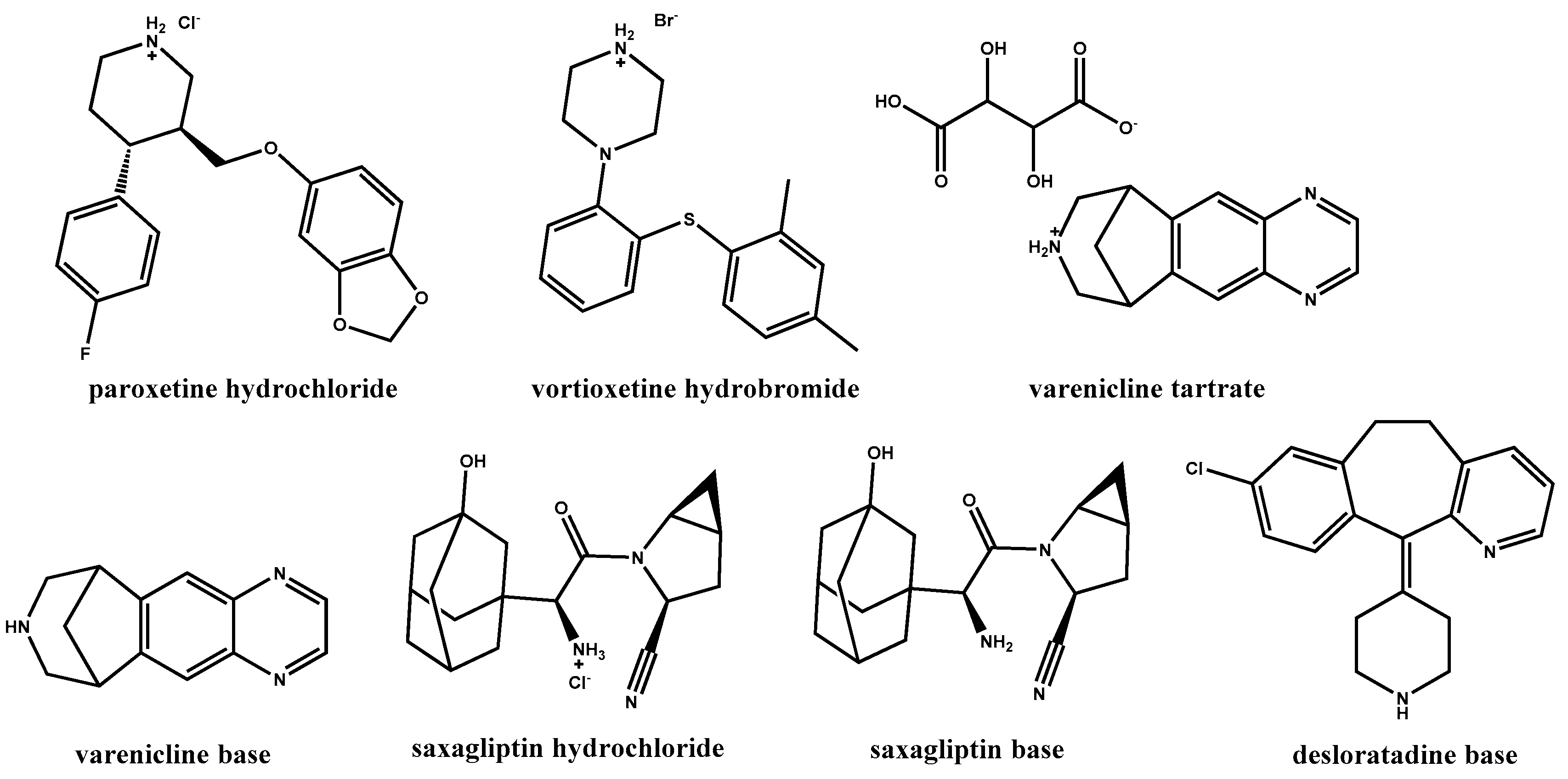
In a recent drug development effort in our laboratories, paroxetine·HCl [
45], a drug substance containing a secondary amine moiety, was formulated as a tablet. It was found that a significant amount of
N-methyl paroxetine impurity, a product of an Eschweiler–Clarke reaction (
Scheme 1) [
25,
31], was formed in packed tablets protected from moisture and light under accelerated stability conditions (data not reported here). The tablet formulation consisted of (1) tablet core components—paroxetine·HCl, hypromellose 2208 (hydroxypropyl methylcellulose, HPMC), povidone K30 (polyvinylpyrrolidone, PVP), lactose monohydrate, silicon dioxide, magnesium stearate; (2) the enteric coat components—methacrylic acid and ethyl acrylate copolymer, talc, triethyl citrate; and (3) the over coat components—hypromellose 2910 (hydroxypropyl methylcellulose, HPMC), polyethylene glycol (PEG) 6000, titanium dioxide, and iron oxide (Fe
2O
3) yellow. An investigation to determine the origin of formaldehyde and formic acid responsible for
N-methylation was conducted.
3.1.1. Origin of Reactive Impurities in Tablet Formulation
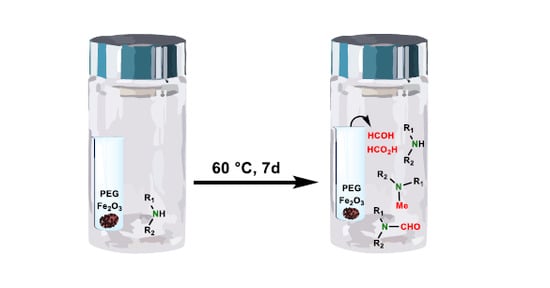
Tablet composition components with the potential to contain trace amounts of reactive impurities, were studied in the form of binary and tertiary mixtures with the drug substance in a sealed vial setup. Povidone K30, due to its direct contact with paroxetine·HCl, as well as triethyl citrate (TEC) and PEG 6000, found in the tablet coat, were considered for testing in a direct mixture setup. Surprisingly, when subjected to 60 °C for seven days, none of these mixtures resulted in an increase in
N-methyl paroxetine (
Table 3, Experiment 1). In the next step, we added Fe
2O
3 to PEG 6000 as a potential oxidation catalyst and constructed a direct and separate mixture design. In a separate mixture, design excipients were transferred into glass inserts within the large vials containing paroxetine·HCl. This setup was introduced to best mimic the conditions in the tablet formulation, where there is also no direct contact between components. Interestingly, the reaction for
N-methyl paroxetine was positive, but only where PEG 6000 and Fe
2O
3 are separated from paroxetine·HCl (
Table 3, Experiment 2). Tablet core excipients were first thought to be responsible for the impurity increase; therefore, such an outcome was unexpected. Similarly to tablet stability results, no other paroxetine degradation was observed, indicating a very good resemblance of this stress test compared to the actual conditions within tablets. Based on the stress test results, it is hypothesized that volatile low molecular weight organic impurities originating from oxidative degradation of plasticizer PEG 6000 in contact with Fe
2O
3 permeated from the tablet coat into the tablet core through the enteric coating and concurrently reacted with the drug substance. The same stress test design was applied to an alternative plasticizer and alternative colorant (
Table 3, Experiment 3). Both results were negative, which confirmed that the combination of PEG and Fe
2O
3 is accountable for incompatibility with the drug substance. After 14 days, the level of
N-methyl paroxetine increased from 0.63% to 0.84% in the sample with PEG 6000 and Fe
2O
3 yellow; in other samples, this increase did not occur.
To confirm that the cause of
N-methylation are in fact reactive impurities formaldehyde and formic acid, paroxetine·HCl in a solid state was spiked with formalin, a 37% formaldehyde solution, and with 99.8% formic acid, prior to elevated temperature exposure. All instances of formalin addition, direct and in separate insert, resulted in
N-methyl paroxetine impurity formation, on the contrary to formic acid addition, which is not capable of forming
N-methylated product (
Table 4). Nonetheless, repeatability of the results with reactive impurity spiking is poor. The determined
N-methyl paroxetine amounts for repetitions of formalin added to paroxetine·HCl showed extensive variability (average: 0.98%, rsd: 76%,
n = 3) and poor specificity, with multiple by-products formed and high drug substance degradation. In practice, it is very challenging to add low volumes of reactive impurities in an accurate and precise manner. What is more, added liquids cannot be homogeneously distributed across the entire sample, regardless of whether they are added directly to the sample and mixed or if they are in a separate container, which presents sampling issues. Reactive impurities solutions often also contain other impurities. Formalin for instance contains methanol, formic acid, aldehydes, and ketones, which have an influence on the outcome of the experiment. On the other hand, combination of PEG and Fe
2O
3, separated from paroxetine·HCl, resulted only in
N-methyl impurity formation (more than 99% of drug substance remaining), with lower variability (average: 0.63%, rsd: 13%,
n = 3). For more in-depth understanding of PEG degradation and iron oxide involvement in
N-methylation of paroxetine, a kinetics study was planned.
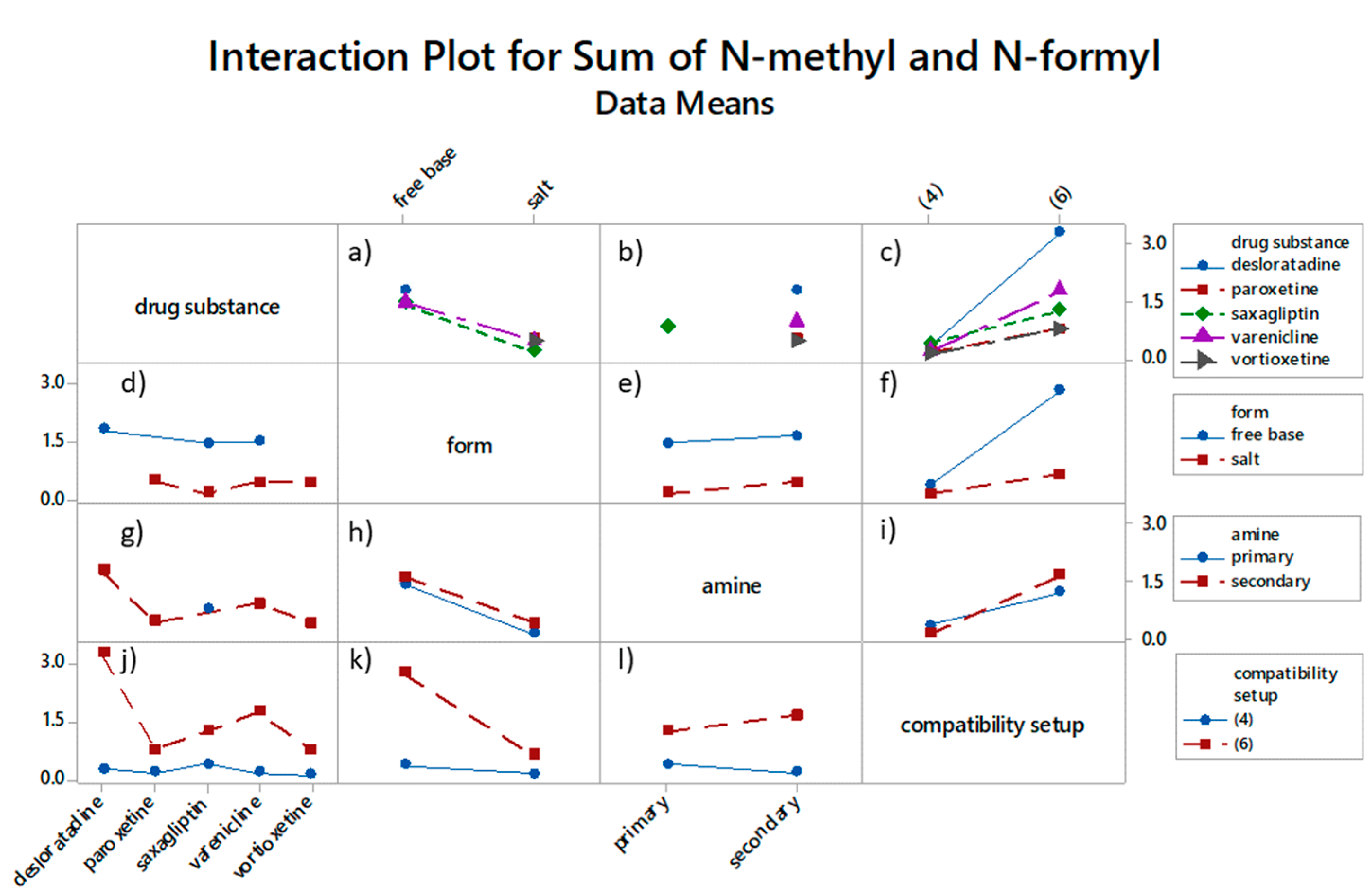
3.1.2. PEG Degradation and N-Methyl Formation Kinetic Study
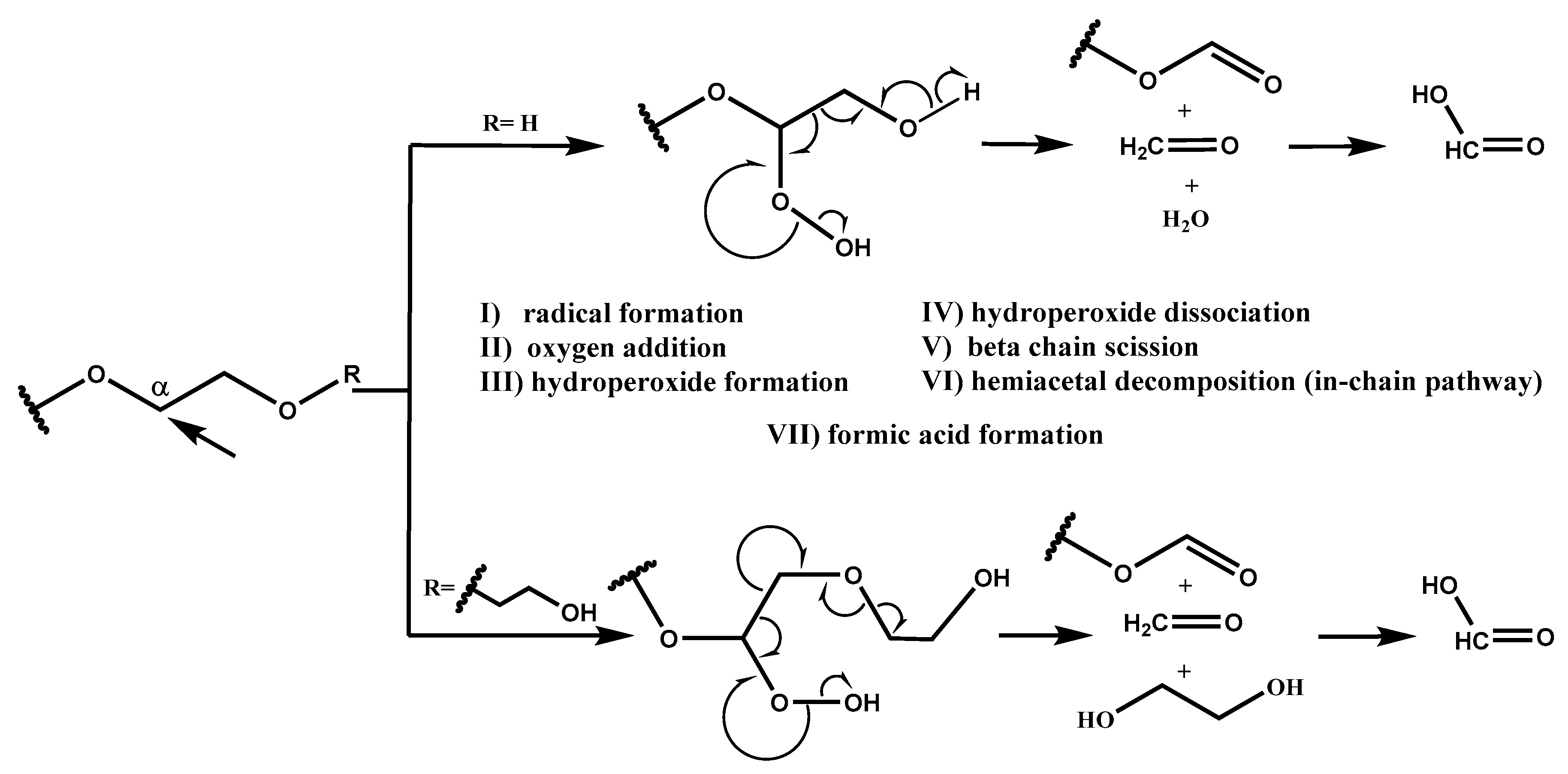
Based on the radical chain scission mechanism (
Scheme 2) [
3,
14], very low concentration of radical sites is needed to initiate the PEG degradation process. These sites can form under the influence of heat, near-UV light, chemical initiators, or transition metals (step I) [
7]. In the propagation phase (steps II, III, IV, V, and VI), first the addition of oxygen occurs. PEG requires molecular oxygen to degrade into formaldehyde and formic acid. Then, peroxy group abstracts hydrogen from a nearby RH, and this is followed by hydroperoxide dissociation and alkoxy radical (RO·) formation, resulting in C–C bond scission and, finally, the formation of formaldehyde and formate ester [
10], which can hydrolyze into formic acid. In-depth NMR studies performed by Mkhatresh et al. [
5,
8] identified, apart from formaldehyde and formate ester end-groups, also the following species: In-chain esters, methyl end-groups, ethanoate end-groups, acetal links, and multiple small molecular species. Gallet et al. added to these lists also ketones and alcohols [
3,
4]. Nevertheless, our study design focused on monitoring formaldehyde and formic acid, as the main PEG degradation products. Each ethylene glycol unit in theory can produce one formaldehyde molecule, which can react with amine functionality of paroxetine. Proposed amounts and ratios of components in 10 mL glass vials (i.e., 1:0.1:0.01), present sufficient reactive reactant availability on one hand, since 68.6% of
N-methylated impurity at complete conversion is formed, and realistic excipient-drug substance ratio on the other.
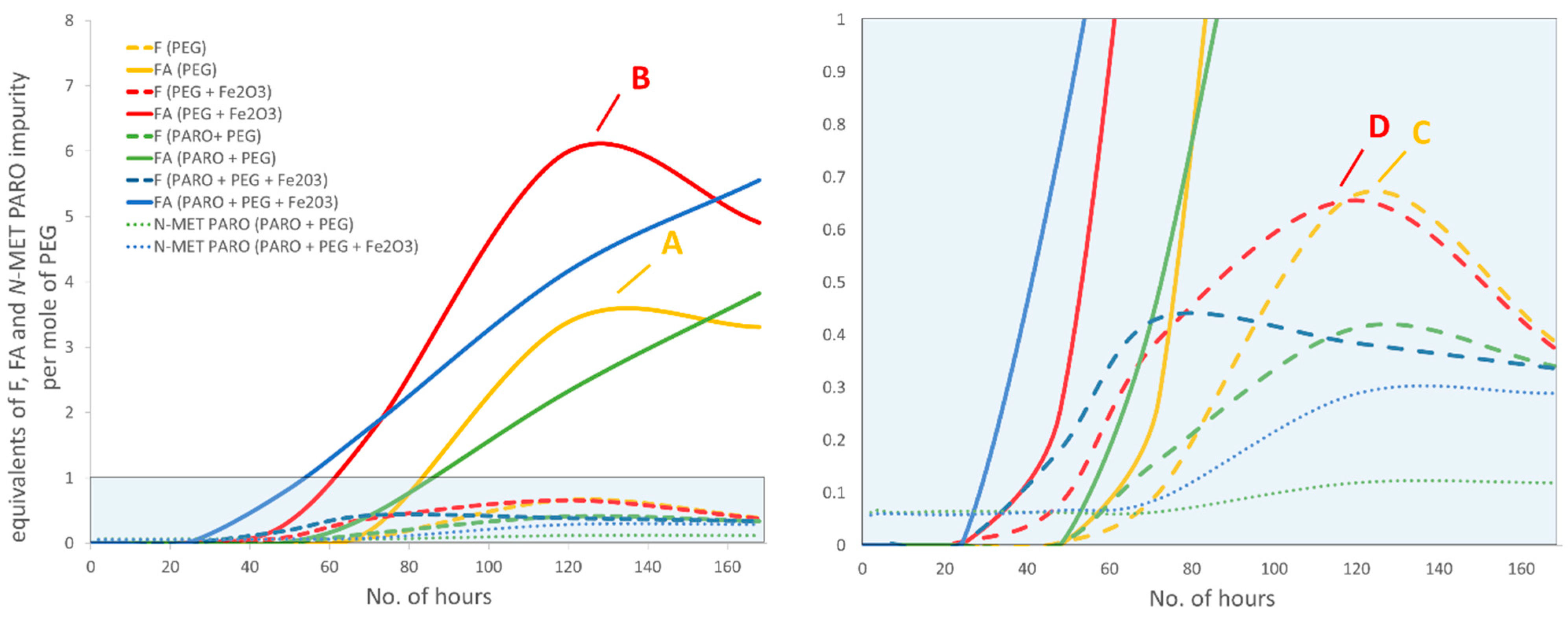
In our kinetic study we paid special attention to events occurring in the early hours of the stress test, to evaluate potential formaldehyde to formic acid conversion and study the lag time between PEG and drug substance degradation. Results of the seven-day stress test study are presented in
Figure 3.
In the first 24 h, no increase in impurities was observed. This is in accordance with Bergh et al. who also observed some lag time prior to formaldehyde detection [
9]. The lag time in our case correlated with the onset of melting. PEG 6000 is a solid powder with a melting point of 55–63 °C [
1]. Visual inspection of samples revealed that solid particles could be observed for up to 24 h at an elevated temperature but were no longer visible after 48 h (
Figure 4). At this time, a smooth film formed at the bottom of the vial. This indicates that degradation kinetics are connected to the mobility of PEG chains and oxygen molecules and are significantly increased when the sample is converted to melt. It is possible that transition to a liquid state is observed only when the molecular weight of longer PEG chains is reduced, which causes a lag time in the melting onset.
The maximum formaldehyde and formic acid concentrations in samples with PEG alone were reached after five days (points A and C,
Figure 3): 3.38 mg of formaldehyde per g of PEG, which is 0.68 equivalents per mole of PEG, and 26.3 mg of formic acid per g of PEG, which is 3.43 equivalents per mole of PEG. The maximum concentrations of formaldehyde and formic acid in samples with PEG and Fe
2O
3 were also reached after five days (points B and D,
Figure 3): 3.29 mg of formaldehyde per g of PEG, which is 0.66 equivalents per mole of PEG, and 46.1 mg of formic acid per g of PEG, which is 6.01 equivalents per mole of PEG. This suggests that in the design where PEG is combined with Fe
2O
3, at least six molecules of formic acid are formed per PEG chain, which is one per every 23 ethylene glycol units (4.4%). After five days, the concentrations of both degradants began to decrease (
Figure 3). Maximal observed levels were up to 10-fold higher compared to the levels determined in similar studies performed by other groups [
14,
46]. These differences are especially ascribed to the temperature difference, which, in our case, was purposely set at the PEG 6000 melting point and the time from the stress test start to sampling, since considerable curvature was observed.
The observed formation of formaldehyde was unproportioned to the formation of formic acid, which was present in significantly higher concentrations. A comparison of the curves (solid red and yellow line compared to dashed yellow and red line in a range from ca. one day three days,
Figure 3) suggests that at least some, if not the majority, of formic acid is formed from rapid formaldehyde consumption [
14]. This observation is also confirmed by our previously mentioned experiments (
Section 3.1.1.), where a significant increase in
N-methyl impurity was observed in direct spikes of formaldehyde solution, where no formic acid was added. The maximum formaldehyde concentration was at an approximately equal level with PEG alone compared to when iron oxide was added (compare points C and D in
Figure 3); however, the formic acid concentration was much higher in the latter (compare points A and B in
Figure 3). This means that iron oxide has to be responsible for the accelerated formation of formaldehyde; otherwise, the molar sum of these two impurities would be the same in both experiments. The concentration of formaldehyde (dashed yellow and red lines,
Figure 3) and formic acid (solid yellow and red lines,
Figure 3) in samples without paroxetine·HCl started to fall after a five-day time point. A decrease in formaldehyde was expected due to oxidation to formic acid, however, the formic acid decrease was not expected. The reactant availability was initially considered. In the 10 mL glass vials used for our stress tests, there was 93.75 µmoles of O
2 molecules, calculated based on the ideal gas law. Five milligrams of PEG 6000 placed in every vial is equivalent to 0.833 µmoles of chains with a molecular weight of 6000 g/mol. Considering there is, on average, 136 ethylene glycol monomer units in PEG 6000, the amount of oxygen available would suffice for the degradation of 82.8% of all ethylene glycolic units. Thus, the concentration of oxygen is sufficient for oxidation to proceed. Formaldehyde, as second essential reactant, is also available as long as there are unoxidized ethylene glycol units left, unless the material itself or the environmental conditions within the vial change, so that the PEG degradation rate is affected. Potential causes for more substantial reduction of the PEG degradation rate could be associated with the change in relative humidity (RH), pH, molecular weight reduction of PEG chains, and oxidative changes to the PEG backbone, resulting in inactivity. To our knowledge, there is no reported evidence in the literature that a drop in pH or increase in RH, through formed molecules of organic acids and water, could significantly decrease the PEG degradation rate. On the other hand, material properties change, such as a decrease in the length of PEG chains [
47] or some other oxidative transformation, could influence the degradation rate. However, one would expect maximum formic acid concentration for samples with PEG alone (point A,
Figure 3) compared to PEG with Fe
2O
3 (point B,
Figure 3) to reach the same level if they are conditioned by material properties or environmental conditions change. Yet, the maximum formic acid concentration could be dependent on the presence of iron oxides, whereas a decrease in the PEG degradation rate after a five-day time point is related to some other physical change.
In our opinion, it is more likely that formic acid will react further, forming a tertiary degradation product, e.g., formate ester [
4,
14], creating a system of three consecutive reactions. Formate esters are expected to have similar reactivity to formic acid; however, their mobility is restrained due to long residual PEG chains. Similar reactivity is also expected for tablet formulations with PEG present in the tablet coat. Formic acid in formate ester form would not be detected with our method for the determination of small molecular weight organic molecules due to the inability to react with an EDC condensing agent.
The relevance of the proposed three consecutive reaction systems was evaluated via kinetic modelling. The reaction stoichiometry is described in Equation (1). “r” represents the reaction rate (change in concentration over time), “k” represents the rate constant, and “c” represents the concentrations of individual species. Experimental results for formaldehyde (F) and formic acid (FA) in the sample with PEG 6000 alone were fitted to the model described in Equations (2)–(5) (lag time was disregarded).
Figure 5 graphically depicts the PEG degradation kinetics of a model fitted to the degradation data of PEG alone (yellow lines). The determined rate constants for the “fitted model” were k
1 = 4.32 × 10
−4 ± 1.68 × 10
−4 h
−1, k
2 = 0.134 ± 0.0730 h
−1, and k
3 = 0.0117 ± 0.00976 h
−1, implicating fast formaldehyde to formic acid conversion (this explains the absence of lag time between both degradants) and an approximately 10-fold slower third consecutive reaction of tertiary degradation product formation from formic acid. Two hypothetical types of PEG degradation kinetics, where we simulated increases in rate constants, are depicted with red and blue lines. In “simulation 1”, the rate constant k
1 was increased by 2-fold (blue lines), and in “simulation 2”, both, k
1 and k
2 were increased by 2-fold (red lines). It was previously established in our experiment that iron oxide has to be responsible for increasing k
1 based on the increased molar sum of determined formaldehyde and formic acid in the sample with PEG and Fe
2O
3 (
Figure 3). Simulations in
Figure 5 show that if along with k
1, k
2 is also increased by the same factor, formic acid’s maximum is higher compared to the fitted model (solid red versus solid yellow), whereas maximum formaldehyde concentration is equal (dashed red versus dashed yellow line). Increasing only k
1 results in an increased maximum formaldehyde concentration in the simulated model compared with the fitted model (dashed yellow versus dashed blue line). Since the simulated kinetics for increased k
1 and k
2 (red lines) reflect the behavior of the experiment depicted in
Figure 3, we confirmed that the degradation of PEG to formaldehyde and formaldehyde to formic acid are both catalyzed by iron oxide. Formate ester formation, which could also influence PEG degradation kinetics by protecting terminal hydroxyl groups from oxidation, was not accounted for in the model [
6].
Iron oxide increases the amount of formed
N-methyl impurity through an increase in the PEG degradation rate and also through an earlier onset of small volatile PEG degradation products. Iron oxide is generally insoluble in water; however, only trace amounts are needed to trigger the formation of free radicals capable of initiating PEG chain degradation. Furthermore, PEG solutions are slightly acidic, which potentially increases the amount of free/dissolved iron in the sample. Transition metal ions may cause the formation of very reactive hydroxyl radicals in a well-known Fenton reaction [
42,
48], but there are also reports that they may act as direct oxidants in the absence of hydroperoxide impurities [
49]. By means of radical formation, Fe
3+ ions participate in formaldehyde oxidation and, in a similar way, in PEG oxidation. Another potential interpretation of an iron-oxide-induced increase in the degradation rate is through the formation of complexes. The oxidized form of the iron ion (Fe
3+) is most likely to form complexes with oxygen and molecules containing oxygen, facilitating electron transfer [
18]. Sakharov et al. suggested that catalytic oxidative deformylation, with Cu
2+ as the catalyst, is the predominant degradation mechanism instead of radical chain scission [
17]. The transition metal in this case does not carry a redox function but acts as a complexing agent, aiding in electron transfer. It is presumed that Cu
2+ preferably forms complexes with longer PEG chains, which could be reflected by a decreased PEG degradation rate over time. Nevertheless, this mechanism is not the likeliest of the Fe mediated degradations, since extreme basic conditions are required.
When paroxetine, separated from excipient(s), was added to the samples, a sigmoid curve shape for formic acid concentration was no longer observed (solid blue and green lines,
Figure 3). Faster initial formaldehyde and formic acid formation was associated with the smaller compartment where PEG was held, resulting in better heat transfer and an earlier onset of melting. Slower formation in the second part of the stress test was associated with the consumption of reactive impurities for the
N-methylation reaction. The difference in formaldehyde concentration in samples with compared to those without paroxetine is in line with the concentration of formed impurities. However, it is also evident that not all formaldehyde reacts with paroxetine, and unexpectedly a high difference between
N-formyl amounts in samples with and without Fe
2O
3 were observed (
Figure 3, dotted blue versus green line). Based on the difference in levels of reactive impurities, this is ascribed to the critical concentration of formic acid needed for the reaction to proceed. Another reaction specific is plateau formation (
Figure 3, dotted blue and green line). We believe that this plateau is related to two low concentrations of formaldehyde and formic acid in the vial. Equivalently, in the early phase of the stress test, formaldehyde and formic acid are already available, but the reaction with paroxetine does not start until they reach critical concentrations. Alternative interpretations for the plateau are (1) a drop in pH that would disable formic acid to release a proton and, consequently, hydride, (2) an increase in the concentration of water molecules that destabilizes the imine intermediate, or (3) an excessively high gas pressure, disabling carbon dioxide release. The paroxetine used in this experiment was already in salt form (hydrochloride); however, amine functionality protonation due to acid formation also decrease compounds’ nucleophilic properties and affects the reaction rate.
To conclude this evaluation, we compared the molar ratios of formed
N-methyl impurities and the formaldehyde content in samples with paroxetine·HCl subjected to stress tests. A 100-fold higher molar ratio of
N-methyl paroxetine to formaldehyde was observed in the experiment where the combination of neat PEG 6000 and Fe
2O
3 is stressed, compared with the direct addition of reactive impurities (i.e., formalin, 37% formaldehyde solution,
Table 4), suggesting a more appropriate stressor for these types of reactions.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}