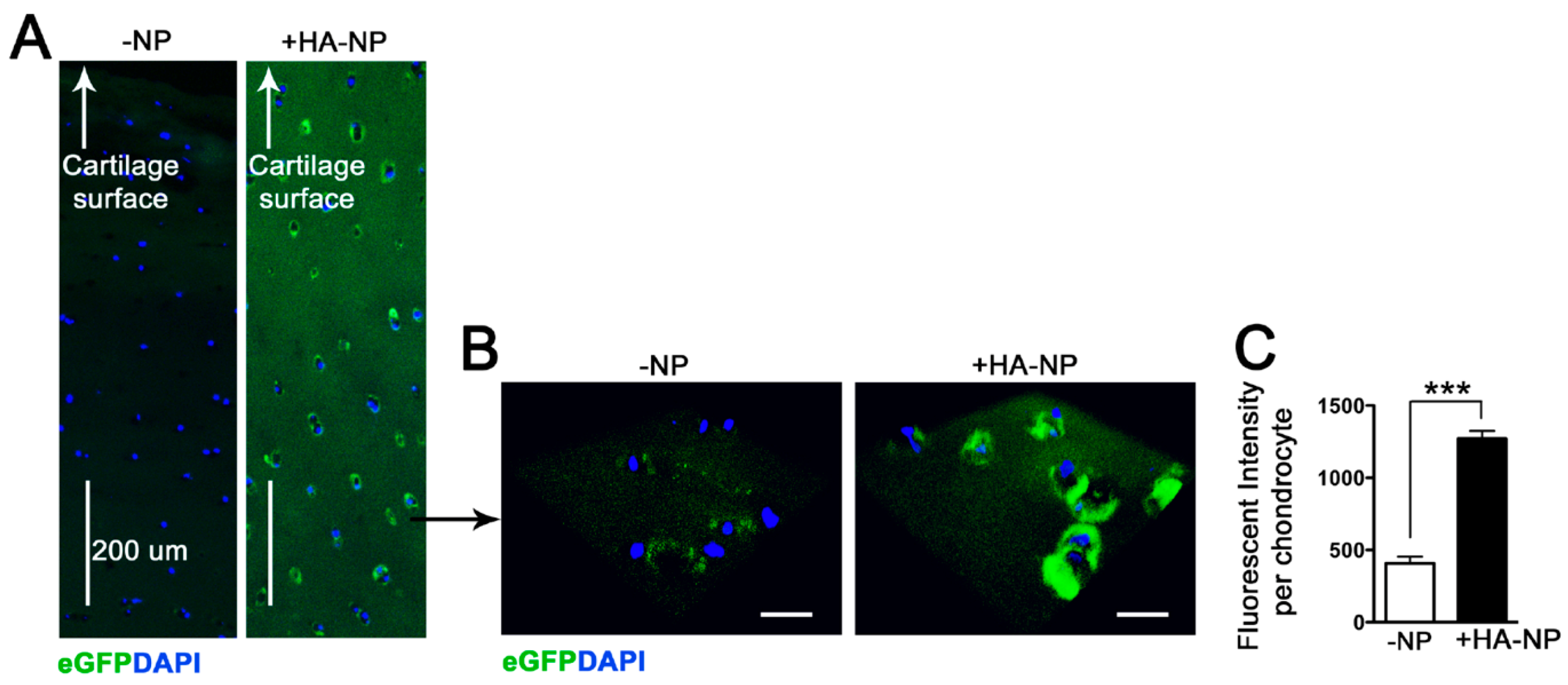
2.3. Delivery of Enhanced Green Fluorescent Protein (eGFP) mRNA in Cartilage Explants
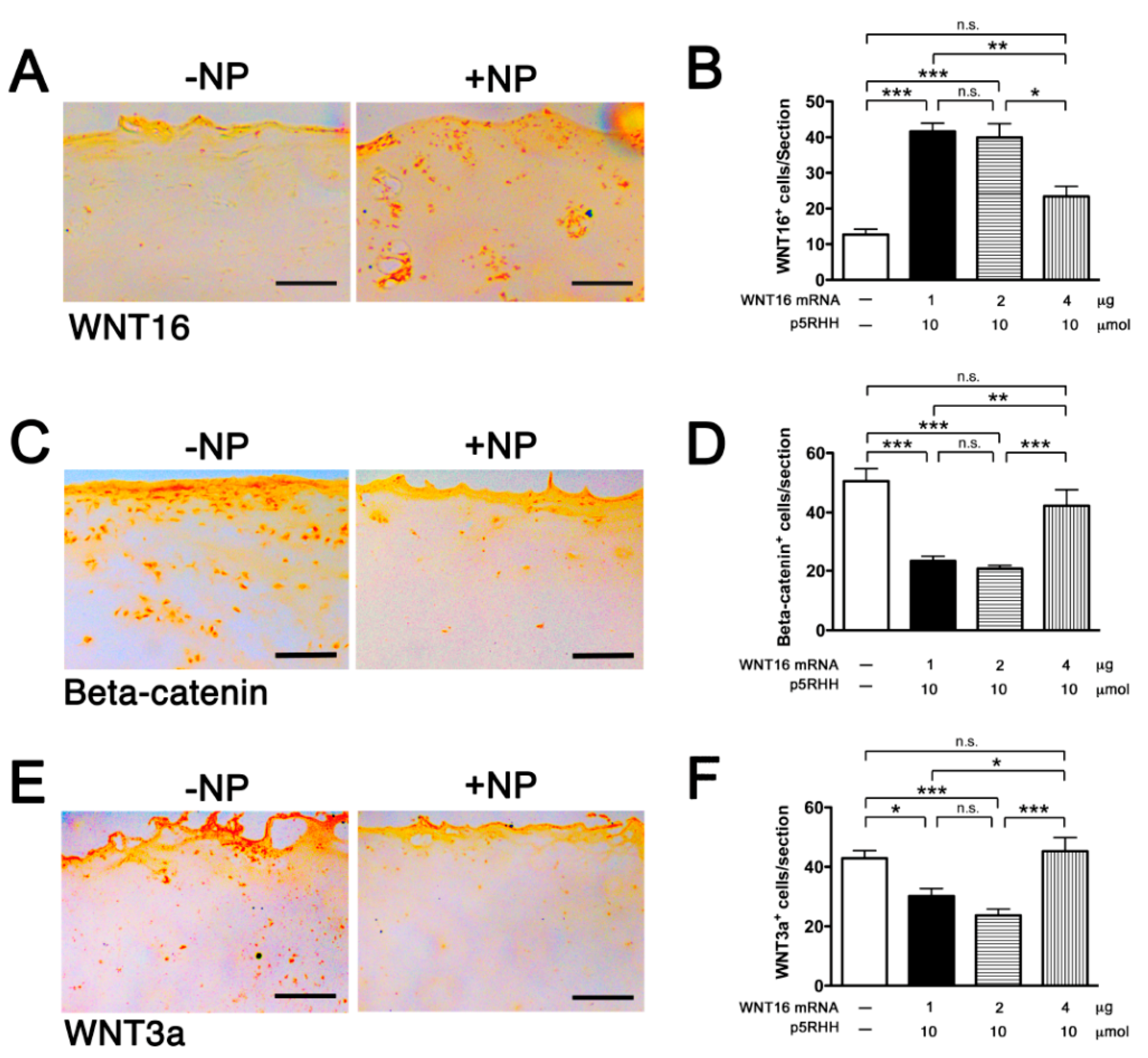
2.4. Delivery of WNT16 mRNA in Cartilage Explants
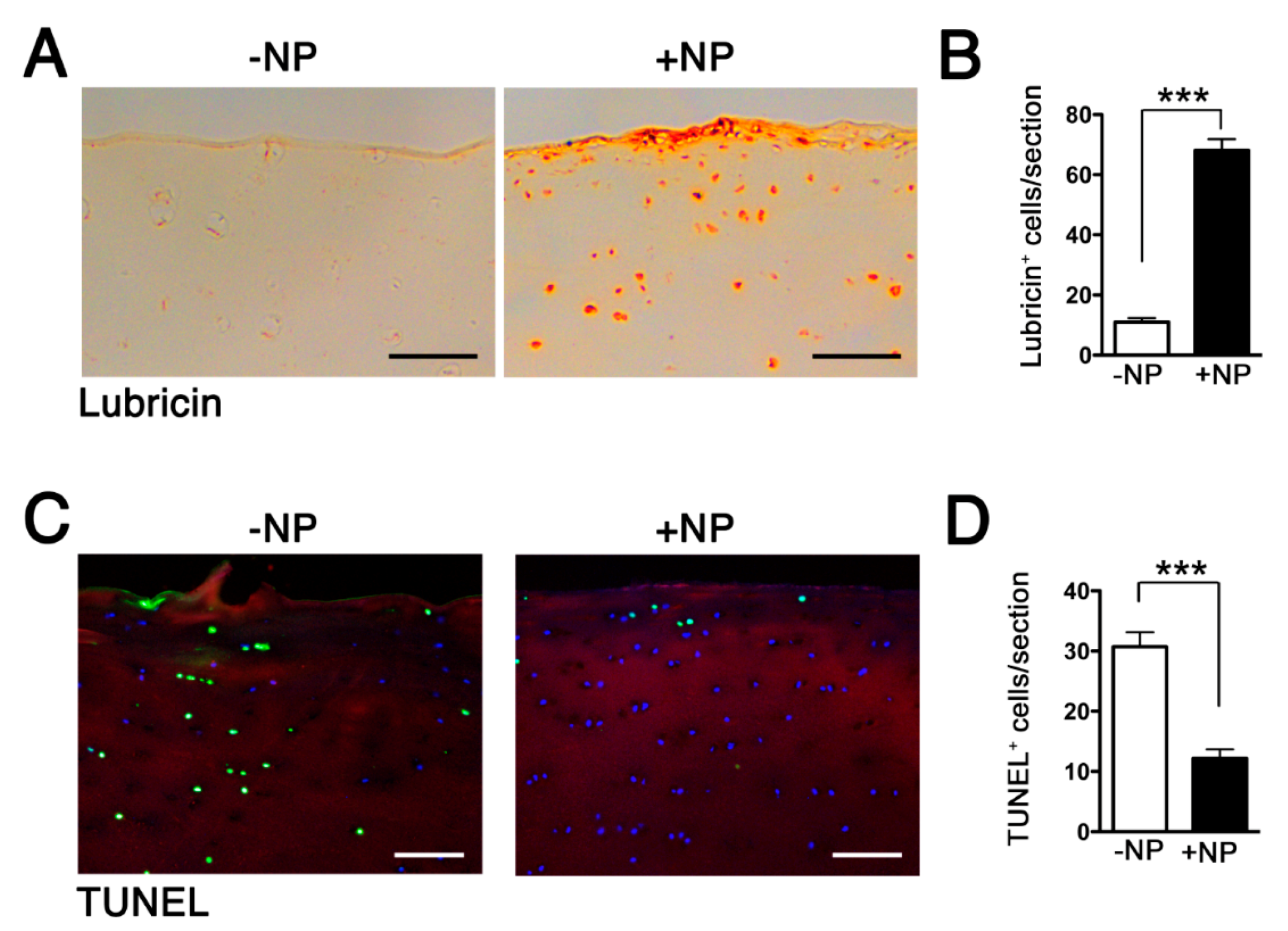
2.5. Effect of WNT16 Overexpression on Cartilage Homeostasis
3. Discussion
4. Materials and Methods
4.1. Preparation of HA-Coated p5RHH-mRNA NPs
4.2. Human Cartilage Explant Culture
4.3. In Vitro NP Uptake by Bone-Marrow-Derived Macrophages (BMMϕ)
4.4. TUNEL Assay
4.5. Confocal Microscopy
4.6. Immunohistochemistry
4.7. Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMMϕ | Bone Marrow-derived Macrophage |
| DAPI | 4’,6-diamidino-2-phenylindole |
| DLS | Dynamic Light Scattering |
| DMOAD | Disease Modifying Osteoarthritis Drug |
| EDTA | Ethylenediaminetetraacetic acid |
| eGFP | enhanced Green Fluorescent Protein |
| ECM | Extracellular Matrix |
| FBS | Fetal Bovine Serum |
| FGF | Fibroblast Growth Factor |
| GM-CSF | Granulocyte-Macrophage Colony Stimulating Factor |
| HA | Hyaluronic acid |
| IA | Intra-articular |
| IL | Interleukin |
| IRB | Institutional Review Board |
| mRNA | messenger RNA |
| NP | Nanoparticle |
| nt | Nucleotide |
| OA | Osteoarthritis |
| OCT | Optimum Cutting Temperature |
| RNA | Ribonucleic acid |
| siRNA | small interfering RNA |
| TEM | Transmission Electron Microscopy |
| TNF | Tumor Necrosis Factor |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling |
| WNT | Wingless and the name Int-1 |
References
- Rai, M.F.; Pham, C.T. Intra-articular drug delivery systems for joint diseases. Curr. Opin. Pharmacol. 2018, 40, 67–73. [Google Scholar] [CrossRef]
- Allen, K.D.; Golightly, Y.M. State of the evidence. Curr. Opin. Rheumatol. 2015, 27, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.A.; Furman, B.D.; Kraus, V.B.; Huebner, J.L.; Guilak, F. Therapeutic opportunities to prevent post-traumatic arthritis: Lessons from the natural history of arthritis after articular fracture. J. Orthop. Res. 2015, 33, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheumatol. 2009, 61, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Kraus, V.B.; Birmingham, J.; Stabler, T.V.; Feng, S.; Taylor, D.C.; Moorman, C.T., 3rd; Garrett, W.E.; Toth, A.P. Effects of intraarticular IL1-Ra for acute anterior cruciate ligament knee injury: A randomized controlled pilot trial (NCT00332254). Osteoarthr. Cartil. 2012, 20, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R. Alternative Wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Corr, M. Wnt-beta-catenin signaling in the pathogenesis of osteoarthritis. Nat. Clin. Pract. Rheumatol. 2008, 4, 550–556. [Google Scholar] [CrossRef]
- Yuasa, T.; Otani, T.; Koike, T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt/beta-catenin signaling stimulates matrix catabolic genes and activity in articular chondrocytes: Its possible role in joint degeneration. Lab. Investig. 2008, 88, 264–274. [Google Scholar] [CrossRef]
- Nalesso, G.; Sherwood, J.; Bertrand, J.; Pap, T.; Ramachandran, M.; De Bari, C.; Pitzalis, C.; Dell’accio, F. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J. Cell Biol. 2011, 193, 551–564. [Google Scholar] [CrossRef]
- Nalesso, G.; Thomas, B.L.; Sherwood, J.C.; Yu, J.; Addimanda, O.; Eldridge, S.E.; Thorup, A.S.; Dale, L.; Schett, G.; Zwerina, J.; et al. WNT16 antagonises excessive canonical WNT activation and protects cartilage in osteoarthritis. Ann. Rheum. Dis. 2017, 76, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Waller, K.A.; Zhang, L.X.; Elsaid, K.A.; Fleming, B.C.; Warman, M.L.; Jay, G.D. Role of lubricin and boundary lubrication in the prevention of chondrocyte apoptosis. Proc. Natl. Acad. Sci. USA 2013, 110, 5852–5857. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, V.P. Intracellular RNA delivery by lipid nanoparticles: Diffusion, degradation, and release. Biosystems 2019, 185, 104032. [Google Scholar] [CrossRef] [PubMed]
- Partlow, K.C.; Lanza, G.M.; Wickline, S.A. Exploiting lipid raft transport with membrane targeted nanoparticles: A strategy for cytosolic drug delivery. Biomaterials 2008, 29, 3367–3375. [Google Scholar] [CrossRef] [PubMed]
- Soman, N.R.; Baldwin, S.L.; Hu, G.; Marsh, J.N.; Lanza, G.M.; Heuser, J.E.; Arbeit, J.M.; Wickline, S.A.; Schlesinger, P.H. Molecularly targeted nanocarriers deliver the cytolytic peptide melittin specifically to tumor cells in mice, reducing tumor growth. J. Clin. Investig. 2009, 119, 2830–2842. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Sasaki, Y.; Lanza, G.M.; Milbrandt, J.; Wickline, S.A. Mechanisms of nucleotide trafficking during siRNA delivery to endothelial cells using perfluorocarbon nanoemulsions. Biomaterials 2010, 31, 3079–3086. [Google Scholar] [CrossRef]
- Hou, K.K.; Pan, H.; Lanza, G.M.; Wickline, S.A. Melittin derived peptides for nanoparticle based siRNA transfection. Biomaterials 2013, 34, 3110–3119. [Google Scholar] [CrossRef]
- Hou, K.K.; Pan, H.; Ratner, L.; Schlesinger, P.H.; Wickline, S.A. Mechanisms of nanoparticle-mediated siRNA transfection by melittin-derived peptides. ACS Nano 2013, 7, 8605–8615. [Google Scholar] [CrossRef]
- Zhou, H.F.; Yan, H.; Pan, H.; Hou, K.K.; Akk, A.; Springer, L.E.; Hu, Y.; Allen, J.S.; Wickline, S.A.; Pham, C.T. Peptide-siRNA nanocomplexes targeting NF-kappaB subunit p65 suppress nascent experimental arthritis. J. Clin. Investig. 2014, 124, 4363–4374. [Google Scholar] [CrossRef]
- Yan, H.; Duan, X.; Pan, H.; Holguin, N.; Rai, M.F.; Akk, A.; Springer, L.E.; Wickline, S.A.; Sandell, L.J.; Pham, C.T. Suppression of NF-kappaB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc. Natl. Acad. Sci. USA 2016, 113, E6199–E6208. [Google Scholar] [CrossRef]
- Knudson, W.; Loeser, R.F. CD44 and integrin matrix receptors participate in cartilage homeostasis. Cell Mol. Life Sci. 2002, 59, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Setton, L. Polymer therapeutics: Reservoir drugs. Nat. Mater. 2008, 7, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Rai, M.F.; Pan, H.; Yan, H.; Sandell, L.J.; Pham, C.T.N.; Wickline, S.A. Applications of RNA interference in the treatment of arthritis. Transl. Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Hom, K.; Zhang, D.; Leng, Q.; Tricoli, L.J.; Hustedt, J.M.; Lee, A.; Shapiro, M.J.; Seog, J.; Kahn, J.D.; et al. Enhanced silencing and stabilization of siRNA polyplexes by histidine-mediated hydrogen bonds. Biomaterials 2014, 35, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Ellman, M.B.; An, H.S.; Muddasani, P.; Im, H.J. Biological impact of the fibroblast growth factor family on articular cartilage and intervertebral disc homeostasis. Gene 2008, 420, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.E.; Bendele, A.M.; Thompson, D.L.; Littau, A.; Waggie, K.S.; Reardon, B.; Ellsworth, J.L. Fibroblast growth factor-18 stimulates chondrogenesis and cartilage repair in a rat model of injury-induced osteoarthritis. Osteoarthr. Cartil. 2005, 13, 623–631. [Google Scholar] [CrossRef]
- Hochberg, M.C.; Guermazi, A.; Guehring, H.; Aydemir, A.; Wax, S.; Fleuranceau-Morel, P.; Reinstrup Bihlet, A.; Byrjalsen, I.; Ragnar Andersen, J.; Eckstein, F. Effect of Intra-Articular Sprifermin vs Placebo on Femorotibial Joint Cartilage Thickness in Patients with Osteoarthritis: The FORWARD Randomised Clinical Trial. JAMA 2019, 322, 1360–1370. [Google Scholar] [CrossRef]
- Zhu, M.; Tang, D.; Wu, Q.; Hao, S.; Chen, M.; Xie, C.; Rosier, R.N.; O’Keefe, R.J.; Zuscik, M.; Chen, D. Activation of beta-catenin signaling in articular chondrocytes leads to osteoarthritis-like phenotype in adult beta-catenin conditional activation mice. J. Bone Miner. Res. 2009, 24, 12–21. [Google Scholar] [CrossRef]
- Luyten, F.P.; Tylzanowski, P.; Lories, R.J. Wnt signaling and osteoarthritis. Bone 2009, 44, 522–527. [Google Scholar] [CrossRef]
- Deshmukh, V.; Hu, H.; Barroga, C.; Bossard, C.; Kc, S.; Dellamary, L.; Stewart, J.; Chiu, K.; Ibanez, M.; Pedraza, M.; et al. A small-molecule inhibitor of the Wnt pathway (SM04690) as a potential disease modifying agent for the treatment of osteoarthritis of the knee. Osteoarthr. Cartil. 2018, 26, 18–27. [Google Scholar] [CrossRef]
- Yan, H.; Duan, X.; Pan, H.; Akk, A.; Sandell, L.J.; Wickline, S.A.; Rai, M.F.; Pham, C.T.N. Development of a peptide-siRNA nanocomplex targeting NF- kappaB for efficient cartilage delivery. Sci. Rep. 2019, 9, 442. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Composition Peptide:mRNA (mol:mol) | 3500:1 | 1750:1 | 875:1 |
|---|---|---|---|
| Average size (nm) TEM | 64.78 ± 8.344 (n = 50) | 105.5 ± 6.959 (n = 150) | 205.1 ± 6.109 (n = 500) |
| Average size (nm) DLS | 181 | 177 | 3000 |
| Zeta potential (mV) | −30.06 ± 0.82 | −28.79 ± 1.77 | −31.48 ± 2.59 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, H.; Hu, Y.; Akk, A.; Rai, M.F.; Pan, H.; Wickline, S.A.; Pham, C.T.N. Induction of WNT16 via Peptide-mRNA Nanoparticle-Based Delivery Maintains Cartilage Homeostasis. Pharmaceutics 2020, 12, 73. https://doi.org/10.3390/pharmaceutics12010073
Yan H, Hu Y, Akk A, Rai MF, Pan H, Wickline SA, Pham CTN. Induction of WNT16 via Peptide-mRNA Nanoparticle-Based Delivery Maintains Cartilage Homeostasis. Pharmaceutics. 2020; 12(1):73. https://doi.org/10.3390/pharmaceutics12010073
Chicago/Turabian StyleYan, Huimin, Ying Hu, Antonina Akk, Muhammad Farooq Rai, Hua Pan, Samuel A. Wickline, and Christine T.N. Pham. 2020. "Induction of WNT16 via Peptide-mRNA Nanoparticle-Based Delivery Maintains Cartilage Homeostasis" Pharmaceutics 12, no. 1: 73. https://doi.org/10.3390/pharmaceutics12010073
APA StyleYan, H., Hu, Y., Akk, A., Rai, M. F., Pan, H., Wickline, S. A., & Pham, C. T. N. (2020). Induction of WNT16 via Peptide-mRNA Nanoparticle-Based Delivery Maintains Cartilage Homeostasis. Pharmaceutics, 12(1), 73. https://doi.org/10.3390/pharmaceutics12010073