ABC Transporters at the Blood–Brain Interfaces, Their Study Models, and Drug Delivery Implications in Gliomas
 ,
,  and
and
Abstract
1. Introduction
2. Brain Barriers and Their Implication in Drug Delivery
2.1. The Blood–Brain Barrier and the Neurovascular Unit
2.2. The Blood–Cerebrospinal Fluid Barrier
2.3. The Blood–Arachnoid Barrier
2.4. Conclusion on the Blood–Brain Interfaces and Their Implication in Drug Delivery
3. Drug-Related ABC Transporters and Their Role at the Blood–Brain Interfaces
3.1. The ABC Superfamily
3.2. ABC Transporters Related to Drug Transport
3.3. ABC Transporters Expressed at the BBB and the NVU
3.4. ABC Transporters Expression at the BCSFB
3.5. ABC Transporters Expression at the BAB
3.6. ABC Transporters Expression Differences between Animals
3.7. Conclusion on the Multidrug Resistance Related to ABC Transporters at the Blood–Brain Interfaces
4. Methods to Study the Blood–Brain Interfaces
4.1. In Vitro Models and Assays for Drug Evaluation
4.2. In Vitro Models of the BBB
4.3. Dynamic In Vitro Model, Toward the BBB-on-Chip
4.4. In Vitro Models of the BCSFB and BAB
4.5. Ex Vivo Models
4.6. In Vivo Models and Assays
4.7. Imaging Methods
4.8. In Silico Models
4.9. Recent Molecular Characterization Techniques
4.10. Conclusion on Methods to Study the Blood–Brain Interfaces
5. Implication of ABC Transporters in the Multidrug Resistance of Glioma
5.1. Glioma Classification
5.2. Epidemiology and Prognosis
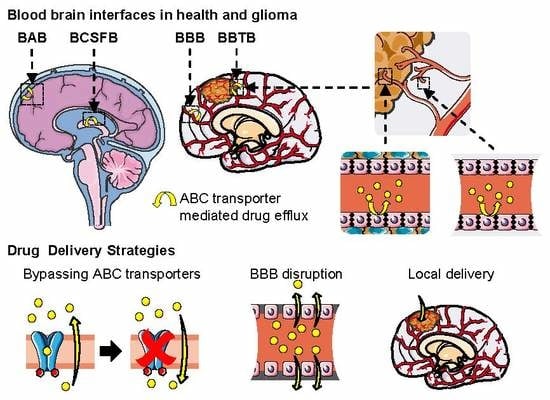
5.3. Multidrug Resistance in Glioma and the Blood–Brain Tumor Barrier (BBTB)
5.4. ABC Transporters Role in Glioma Drug Resistance
5.5. Glioma Models to Study Drug Transport and Delivery
5.6. Conclusion on the Implication of ABC Transporters in the Multidrug Resistance of Glioma
6. Strategies to Improve CNS Drug Delivery in Brain Cancer
6.1. Inhibition of ABC Transporters
6.2. Other Modulators of ABC Transporter-Dependent Multidrug Resistance
6.3. Rational Drug Design
6.4. Local Delivery: Polymeric Drug Delivery Systems and Convection-Enhanced Delivery
6.5. BBB Disruption: Osmotic Disruption and Ultrasound-Enhanced Delivery
6.6. Nanoparticles and Targeting Nanocarriers
6.7. Conclusion on Strategies to Improve CNS Drug Delivery in Brain Cancer
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease. Epilepsia 2012, 53, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Scherrmann, J.-M. Drug delivery to brain via the blood–brain barrier. Vascul. Pharmacol. 2002, 38, 349–354. [Google Scholar] [CrossRef]
- Chaves, C.; Shawahna, R.; Jacob, A.; Scherrmann, J.-M.; Declèves, X. Human ABC Transporters at blood-CNS Interfaces as Determinants of CNS Drug Penetration. Curr. Pharm. Des. 2014, 20, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood-brain barrier: Molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef]
- Palmer, A.M.; Alavijeh, M.S. Overview of experimental models of the blood-brain barrier in CNS drug discovery. Curr. Protoc. Pharmacol. 2013, 1, 1–30. [Google Scholar] [CrossRef]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood–brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Amore, B.; Gibbs, J.; Emery, M. Application of In Vivo Animal Models to Characterize the Pharmacokinetic and Pharmacodynamic Properties of Drug Candidates in Discovery Settings. Comb. Chem. High Throughput Screen. 2010, 13, 207–218. [Google Scholar] [CrossRef]
- Patel, A.P.; Fisher, J.L.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Abraha, H.N.; Agius, D.; Alahdab, F.; Alam, T.; et al. Global, regional, and national burden of brain and other CNS cancer, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 376–393. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 1–18. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Prim. 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, G.; Wirsching, H.G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas-implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef] [PubMed]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Calatozzolo, C.; Gelati, M.; Ciusani, E.; Sciacca, F.L.; Pollo, B.; Cajola, L.; Marras, C.; Silvani, A.; Vitellaro-Zuccarello, L.; Croci, D.; et al. Expression of Drug Resistance Proteins Pgp, MRP1, MRP3, MRP5 AND GST-π in Human Glioma. J. Neurooncol. 2005, 74, 113–121. [Google Scholar] [CrossRef]
- Declèves, X.; Amiel, A.; Delattre, J.-Y.; Scherrmann, J.-M.; Decleves, X.; Amiel, A.; Delattre, J.-Y.; Scherrmann, J.-M. Role of ABC Transporters in the Chemoresistance of Human Gliomas. Curr. Cancer Drug Targets 2006, 6, 433–445. [Google Scholar] [CrossRef]
- Ghersi-Egea, J.F.; Strazielle, N.; Catala, M.; Silva-Vargas, V.; Doetsch, F.; Engelhardt, B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 2018, 135, 337–361. [Google Scholar] [CrossRef]
- Saunders, N.R.; Habgood, M.D.; Møllgård, K.; Dziegielewska, K.M. The biological significance of brain barrier mechanisms: Help or hindrance in drug delivery to the central nervous system? F1000Research 2016, 5, 313. [Google Scholar] [CrossRef]
- Ueno, M.; Chiba, Y.; Murakami, R.; Matsumoto, K.; Kawauchi, M.; Fujihara, R. Blood–brain barrier and blood–cerebrospinal fluid barrier in normal and pathological conditions. Brain Tumor Pathol. 2016, 33, 89–96. [Google Scholar] [CrossRef]
- Morris, M.E.; Rodriguez-Cruz, V.; Felmlee, M.A. SLC and ABC Transporters: Expression, Localization, and Species Differences at the Blood-Brain and the Blood-Cerebrospinal Fluid Barriers. AAPS J. 2017, 19, 1317–1331. [Google Scholar] [CrossRef]
- Dauchy, S.; Dutheil, F.; Weaver, R.J.; Chassoux, F.; Daumas-Duport, C.; Couraud, P.-O.; Scherrmann, J.-M.; De Waziers, I.; Declèves, X. ABC transporters, cytochromes P450 and their main transcription factors: Expression at the human blood-brain barrier. J. Neurochem. 2008, 107, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Shawahna, R.; Uchida, Y.; Declèves, X.; Ohtsuki, S.; Yousif, S.; Dauchy, S.; Jacob, A.; Chassoux, F.; Daumas-Duport, C.; Couraud, P.-O.; et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol. Pharm. 2011, 8, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, F.; Jacob, A.; Dauchy, S.; Beaune, P.; Scherrmann, J.-M.; Declèves, X.; Loriot, M.-A. ABC transporters and cytochromes P450 in the human central nervous system: Influence on brain pharmacokinetics and contribution to neurodegenerative disorders. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Kutuzov, N.; Flyvbjerg, H.; Lauritzen, M. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood–brain barrier. Proc. Natl. Acad. Sci. USA 2018, 115, E9429–E9438. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, S.; Blaney, S. New Agents for Intrathecal Administration. Cancer Invest. 2006, 24, 528–534. [Google Scholar] [CrossRef]
- Liu, Y.X.; Liu, W.J.; Zhang, H.R.; Zhang, Z.W. Delivery of bevacizumab by intracranial injection: Assessment in glioma model. Onco. Targets. Ther. 2018, 11, 2673–2683. [Google Scholar] [CrossRef]
- Nag, S.; Begley, D.J. Blood–brain barrier, exchange of metabolites and gases. In Pathology and Genetics. Cerebrovascular Diseases; Kalimo, H., Ed.; ISN Neuropath Press: Basel, Switzerland, 2005; pp. 22–29. [Google Scholar]
- Schulze, C.; Firth, J.A. Immunohistochemical localization of adherens junction components in blood-brain barrier microvessels of the rat. J. Cell Sci. 1993, 104, 773–782. [Google Scholar]
- Wang, Q.; Zuo, Z. Impact of transporters and enzymes from blood–cerebrospinal fluid barrier and brain parenchyma on CNS drug uptake. Expert Opin. Drug Metab. Toxicol. 2018, 14, 961–972. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.-F. Demonstration of a Coupled Metabolism–Efflux Process at the Choroid Plexus as a Mechanism of Brain Protection Toward Xenobiotics. J. Neurosci. 1999, 19, 6275–6289. [Google Scholar] [CrossRef]
- Keep, R.F.; Jones, H.C. A morphometric study on the development of the lateral ventricle choroid plexus, choroid plexus capillaries and ventricular ependyma in the rat. Dev. Brain Res. 1990, 56, 47–53. [Google Scholar] [CrossRef]
- Weller, R.O.; Sharp, M.M.; Christodoulides, M.; Carare, R.O.; Møllgård, K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol. 2018, 135, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Cline, C.; Vogel, P.; Onciu, M.; Fatima, S.; Sorrentino, B.P.; Thirumaran, R.K.; Ekins, S.; Urade, Y.; Fujimori, K.; et al. Drug transporters on arachnoid barrier cells contribute to the blood-cerebrospinal fluid barrier. Drug Metab. Dispos. 2013, 41, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tachikawa, M.; Uchida, Y.; Terasaki, T. Drug Clearance from Cerebrospinal Fluid Mediated by Organic Anion Transporters 1 (Slc22a6) and 3 (Slc22a8) at Arachnoid Membrane of Rats. Mol. Pharm. 2018, 15, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, Y.; Tachikawa, M.; Zhang, Z.; Terasaki, T. Organic Anion-Transporting Polypeptide 1a4 (Oatp1a4/Slco1a4) at the Blood-Arachnoid Barrier is the Major Pathway of Sulforhodamine-101 Clearance from Cerebrospinal Fluid of Rats. Mol. Pharm. 2019, 16, 2021–2027. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Kodaira, H.; Kusuhara, H.; Fujita, T.; Ushiki, J.; Fuse, E.; Sugiyama, Y. Quantitative Evaluation of the Impact of Active Efflux by P-Glycoprotein and Breast Cancer Resistance Protein at the Blood-Brain Barrier on the Predictability of the Unbound Concentrations of Drugs in the Brain Using Cerebrospinal Fluid Concentration as a. J. Pharmacol. Exp. Ther. 2011, 339, 935–944. [Google Scholar] [CrossRef]
- Pui, C.-H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef]
- Mahringer, A.; Fricker, G. ABC transporters at the blood–brain barrier. Expert Opin. Drug Metab. Toxicol. 2016, 5255, 1–10. [Google Scholar] [CrossRef]
- Mohammad, I.S.; He, W.; Yin, L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed. Pharmacother. 2018, 100, 335–348. [Google Scholar] [CrossRef]
- Wijaya, J.; Fukuda, Y.; Schuetz, J.D. Obstacles to brain tumor therapy: Key ABC transporters. Int. J. Mol. Sci. 2017, 18, 2544. [Google Scholar] [CrossRef] [PubMed]
- HUGO Gene Nomenclature Committee: ABC Family. Available online: http://www.genenames.org/cgi-bin/genefamilies/set/417 (accessed on 5 August 2016).
- DeGorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug Transporters in Drug Efficacy and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Ambudkar, S.V. Overview: ABC Transporters and Human Disease. J. Bioenerg. Biomembr. 2001, 33, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Jedlitschky, G.; König, J.; Herold-Mende, C.; Steiner, H.H.; Schmitt, H.P.; Keppler, D. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience 2004, 129, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.; Miller, D.S.; Bendayan, R. Multidrug Resistance-Associated Proteins: Expression and Function in the Central Nervous System. Pharmacol. Rev. 2006, 58, 140–161. [Google Scholar] [CrossRef]
- Begley, D. ABC Transporters and the Blood-Brain Barrier. CPD 2004, 10, 1295–1312. [Google Scholar] [CrossRef]
- Scherrmann, J.-M. ABC Superfamily Transporters at the Human Blood–Brain Barrier. In ABC Transporters and Multidrug Resistance; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005; pp. 363–384. ISBN 9780470495131. [Google Scholar]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Sharom, F.J. The P-glycoprotein multidrug transporter. Essays Biochem. 2011, 50, 161–178. [Google Scholar] [CrossRef]
- Stacy, A.E.; Jansson, P.J.; Richardson, D.R. Molecular Pharmacology of ABCG2 and Its Role in Chemoresistance. Mol. Pharmacol. 2013, 84, 655–669. [Google Scholar] [CrossRef]
- Agarwal, S.; Hartz, A.M.S.; Elmquist, W.F.; Bauer, B. Breast cancer resistance protein and P-glycoprotein in brain cancer: Two gatekeepers team up. Curr. Pharm. Des. 2011, 17, 2793–2802. [Google Scholar] [CrossRef]
- Robey, W.R.; Ierano, C.; Zhan, Z.; Bates, E.S. The Challenge of Exploiting ABCG2 in the Clinic. Curr. Pharm. Biotechnol. 2011, 12, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Hagiya, Y.; Adachi, T.; Hoshijima, K.; Kuo, M.T.; Ishikawa, T. MRP class of human ATP binding cassette (ABC) transporters: Historical background and new research directions. Xenobiotica 2008, 38, 833–862. [Google Scholar] [CrossRef] [PubMed]
- Slot, A.J.; Molinski, S.V.; Cole, S.P.C. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011, 50, 179–207. [Google Scholar] [PubMed]
- Zhou, S.-F.; Wang, L.-L.; Di, Y.; Xue, C.; Duan, W.; Li, C.; Li, Y. Substrates and Inhibitors of Human Multidrug Resistance Associated Proteins and the Implications in Drug Development. Curr. Med. Chem. 2008, 15, 1981–2039. [Google Scholar] [CrossRef]
- Beaulieu, E.; Demeule, M.; Ghitescu, L.; Béliveau, R. P-glycoprotein is strongly expressed in the luminal membranes of the endothelium of blood vessels in the brain. Biochem. J. 1997, 326 Pt 2, 539–544. [Google Scholar] [CrossRef]
- Virgintino, D.; Robertson, D.; Errede, M.; Benagiano, V.; Girolamo, F.; Maiorano, E.; Roncali, L.; Bertossi, M. Expression of P-Glycoprotein in Human Cerebral Cortex Microvessels. J. Histochem. Cytochem. 2002, 50, 1671–1676. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Golden, P.L.; Kang, Y.-S.; Bickel, U. Brain Microvascular and Astrocyte Localization of P-Glycoprotein. J. Neurochem. 2002, 68, 1278–1285. [Google Scholar] [CrossRef]
- Soontornmalai, A.; Vlaming, M.L.H.; Fritschy, J.-M. Differential, strain-specific cellular and subcellular distribution of multidrug transporters in murine choroid plexus and blood-brain barrier. Neuroscience 2006, 138, 159–169. [Google Scholar] [CrossRef]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In Situ Localization of P-glycoprotein (ABCB1) in Human and Rat Brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef]
- Kubo, Y.; Ohtsuki, S.; Uchida, Y.; Terasaki, T. Quantitative Determination of Luminal and Abluminal Membrane Distributions of Transporters in Porcine Brain Capillaries by Plasma Membrane Fractionation and Quantitative Targeted Proteomics. J. Pharm. Sci. 2015, 104, 3060–3068. [Google Scholar] [CrossRef]
- Boulay, A.C.; Saubameá, B.; Adam, N.; Chasseigneaux, S.; Mazaré, N.; Gilbert, A.; Bahin, M.; Bastianelli, L.; Blugeon, C.; Perrin, S.; et al. Translation in astrocyte distal processes sets molecular heterogeneity at the gliovascular interface. Cell Discov. 2017, 3, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Spiegl-Kreinecker, S.; Buchroithner, J.; Elbling, L.; Steiner, E.; Wurm, G.; Bodenteich, A.; Fischer, J.; Micksche, M.; Berger, W. Expression and functional activity of the ABC-transporter proteins P-glycoprotein and multidrug-resistance protein 1 in human brain tumor cells and astrocytes. J. Neurooncol. 2002, 57, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Ramkema, M.; Redeker, S.; Ozbas-Gerçeker, F.; van Vliet, E.A.; Scheffer, G.L.; Scheper, R.J.; van der Valk, P.; Baayen, J.C.; et al. Expression and cellular distribution of multidrug resistance-related proteins in the hippocampus of patients with mesial temporal lobe epilepsy. Epilepsia 2004, 45, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Marroni, M.; Agrawal, M.; Kight, K.; Hallene, K.; Hossain, M.; Cucullo, L.; Signorelli, K.; Namura, S.; Bingaman, W.; Janigro, D. Relationship between expression of multiple drug resistance proteins and p53 tumor suppressor gene proteins in human brain astrocytes. Neuroscience 2003, 121, 605–617. [Google Scholar] [CrossRef]
- Uchida, Y.; Zhang, Z.; Tachikawa, M.; Terasaki, T. Quantitative targeted absolute proteomics of rat blood-cerebrospinal fluid barrier transporters: Comparison with a human specimen. J. Neurochem. 2015, 134, 1104–1115. [Google Scholar] [CrossRef]
- Rao, V.V.; Dahlheimer, J.L.; Bardgett, M.E.; Snyder, A.Z.; Finch, R.A.; Sartorelli, A.C.; Piwnica-Worms, D. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 3900–3905. [Google Scholar] [CrossRef]
- Matsumoto, K.; Chiba, Y.; Fujihara, R.; Kubo, H.; Sakamoto, H.; Ueno, M. Immunohistochemical analysis of transporters related to clearance of amyloid-β peptides through blood–cerebrospinal fluid barrier in human brain. Histochem. Cell Biol. 2015, 144, 597–611. [Google Scholar] [CrossRef]
- Mercier, C.; Masseguin, C.; Roux, F.; Gabrion, J.; Scherrmann, J.-M. Expression of P-glycoprotein (ABCB1) and Mrp1 (ABCC1) in adult rat brain: Focus on astrocytes. Brain Res. 2004, 1021, 32–40. [Google Scholar] [CrossRef]
- GENSAT. Available online: http://www.gensat.org/index.html (accessed on 16 October 2019).
- Allen Brain Atlas. Available online: http://mouse.brain-map.org/ (accessed on 16 October 2019).
- Gong, S.; Zheng, C.; Doughty, M.L.; Losos, K.; Didkovsky, N.; Schambra, U.B.; Nowak, N.J.; Joyner, A.; Leblanc, G.; Hatten, M.E.; et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 2003, 425, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.; Ng, L.; Thompson, C.; Pathak, S.; Kuan, L.; Jones, A.; Hawrylycz, M. Exploration and visualization of gene expression with neuroanatomy in the adult mouse brain. BMC Bioinformatics 2008, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Eisenblätter, T.; Hüwel, S.; Galla, H.-J. Characterisation of the brain multidrug resistance protein (BMDP/ABCG2/BCRP) expressed at the blood–brain barrier. Brain Res. 2003, 971, 221–231. [Google Scholar] [CrossRef]
- Cooray, H.C.; Blackmore, C.G.; Maskell, L.; Barrand, M.A. Localisation of breast cancer resistance protein in microvessel endothelium of human brain. Neuroreport 2002, 13, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Redeker, S.; van Vliet, E.A.; Ramkema, M.; Scheffer, G.L.; Scheper, R.J.; van der Valk, P.; Leenstra, S.; Baayen, J.C.; et al. Localization of Breast Cancer Resistance Protein (BCRP) in Microvessel Endothelium of Human Control and Epileptic Brain. Epilepsia 2005, 46, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Watanabe, M.; Hori, S.; Fukaya, M.; Ohtsuki, S.; Asashima, T.; Terasaki, T. Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J. Neurochem. 2005, 95, 294–304. [Google Scholar] [CrossRef]
- Hori, S.; Ohtsuki, S.; Tachikawa, M.; Kimura, N.; Kondo, T.; Watanabe, M.; Nakashima, E.; Terasaki, T. Functional expression of rat ABCG2 on the luminal side of brain capillaries and its enhancement by astrocyte-derived soluble factor(s). J. Neurochem. 2004, 90, 526–536. [Google Scholar] [CrossRef]
- Roberts, L.M.M.; Black, D.S.S.; Raman, C.; Woodford, K.; Zhou, M.; Haggerty, J.E.E.; Yan, A.T.T.; Cwirla, S.E.E.; Grindstaff, K.K.K. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience 2008, 155, 423–438. [Google Scholar] [CrossRef]
- Lee, G.; Babakhanian, K.; Ramaswamy, M.; Prat, A.; Wosik, K.; Bendayan, R. Expression of the ATP-binding Cassette Membrane Transporter, ABCG2, in Human and Rodent Brain Microvessel Endothelial and Glial Cell Culture Systems. Pharm. Res. 2007, 24, 1262–1274. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Warren, M.S.; Zerangue, N.; Woodford, K.; Roberts, L.M.; Tate, E.H.; Feng, B.; Li, C.; Feuerstein, T.J.; Gibbs, J.; Smith, B. Comparative gene expression profiles of ABC transporters in brain microvessel endothelial cells and brain in five species including human. Pharmacol. Res. 2009, 59, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Niehof, M.; Borlak, J. Expression of HNF4alpha in the human and rat choroid plexus—Implications for drug transport across the blood-cerebrospinal-fluid (CSF) barrier. BMC Mol. Biol. 2009, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, S.; Cherrington, N.J.; Li, N.; Klaassen, C.D. Constitutive expression of various xenobiotic and endobiotic transporter mRNAs in the choroid plexus of rats. Drug Metab. Dispos. 2003, 31, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Wijnholds, J.; de Lange, E.C.M.; Scheffer, G.L.; van den Berg, D.-J.; Mol, C.A.A.M.; van der Valk, M.; Schinkel, A.H.; Scheper, R.J.; Breimer, D.D.; Borst, P. Multidrug resistance protein 1 protects the choroid plexus epithelium and contributes to the blood-cerebrospinal fluid barrier. J. Clin. Invest. 2000, 105, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.S.; Nobmann, S.N.; Gutmann, H.; Toeroek, M.; Drewe, J.; Fricker, G. Xenobiotic Transport across Isolated Brain Microvessels Studied by Confocal Microscopy. Mol. Pharmacol. 2000, 58, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Bauer, B.; Hartz, A.M.S.; Lucking, J.R.; Yang, X.; Pollack, G.M.; Miller, D.S. Coordinated nuclear receptor regulation of the efflux transporter, Mrp2, and the phase-II metabolizing enzyme, GSTπ, at the blood-brain barrier. J. Cereb. Blood Flow Metab. 2008, 28, 1222–1234. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, Y.; Uchida, Y.; Tachikawa, M.; Inoue, T.; Ohtsuki, S.; Terasaki, T. Quantitative atlas of blood-brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J. Pharm. Sci. 2013, 102, 3343–3355. [Google Scholar] [CrossRef]
- Kamiie, J.; Ohtsuki, S.; Iwase, R.; Ohmine, K.; Katsukura, Y.; Yanai, K.; Sekine, Y.; Uchida, Y.; Ito, S.; Terasaki, T. Quantitative Atlas of Membrane Transporter Proteins: Development and Application of a Highly Sensitive Simultaneous LC/MS/MS Method Combined with Novel In-silico Peptide Selection Criteria. Pharm. Res. 2008, 25, 1469–1483. [Google Scholar] [CrossRef]
- Ito, K.; Uchida, Y.; Ohtsuki, S.; Aizawa, S.; Kawakami, H.; Katsukura, Y.; Kamiie, J.; Terasaki, T. Quantitative membrane protein expression at the blood–brain barrier of adult and younger cynomolgus monkeys. J. Pharm. Sci. 2011, 100, 3939–3950. [Google Scholar] [CrossRef]
- Leggas, M.; Adachi, M.; Scheffer, G.; Sun, D.; Wielinga, P.; Du, G.; Mercer, K.; Zhuang, Y.; Panetta, J.; Johnston, B.; et al. MRP4 confers resistance to topotecan and protects the brain from chemotherapy. Mol. Cell. Biol. 2004, 24, 7612–7621. [Google Scholar] [CrossRef] [PubMed]
- Potschka, H. Transporter hypothesis of drug-resistant epilepsy: Challenges for pharmacogenetic approaches. Pharmacogenomics 2010, 11, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- de Vries, N.A.; Zhao, J.; Kroon, E.; Buckle, T.; Beijnen, J.H.; van Tellingen, O. P-Glycoprotein and Breast Cancer Resistance Protein: Two Dominant Transporters Working Together in Limiting the Brain Penetration of Topotecan. Clin. Cancer Res. 2007, 13, 6440–6449. [Google Scholar] [CrossRef] [PubMed]
- Dufour, R.; Daumar, P.; Mounetou, E.; Aubel, C.; Kwiatkowski, F.; Abrial, C.; Vatoux, C.; Penault-Llorca, F.; Bamdad, M. BCRP and P-gp relay overexpression in triple negative basal-like breast cancer cell line: A prospective role in resistance to Olaparib. Sci. Rep. 2015, 5, 12670. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. BBA - Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Regina, A.; Koman, A.; Piciotti, M.; El Hafny, B.; Center, M.S.; Bergmann, R.; Couraud, P.O.; Roux, F. Mrp1 multidrug resistance-associated protein and P-glycoprotein expression in rat brain microvessel endothelial cells. J. Neurochem. 1998, 71, 705–715. [Google Scholar] [CrossRef]
- Demeule, M.; Régina, A.; Jodoin, J.; Laplante, A.; Dagenais, C.; Berthelet, F.; Moghrabi, A.; Béliveau, R. Drug transport to the brain: Key roles for the efflux pump P-glycoprotein in the blood–brain barrier. Vascul. Pharmacol. 2002, 38, 339–348. [Google Scholar] [CrossRef]
- Saraswathy, M.; Gong, S. Different strategies to overcome multidrug resistance in cancer. Biotechnol. Adv. 2013, 31, 1397–1407. [Google Scholar] [CrossRef]
- Pavan, B.; Paganetto, G.; Rossi, D.; Dalpiaz, A. Multidrug resistance in cancer or inefficacy of neuroactive agents: Innovative strategies to inhibit or circumvent the active efflux transporters selectively. Drug Discov. Today 2014, 19, 1563–1571. [Google Scholar] [CrossRef]
- Mo, W.; Zhang, J.T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 2012, 3, 1–27. [Google Scholar]
- Tivnan, A. Targeting Chemotherapy Resistance in Glioblastoma Through Modulation of ABC Transporters. In Resistance to Targeted Anti-Cancer Therapeutics; Tivnan, A., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 25–54. ISBN 978-3-319-46504-3. [Google Scholar]
- Mercier, C.; Declèves, X.; Masseguin, C.; Fragner, P.; Tardy, M.; Roux, F.; Gabrion, J.; Scherrmann, J.-M. P-glycoprotein (ABCB1) but not multidrug resistance-associated protein 1 (ABCC1) is induced by doxorubicin in primary cultures of rat astrocytes. J. Neurochem. 2003, 87, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T. The role of membrane transporters in drug delivery to brain tumors. Cancer Lett. 2007, 254, 11–29. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P.C.C. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Wielinga, P.R.; van der Heijden, I.; Reid, G.; Beijnen, J.H.; Wijnholds, J.; Borst, P. Characterization of the MRP4- and MRP5-mediated Transport of Cyclic Nucleotides from Intact Cells. J. Biol. Chem. 2003, 278, 17664–17671. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, J.D.; Connelly, M.C.; Sun, D.; Paibir, S.G.; Flynn, P.M.; Srinivas, R.V.; Kumar, A.; Fridland, A. MRP4: A previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nat. Med. 1999, 5, 1048–1051. [Google Scholar] [CrossRef]
- Jedlitschky, G.; Burchell, B.; Keppler, D. The Multidrug Resistance Protein 5 Functions as an ATP-dependent Export Pump for Cyclic Nucleotides. J. Biol. Chem. 2000, 275, 30069–30074. [Google Scholar] [CrossRef]
- Lai, L.; Tan, T.M.C. Role of glutathione in the multidrug resistance protein 4 (MRP4/ABCC4)-mediated efflux of cAMP and resistance to purine analogues. Biochem. J. 2002, 361, 497–503. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735–7738. [Google Scholar] [CrossRef]
- Yousif, S.; Marie-Claire, C.; Roux, F.; Scherrmann, J.-M.; Declèves, X. Expression of drug transporters at the blood–brain barrier using an optimized isolated rat brain microvessel strategy. Brain Res. 2007, 1134, 1–11. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; O’Brien, J.P.; Casals, D.; Rittman-Grauer, L.; Biedler, J.L.J.L.; Melamed, M.R.; Bertino, J.R.; Peschanski, M.; Cordon-Cardo, C.; O’Brien, J.P.; et al. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. USA 1989, 86, 695–698. [Google Scholar] [CrossRef]
- Decleves, X.; Regina, A.; Laplanche, J.-L.; Roux, F.; Boval, B.; Launay, J.-M.; Scherrmann, J.-M. Functional expression of P-glycoprotein and multidrug resistance-associated protein (Mrp1) in primary cultures of rat astrocytes. J. Neurosci. Res. 2000, 60, 594–601. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Bendayan, M.; Gingras, D.; Piquette-Miller, M.; Bendayan, R. Cellular localization and functional expression of P-glycoprotein in rat astrocyte cultures. J. Neurochem. 2004, 89, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Schlichter, L.; Bendayan, M.; Bendayan, R. Functional expression of P-glycoprotein in rat brain microglia. J. Pharmacol. Exp. Ther. 2001, 299, 204–212. [Google Scholar] [PubMed]
- Shimizu, F.; Sano, Y.; Maeda, T.; Abe, M.; Nakayama, H.; Takahashi, R.; Ueda, M.; Ohtsuki, S.; Terasaki, T.; Obinata, M.; et al. Peripheral Nerve pericytes originating from the blood-nerve barrier expresses tight junctional molecules and transporters as barrier-forming cells. J. Cell. Physiol. 2008, 217, 388–399. [Google Scholar] [CrossRef]
- Bronger, H.; König, J.; Kopplow, K.; Steiner, H.-H.; Ahmadi, R.; Herold-Mende, C.; Keppler, D.; Nies, A.T. ABCC Drug Efflux Pumps and Organic Anion Uptake Transporters in Human Gliomas and the Blood-Tumor Barrier. Cancer Res. 2005, 65, 11419–11428. [Google Scholar] [CrossRef]
- Cisternino, S.; Rousselle, C.; Lorico, A.; Rappa, G.; Scherrmann, J.M. Apparent lack of mrp1-mediated efflux at the luminal side of mouse blood-brain barrier endothelial cells. Pharm. Res. 2003, 20, 904–909. [Google Scholar] [CrossRef]
- Hirrlinger, J.; König, J.; Dringen, R. Expression of mRNAs of multidrug resistance proteins (Mrps) in cultured rat astrocytes, oligodendrocytes, microglial cells and neurones. J. Neurochem. 2002, 82, 716–719. [Google Scholar] [CrossRef]
- Dallas, S.; Schlichter, L.; Bendayan, R. Multidrug resistance protein (MRP) 4-and MRP 5-mediated efflux of 9-(2-phosphonylmethoxyethyl) adenine by microglia. J. Pharmacol. Exp. Ther. 2004, 309, 1221. [Google Scholar] [CrossRef]
- Uchida, Y.; Goto, R.; Takeuchi, H.; Łuczak, M.; Usui, T.; Tachikawa, M.; Terasaki, T. Abundant expression of OCT2, MATE1, OAT1, OAT3, PEPT2, BCRP, MDR1 and xCT transporters in blood-arachnoid barrier of pig, and polarized localizations at CSF- and blood-facing plasma membranes. Drug Metab. Dispos. 2019, 53, dmd.119.089516. [Google Scholar] [CrossRef]
- Ball, K.; Bouzom, F.; Scherrmann, J.-M.; Walther, B.; Declèves, X. Physiologically based pharmacokinetic modelling of drug penetration across the blood-brain barrier--towards a mechanistic IVIVE-based approach. AAPS J. 2013, 15, 913–932. [Google Scholar] [CrossRef]
- Syvänen, S.; Lindhe, O.; Palner, M.; Kornum, B.R.; Rahman, O.; Långström, B.; Knudsen, G.M.; Hammarlund-Udenaes, M.; Lindhe, Ö.; Palner, M. Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab. 2009, 37, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.; Bauer, B. ABC transporters in the CNS—an inventory. Curr. Pharm. Biotechnol. 2011, 12, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Tamiya, T.; Nagao, S. Resistance to topoisomerase II inhibitors in human glioma cell lines overexpressing multidrug resistant associated protein (MRP) 2. J. Med. Invest. 2005, 52, 41–48. [Google Scholar] [CrossRef] [PubMed][Green Version]
- U.S. Department of Health and Human Services Administration, Food and Drug (CDER), Center for Drug Evaluation and Research. In vitro metabolism- and transporter-mediated drug-drug interaction studies guidance for industry. FDA Guidel. 2017, 1–45. [Google Scholar]
- European Medicines Agency ICH topic S 7 A Safety Pharmacology Studies for Human Pharmaceuticals. Eur. Med. Agency 2002, 16, 79–81.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Abbott, N.J. In Vitro Models for Examining and Predicting Brain Uptake of Drugs. In Comprehensive Medicinal Chemistry II; Elsevier: Amsterdam, The Netherlands, 2007; Volume 5, pp. 301–320. ISBN 9780080450445. [Google Scholar]
- Bickel, U. How to measure drug transport across the blood-brain barrier. NeuroRX 2005, 2, 15–26. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER Measurement Techniques for In Vitro Barrier Model Systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Wilhelm, I.; Krizbai, I.A. In Vitro Models of the Blood–Brain Barrier for the Study of Drug Delivery to the Brain. Mol. Pharm. 2014, 11, 1949–1963. [Google Scholar] [CrossRef]
- Puech, C.; Delavenne, X.; Perek, N. The expected characteristics of an in vitro human Blood Brain Barrier model derived from cell lines, for studying how ABC transporters influence drug permeability. J. Drug Deliv. Sci. Technol. 2018, 45, 159–167. [Google Scholar] [CrossRef]
- Kuteykin-Teplyakov, K.; Luna-Tortós, C.; Ambroziak, K.; Löscher, W. Differences in the expression of endogenous efflux transporters in MDR1-transfected versus wildtype cell lines affect P-glycoprotein mediated drug transport. Br. J. Pharmacol. 2010, 160, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Phan, D.T.T.; Bender, R.H.F.; Andrejecsk, J.W.; Sobrino, A.; Hachey, S.J.; George, S.C.; Hughes, C.C.W. Blood–brain barrier-on-a-chip: Microphysiological systems that capture the complexity of the blood–central nervous system interface. Exp. Biol. Med. 2017, 242, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.K.; Sharma, A.K.; Gupta, U. Blood brain barrier: An overview on strategies in drug delivery, realistic in vitro modeling and in vivo live tracking. Tissue Barriers 2016, 4, e1129476. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.; Antfolk, M.; Brodin, B.; Tenje, M. In Vitro Blood–Brain Barrier Models—An Overview of Established Models and New Microfluidic Approaches. J. Pharm. Sci. 2015, 104, 2727–2746. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Cucullo, L. In Vitro Blood–Brain Barrier Models: Current and Perspective Technologies. J. Pharm. Sci. 2012, 101, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Kaisar, M.A.; Sajja, R.K.; Prasad, S.; Abhyankar, V.V.; Liles, T.; Cucullo, L. New experimental models of the blood-brain barrier for CNS drug discovery. Expert Opin. Drug Discov. 2017, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Coisne, C.; Dehouck, L.; Faveeuw, C.; Delplace, Y.; Miller, F.; Landry, C.; Morissette, C.; Fenart, L.; Cecchelli, R.; Tremblay, P.; et al. Mouse syngenic in vitro blood–brain barrier model: A new tool to examine inflammatory events in cerebral endothelium. Lab. Investig. 2005, 85, 734–746. [Google Scholar] [CrossRef]
- Roux, F.; Couraud, P.-O. Rat Brain Endothelial Cell Lines for the Study of Blood–Brain Barrier Permeability and Transport Functions. Cell. Mol. Neurobiol. 2005, 25, 41–57. [Google Scholar] [CrossRef]
- Bowman, P.D.; Betz, A.L.; AR, D.; Wolinsky, J.S.; Penney, J.B.; Shivers, R.R.; Goldstein, G.W. Primary culture of capillary endothelium from rat brain. In Vitro 1981, 17, 353–362. [Google Scholar] [CrossRef]
- Perrière, N.; Demeuse, P.; Garcia, E.; Regina, A.; Debray, M.; Andreux, J.-P.; Couvreur, P.; Scherrmann, J.-M.; Temsamani, J.; Couraud, P.-O.; et al. Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J. Neurochem. 2005, 93, 279–289. [Google Scholar] [CrossRef]
- Calabria, A.R.; Weidenfeller, C.; Jones, A.R.; de Vries, H.E.; Shusta, E.V. Puromycin-purified rat brain microvascular endothelial cell cultures exhibit improved barrier properties in response to glucocorticoid induction. J. Neurochem. 2006, 97, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Perrière, N.; Yousif, S.; Cazaubon, S.; Chaverot, N.; Bourasset, F.; Cisternino, S.; Declèves, X.; Hori, S.; Terasaki, T.; Deli, M.; et al. A functional in vitro model of rat blood–brain barrier for molecular analysis of efflux transporters. Brain Res. 2007, 1150, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Culot, M.; Lundquist, S.; Vanuxeem, D.; Nion, S.; Landry, C.; Delplace, Y.; Dehouck, M.-P.; Berezowski, V.; Fenart, L.; Cecchelli, R. An in vitro blood-brain barrier model for high throughput (HTS) toxicological screening. Toxicol. Vitr. 2008, 22, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A.; Skinner, R.A.; Abbott, N.J. Establishment of a simplified in vitro porcine blood–brain barrier model with high transendothelial electrical resistance. Brain Res. 2013, 1521, 1–15. [Google Scholar] [CrossRef]
- MacLean, A.G.; Orandle, M.S.; MacKey, J.; Williams, K.C.; Alvarez, X.; Lackner, A.A. Characterization of an in vitro rhesus macaque blood–brain barrier. J. Neuroimmunol. 2002, 131, 98–103. [Google Scholar] [CrossRef]
- Weksler, B.; Romero, I.A.; Couraud, P.-O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef]
- Weksler, B.B.; Subileau, E.A.; Perrière, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef]
- Bernas, M.; Cardoso, F.; Daley, S. Establishment of primary cultures of human brain microvascular endothelial cells: A new and simplified method to obtain cells for an in vitro model of the blood. Nat. Protoc. 2010, 5, 1265–1272. [Google Scholar]
- Cecchelli, R.; Aday, S.; Sevin, E.; Almeida, C.; Culot, M.; Dehouck, L.; Coisne, C.; Engelhardt, B.; Dehouck, M.-P.; Ferreira, L. A Stable and Reproducible Human Blood-Brain Barrier Model Derived from Hematopoietic Stem Cells. PLoS ONE 2014, 9, e99733. [Google Scholar] [CrossRef]
- Ponio, J.B.-D.; El-Ayoubi, F.; Glacial, F.; Ganeshamoorthy, K.; Driancourt, C.; Godet, M.; Perrière, N.; Guillevic, O.; Couraud, P.O.; Uzan, G. Instruction of Circulating Endothelial Progenitors In Vitro towards Specialized Blood-Brain Barrier and Arterial Phenotypes. PLoS ONE 2014, 9, e84179. [Google Scholar] [CrossRef]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Human Blood-Brain Barrier Endothelial Cells Derived from Pluripotent Stem Cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Lippmann, E.S.; Al-Ahmad, A.; Azarin, S.M.; Palecek, S.P.; Shusta, E.V. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep. 2015, 4, 4160. [Google Scholar] [CrossRef] [PubMed]
- Cecchelli, R.; Berezowski, V.; Lundquist, S.; Culot, M.; Renftel, M.; Dehouck, M.-P.; Fenart, L. Modelling of the blood–brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 2007, 6, 650–661. [Google Scholar] [CrossRef]
- Boulay, A.-C.; Saubaméa, B.; Declèves, X.; Cohen-Salmon, M. Purification of Mouse Brain Vessels. J. Vis. Exp. 2015, e53208. [Google Scholar] [CrossRef] [PubMed]
- Roux, F.; Durieu-Trautmann, O.; Chaverot, N.; Claire, M.; Mailly, P.; Bourre, J.M.; Strosberg, A.D.; Couraud, P.O. Regulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities in immortalized rat brain microvessel endothelial cells. J. Cell. Physiol. 1994, 159, 101–113. [Google Scholar] [CrossRef]
- Noack, A.; Noack, S.; Hoffmann, A.; Maalouf, K.; Buettner, M.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Alms, D.; Römermann, K.; et al. Drug-induced trafficking of p-glycoprotein in human brain capillary endothelial cells as demonstrated by exposure to mitomycin C. PLoS ONE 2014, 9, e88154. [Google Scholar] [CrossRef] [PubMed]
- Vu, K.; Weksler, B.; Romero, I.; Couraud, P.-O.; Gelli, A. Immortalized human brain endothelial cell line HCMEC/D3 as a model of the blood-brain barrier facilitates in vitro studies of central nervous system infection by Cryptococcus neoformans. Eukaryot. Cell 2009, 8, 1803–1807. [Google Scholar] [CrossRef]
- Chapy, H.; Smirnova, M.; Andre, P.; Schlatter, J.; Chiadmi, F.; Couraud, P.-O.; Scherrmann, J.-M.; Decleves, X.; Cisternino, S. Carrier-Mediated Cocaine Transport at the Blood-Brain Barrier as a Putative Mechanism in Addiction Liability. Int. J. Neuropsychopharmacol. 2015, 18, pyu001. [Google Scholar] [CrossRef]
- Yousif, S.; Chaves, C.; Potin, S.; Margaill, I.; Scherrmann, J.-M.; Declèves, X. Induction of P-glycoprotein and Bcrp at the rat blood-brain barrier following a subchronic morphine treatment is mediated through NMDA/COX-2 activation. J. Neurochem. 2012, 123, 491–503. [Google Scholar] [CrossRef]
- Carl, S.M.; Lindley, D.J.; Couraud, P.O.; Weksler, B.B.; Romero, I.; Mowery, S.A.; Knipp, G.T. ABC and SLC Transporter Expression and Pot Substrate Characterization across the Human CMEC/D3 Blood−Brain Barrier Cell Line. Mol. Pharm. 2010, 7, 1057–1068. [Google Scholar] [CrossRef]
- Ohtsuki, S.; Ikeda, C.; Uchida, Y.; Sakamoto, Y.; Miller, F.; Glacial, F.; Decleves, X.; Scherrmann, J.-M.; Couraud, P.-O.; Kubo, Y.; et al. Quantitative Targeted Absolute Proteomic Analysis of Transporters, Receptors and Junction Proteins for Validation of Human Cerebral Microvascular Endothelial Cell Line hCMEC/D3 as a Human Blood–Brain Barrier Model. Mol. Pharm. 2013, 10, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Urich, E.; Lazic, S.E.; Molnos, J.; Wells, I.; Freskgård, P.-O. Transcriptional Profiling of Human Brain Endothelial Cells Reveals Key Properties Crucial for Predictive In Vitro Blood-Brain Barrier Models. PLoS ONE 2012, 7, e38149. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Grifno, G.N.; Farrell, A.M.; Linville, R.M.; Arevalo, D.; Kim, J.H.; Gu, L.; Searson, P.C. Tissue-engineered blood-brain barrier models via directed differentiation of human induced pluripotent stem cells. Sci. Rep. 2019, 9, 13957. [Google Scholar] [CrossRef]
- Le Roux, G.; Jarray, R.; Guyot, A.-C.; Pavoni, S.; Costa, N.; Théodoro, F.; Nassor, F.; Pruvost, A.; Tournier, N.; Kiyan, Y.; et al. Proof-of-Concept Study of Drug Brain Permeability Between in Vivo Human Brain and an in Vitro iPSCs-Human Blood-Brain Barrier Model. Sci. Rep. 2019, 9, 16310. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Abe, M.-A.; Maeda, T.; Ohtsuki, S.; Terasaki, T.; Kanda, T. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J. Cell. Physiol. 2011, 226, 255–266. [Google Scholar] [CrossRef]
- Siddharthan, V.; Kim, Y.V.; Liu, S.; Kim, K.S. Human astrocytes/astrocyte-conditioned medium and shear stress enhance the barrier properties of human brain microvascular endothelial cells. Brain Res. 2007, 1147, 39–50. [Google Scholar] [CrossRef]
- Hartmann, C.; Zozulya, A.; Wegener, J.; Galla, H.-J. The impact of glia-derived extracellular matrices on the barrier function of cerebral endothelial cells: An in vitro study. Exp. Cell Res. 2007, 313, 1318–1325. [Google Scholar] [CrossRef]
- Haseloff, R.F.; Blasig, I.E.; Bauer, H.-C.; Bauer, H. In Search of the Astrocytic Factor(s) Modulating Blood–Brain Barrier Functions in Brain Capillary Endothelial Cells In Vitro. Cell. Mol. Neurobiol. 2005, 25, 25–39. [Google Scholar] [CrossRef]
- Helms, H.C.; Madelung, R.; Waagepetersen, H.S.; Nielsen, C.U.; Brodin, B. In vitro evidence for the brain glutamate efflux hypothesis: Brain endothelial cells cocultured with astrocytes display a polarized brain-to-blood transport of glutamate. Glia 2012, 60, 882–893. [Google Scholar] [CrossRef]
- Cecchelli, R.; Dehouck, B.; Descamps, L.; Fenart, L.; Buée-Scherrer, V.; Duhem, C.; Lundquist, S.; Rentfel, M.; Torpier, G.; Dehouck, M. In vitro model for evaluating drug transport across the blood–brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 165–178. [Google Scholar] [CrossRef]
- Lai, C.-H.; Kuo, K.-H. The critical component to establish in vitro BBB model: Pericyte. Brain Res. Rev. 2005, 50, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Savettieri, G.; Di Liegro, I.; Catania, C.; Licata, L.; Pitarresi, G.L.; D’Agostino, S.; Schiera, G.; De Caro, V.; Giandalia, G.; Giannola, L.I.; et al. Neurons and ECM regulate occludin localization in brain endothelial cells. Neuroreport 2000, 11, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Sumi, N.; Nishioku, T.; Takata, F.; Matsumoto, J.; Watanabe, T.; Shuto, H.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Lipopolysaccharide-activated microglia induce dysfunction of the blood-brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell. Mol. Neurobiol. 2010, 30, 247–253. [Google Scholar] [CrossRef]
- Stone, N.L.; England, T.J.; O’Sullivan, S.E. A Novel Transwell Blood Brain Barrier Model Using Primary Human Cells. Front. Cell. Neurosci. 2019, 13, 1–11. [Google Scholar] [CrossRef]
- Malina, K.C.-K.; Cooper, I.; Teichberg, V.I. Closing the gap between the in-vivo and in-vitro blood–brain barrier tightness. Brain Res. 2009, 1284, 12–21. [Google Scholar] [CrossRef]
- Cantrill, C.A.; Skinner, R.A.; Rothwell, N.J.; Penny, J.I. An immortalised astrocyte cell line maintains the in vivo phenotype of a primary porcine in vitro blood–brain barrier model. Brain Res. 2012, 1479, 17–30. [Google Scholar] [CrossRef]
- Molino, Y.; Jabès, F.; Lacassagne, E.; Gaudin, N.; Khrestchatisky, M. Setting-up an In Vitro Model of Rat Blood-brain Barrier (BBB): A Focus on BBB Impermeability and Receptor-mediated Transport. J. Vis. Exp. 2014, 88, e51278. [Google Scholar] [CrossRef]
- Helms, H.C.; Hersom, M.; Kuhlmann, L.B.; Badolo, L.; Nielsen, C.U.; Brodin, B. An Electrically Tight In Vitro Blood–Brain Barrier Model Displays Net Brain-to-Blood Efflux of Substrates for the ABC Transporters, P-gp, Bcrp and Mrp-1. AAPS J. 2014, 16, 1046–1055. [Google Scholar] [CrossRef]
- Gaillard, P.J.; Van Der Sandt, I.C.J.; Voorwinden, L.H.; Vu, D.; Nielsen, J.L.; De Boer, A.G.; Breimer, D.D. Astrocytes increase the functional expression of P-glycoprotein in an in vitro model of the blood-brain barrier. Pharm. Res. 2000, 17, 1198–1205. [Google Scholar] [CrossRef]
- Brown, J.A.; Pensabene, V.; Markov, D.A.; Allwardt, V.; Neely, M.D.; Shi, M.; Britt, C.M.; Hoilett, O.S.; Yang, Q.; Brewer, B.M.; et al. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 2015, 9, 054124. [Google Scholar] [CrossRef] [PubMed]
- Maoz, B.M.; Herland, A.; FitzGerald, E.A.; Grevesse, T.; Vidoudez, C.; Pacheco, A.R.; Sheehy, S.P.; Park, T.-E.; Dauth, S.; Mannix, R.; et al. A linked organ-on-chip model of the human neurovascular unit reveals the metabolic coupling of endothelial and neuronal cells. Nat. Biotechnol. 2018, 36, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Patrick, C.W., Jr.; McIntire, L.V. Shear Stress and Cyclic Strain Modulation of Gene Expression in Vascular Endothelial Cells. Blood Purif. 1995, 13, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Ballermann, B.J.; Dardik, A.; Eng, E.; Liu, A. Shear stress and the endothelium. Kidney Int. 1998, 54, 100–108. [Google Scholar] [CrossRef] [PubMed]
- OTT, M.; BALLERMANN, B. Shear stress-conditioned, endothelial cell-seeded vascular grafts: Improved cell adherence in response to in vitro shear stress*. Surgery 1995, 117, 334–339. [Google Scholar] [CrossRef]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef]
- Traub, O.; Berk, B.C. Laminar Shear Stress. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 677–685. [Google Scholar] [CrossRef]
- Ando, J.; Yamamoto, K. Vascular Mechanobiology. Circ. J. 2009, 73, 1983–1992. [Google Scholar] [CrossRef]
- Cucullo, L.; Hossain, M.; Puvenna, V.; Marchi, N.; Janigro, D. The role of shear stress in Blood-Brain Barrier endothelial physiology. BMC Neurosci. 2011, 12, 40. [Google Scholar] [CrossRef]
- Walsh, T.G.; Murphy, R.P.; Fitzpatrick, P.; Rochfort, K.D.; Guinan, A.F.; Murphy, A.; Cummins, P.M. Stabilization of brain microvascular endothelial barrier function by shear stress involves VE-cadherin signaling leading to modulation of pTyr-occludin levels. J. Cell. Physiol. 2011, 226, 3053–3063. [Google Scholar] [CrossRef]
- Dewey, C.F.; Bussolari, S.R.; Gimbrone, M.A.; Davies, P.F. The Dynamic Response of Vascular Endothelial Cells to Fluid Shear Stress. J. Biomech. Eng. 1981, 103, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Colgan, O.C.; Ferguson, G.; Collins, N.T.; Murphy, R.P.; Meade, G.; Cahill, P.A.; Cummins, P.M. Regulation of bovine brain microvascular endothelial tight junction assembly and barrier function by laminar shear stress. Am. J. Physiol. Circ. Physiol. 2007, 292, H3190–H3197. [Google Scholar] [CrossRef] [PubMed]
- Stanness, K.A.; Westrum, L.E.; Fornaciari, E.; Mascagni, P.; Nelson, J.A.; Stenglein, S.G.; Myers, T.; Janigro, D. Morphological and functional characterization of an in vitro blood–brain barrier model. Brain Res. 1997, 771, 329–342. [Google Scholar] [CrossRef]
- Cucullo, L.; Hossain, M.; Rapp, E.; Manders, T.; Marchi, N.; Janigro, D. Development of a humanized in vitro blood-brain barrier model to screen for brain penetration of antiepileptic drugs. Epilepsia 2007, 48, 505–516. [Google Scholar] [CrossRef]
- Cucullo, L.; Couraud, P.-O.; Weksler, B.; Romero, I.-A.; Hossain, M.; Rapp, E.; Janigro, D. Immortalized human brain endothelial cells and flow-based vascular modeling: A marriage of convenience for rational neurovascular studies. J. Cereb. Blood Flow Metab. 2008, 28, 312–328. [Google Scholar] [CrossRef]
- Bussolari, S.R.; Dewey, C.F.; Gimbrone, M.A. Apparatus for subjecting living cells to fluid shear stress. Rev. Sci. Instrum. 1982. [Google Scholar] [CrossRef]
- Stanness, K.A.; Guatteo, E.; Janigro, D. A dynamic model of the blood-brain barrier “in vitro”. Neurotoxicology 1996, 17, 481–496. [Google Scholar]
- Janigro, D.; Leaman, S.M.; Stanness, K.A. Dynamic in vitro modeling of the blood–brain barrier: A novel tool for studies of drug delivery to the brain. Pharm. Sci. Technolo. Today 1999, 2, 7–12. [Google Scholar] [CrossRef]
- Jiang, L.; Li, S.; Zheng, J.; Li, Y.; Huang, H. Recent Progress in Microfluidic Models of the Blood-Brain Barrier. Micromachines 2019, 10, 375. [Google Scholar] [CrossRef]
- Brown, J.A.; Codreanu, S.G.; Shi, M.; Sherrod, S.D.; Markov, D.A.; Neely, M.D.; Britt, C.M.; Hoilett, O.S.; Reiserer, R.S.; Samson, P.C.; et al. Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit. J. Neuroinflammation 2016, 13, 306. [Google Scholar] [CrossRef]
- Park, T.-E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef] [PubMed]
- Monnot, A.D.; Zheng, W. Culture of Choroid Plexus Epithelial Cells and In Vitro Model of Blood–CSF Barrier. In Methods in Molecular Biology; Randell, S.H., Fulcher, M.L., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 13–29. ISBN 9781627031240. [Google Scholar]
- Menheniott, T.R.; Charalambous, M.; Ward, A. Derivation of Primary Choroid Plexus Epithelial Cells from the Mouse. In Methods in molecular biology (Clifton, N.J.); Ward, A., Tosh, D., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 207–220. [Google Scholar]
- Kläs, J.; Wolburg, H.; Terasaki, T.; Fricker, G.; Reichel, V. Characterization of immortalized choroid plexus epithelial cell lines for studies of transport processes across the blood-cerebrospinal fluid barrier. Cerebrospinal Fluid Res. 2010, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- ANGELOW, S.; ZENI, P.; GALLA, H. Usefulness and limitation of primary cultured porcine choroid plexus epithelial cells as an in vitro model to study drug transport at the blood?CSF barrier. Adv. Drug Deliv. Rev. 2004, 56, 1859–1873. [Google Scholar] [CrossRef] [PubMed]
- Baehr, C.; Reichel, V.; Fricker, G. Choroid plexus epithelial monolayers—A cell culture model from porcine brain. Cerebrospinal Fluid Res. 2006, 3, 1–14. [Google Scholar] [CrossRef]
- Delery, E.C.; MacLean, A.G. Culture Model for Non-human Primate Choroid Plexus. Front. Cell. Neurosci. 2019, 13, 1–10. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, Q. Establishment and characterization of an immortalized Z310 choroidal epithelial cell line from murine choroid plexus. Brain Res. 2002, 958, 371–380. [Google Scholar] [CrossRef]
- Kitazawa, T.; Hosoya, K.; Watanabe, M.; Takashima, T.; Ohtsuki, S.; Takanaga, H.; Ueda, M.; Yanai, N.; Obinata, M.; Terasaki, T. Characterization of the amino acid transport of new immortalized choroid plexus epithelial cell lines: A novel in vitro system for investigating transport functions at the blood-cerebrospinal fluid barrier. Pharm. Res. 2001, 18, 16–22. [Google Scholar] [CrossRef]
- Janson, C.; Romanova, L.; Hansen, E.; Hubel, A.; Lam, C. Immortalization and functional characterization of rat arachnoid cell lines. Neuroscience 2011, 177, 23–34. [Google Scholar] [CrossRef]
- Holman, D.W.; Grzybowski, D.M.; Mehta, B.C.; Katz, S.E.; Lubow, M. Characterization of cytoskeletal and junctional proteins expressed by cells cultured from human arachnoid granulation tissue. Cerebrospinal Fluid Res. 2005, 2, 1–12. [Google Scholar] [CrossRef]
- Hansen, E.A.; Romanova, L.; Janson, C.; Lam, C.H. The effects of blood and blood products on the arachnoid cell. Exp. Brain Res. 2017, 235, 1749–1758. [Google Scholar] [CrossRef]
- Chen, X.; Loryan, I.; Payan, M.; Keep, R.F.; Smith, D.E.; Hammarlund-Udenaes, M. Effect of transporter inhibition on the distribution of cefadroxil in rat brain. Fluids Barriers CNS 2014, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Wood, A.; Bowlby, M. Brain Slices as Models for Neurodegenerative Disease and Screening Platforms to Identify Novel Therapeutics. Curr. Neuropharmacol. 2007, 5, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ohtsuki, S.; Kamiie, J.; Terasaki, T. Blood-Brain Barrier (BBB) Pharmacoproteomics: Reconstruction of In Vivo Brain Distribution of 11 P-Glycoprotein Substrates Based on the BBB Transporter Protein Concentration, In Vitro Intrinsic Transport Activity, and Unbound Fraction in Plasma and Brain. J. Pharmacol. Exp. Ther. 2011, 339, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Chaves, C.; Gómez-Zepeda, D.; Auvity, S.; Menet, M.-C.; Crété, D.; Labat, L.; Remião, F.; Cisternino, S.; Declèves, X. Effect of Subchronic Intravenous Morphine Infusion and Naloxone-Precipitated Morphine Withdrawal on P-gp and Bcrp at the Rat Blood–Brain Barrier. J. Pharm. Sci. 2016, 105, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Hartz, A.M.; Potin, S.; Coumoul, X.; Yousif, S.; Scherrmann, J.-M.; Bauer, B.; Declèves, X. Aryl hydrocarbon receptor-dependent upregulation of Cyp1b1 by TCDD and diesel exhaust particles in rat brain microvessels. Fluids Barriers CNS 2011, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Gauthier, M.; Tan, X.; Landry, C.; Poupon, J.; Dehouck, M.-P.; Gosselet, F.; Perrière, N.; Bellivier, F.; Cisternino, S.; et al. Sodium Transporters Are Involved in Lithium Influx in Brain Endothelial Cells. Mol. Pharm. 2018, 15, 2528–2538. [Google Scholar] [CrossRef]
- Luo, H.; Rossi, E.; Saubamea, B.; Chasseigneaux, S.; Cochois, V.; Choublier, N.; Smirnova, M.; Glacial, F.; Perrière, N.; Bourdoulous, S.; et al. Cannabidiol Increases Proliferation, Migration, Tubulogenesis, and Integrity of Human Brain Endothelial Cells through TRPV2 Activation. Mol. Pharm. 2019, 16, 1312–1326. [Google Scholar] [CrossRef]
- Dodacki, A.; Wortman, M.; Saubaméa, B.; Chasseigneaux, S.; Nicolic, S.; Prince, N.; Lochus, M.; Raveu, A.-L.; Declèves, X.; Scherrmann, J.-M.; et al. Expression and function of Abcg4 in the mouse blood-brain barrier: Role in restricting the brain entry of amyloid-β peptide. Sci. Rep. 2017, 7, 13393. [Google Scholar] [CrossRef]
- Dayton, J.R.; Franke, M.C.; Yuan, Y.; Cruz-Orengo, L. Straightforward method for singularized and region-specific CNS microvessels isolation. J. Neurosci. Methods 2019, 318, 17–33. [Google Scholar] [CrossRef]
- Rosas-Hernandez, H.; Cuevas, E.; Lantz, S.M.; Paule, M.G.; Ali, S.F. Isolation and Culture of Brain Microvascular Endothelial Cells for In Vitro Blood-Brain Barrier Studies. In Neurotrophic Factors: Methods and Protocols; Skaper, S.D., Ed.; Humana Press: Totowa, NJ, USA, 2018; Volume 1727, pp. 315–331. ISBN 9780128110379. [Google Scholar]
- Steffensen, A.B.; Oernbo, E.K.; Stoica, A.; Gerkau, N.J.; Barbuskaite, D.; Tritsaris, K.; Rose, C.R.; MacAulay, N. Cotransporter-mediated water transport underlying cerebrospinal fluid formation. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Baird, A.; Eliceiri, B.P.; Gonzalez, A.M.; Johanson, C.E.; Leadbeater, W.; Stopa, E.G. Targeting the Choroid Plexus-CSF-Brain Nexus Using Peptides Identified by Phage Display. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: Totowa, NJ, USA, 2011; Volume 686, pp. 483–498. ISBN 9781607619383. [Google Scholar]
- Gonzalez, A.; Leadbeater, W.E.; Burg, M.; Sims, K.; Terasaki, T.; Johanson, C.E.; Stopa, E.G.; Eliceiri, B.P.; Baird, A. Targeting choroid plexus epithelia and ventricular ependyma for drug delivery to the central nervous system. BMC Neurosci. 2011, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Brandhonneur, N.; Dollo, G.; Ratajczak-Enselme, M.; Deniau, A.L.; Chevanne, F.; Estbe, J.P.; Legrand, A.; Le Corre, P. Ex vivo and in vivo diffusion of ropivacaine through spinal meninges: Influence of absorption enhancers. Int. J. Pharm. 2011, 404, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Glimcher, S.A.; Holman, D.W.; Lubow, M.; Grzybowski, D.M. Ex Vivo Model of Cerebrospinal Fluid Outflow across Human Arachnoid Granulations. Investig. Opthalmology Vis. Sci. 2008, 49, 4721. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Holland, E.C. Genetically engineered mouse models of brain cancer and the promise of preclinical testing. Brain Pathol. 2009, 19, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Sadiq, M.W.; Uchida, Y.; Hoshi, Y.; Tachikawa, M.; Terasaki, T.; Hammarlund-Udenaes, M. Validation of a P-Glycoprotein (P-gp) Humanized Mouse Model by Integrating Selective Absolute Quantification of Human MDR1, Mouse Mdr1a and Mdr1b Protein Expressions with In Vivo Functional Analysis for Blood-Brain Barrier Transport. PLoS ONE 2015, 10, e0118638. [Google Scholar] [CrossRef]
- Yamasaki, Y.; Kobayashi, K.; Okuya, F.; Kajitani, N.; Kazuki, K.; Abe, S.; Takehara, S.; Ito, S.; Ogata, S.; Uemura, T.; et al. Characterization of P-glycoprotein humanized mice generated by chromosome engineering technology: Its utility for prediction of drug distribution to the brain in humans. Drug Metab. Dispos. 2018, 46, 1756–1766. [Google Scholar] [CrossRef]
- Dallas, S.; Salphati, L.; Gomez-Zepeda, D.; Wanek, T.; Chu, X.; Kunta, J.; Mezler, M.; Menet, M.M.-C.; Declèves, X.; Langer, O.; et al. Generation and Characterization of a Breast Cancer Resistance Protein Humanized Mouse Model. Mol. Pharmacol. 2016, 89, 492–504. [Google Scholar] [CrossRef]
- Krohn, M.; Zoufal, V.; Mairinger, S.; Wanek, T.; Paarmann, K.; Brüning, T.; Eiriz, I.; Brackhan, M.; Langer, O.; Pahnke, J. Generation and Characterization of an Abcc1 Humanized Mouse Model (hABCC1 flx/flx) with Knockout Capability. Mol. Pharmacol. 2019, 96, 138–147. [Google Scholar] [CrossRef]
- Kusuhara, H.; Sugiyama, Y. Active efflux across the blood-brain barrier: Role of the solute carrier family. NeuroRx 2005, 2, 73–85. [Google Scholar] [CrossRef]
- Takasato, Y.; Rapoport, S.I.; Smith, Q.R. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am. J. Physiol. Circ. Physiol. 1984, 247, H484–H493. [Google Scholar] [CrossRef]
- Cattelotte, J.; André, P.; Ouellet, M.; Bourasset, F.; Scherrmann, J.-M.; Cisternino, S. In situ mouse carotid perfusion model: Glucose and cholesterol transport in the eye and brain. J. Cereb. Blood Flow Metab. 2008, 28, 1449–1459. [Google Scholar] [CrossRef]
- Chapy, H.; Saubaméa, B.; Tournier, N.; Bourasset, F.; Behar-Cohen, F.; Declèves, X.; Scherrmann, J.-M.; Cisternino, S. Blood-brain and retinal barriers show dissimilar ABC transporter impacts and concealed effect of P-glycoprotein on a novel verapamil influx carrier. Br. J. Pharmacol. 2016, 173, 497–510. [Google Scholar] [CrossRef]
- Taccola, C.; Cartot-Cotton, S.; Valente, D.; Barneoud, P.; Aubert, C.; Boutet, V.; Gallen, F.; Lochus, M.; Nicolic, S.; Dodacki, A.; et al. High brain distribution of a new central nervous system drug candidate despite its P-glycoprotein-mediated efflux at the mouse blood-brain barrier. Eur. J. Pharm. Sci. 2018, 117, 68–79. [Google Scholar] [CrossRef]
- Tournier, N.; Stieger, B.; Langer, O. Imaging techniques to study drug transporter function in vivo. Pharmacol. Ther. 2018, 189, 104–122. [Google Scholar] [CrossRef]
- Langer, O. Use of PET Imaging to Evaluate Transporter-Mediated Drug-Drug Interactions. J. Clin. Pharmacol. 2016, 56, S143–S156. [Google Scholar] [CrossRef]
- Suridjan, I.; Comley, R.A.; Rabiner, E.A. The application of positron emission tomography (PET) imaging in CNS drug development. Brain Imaging Behav. 2019, 13, 354–365. [Google Scholar] [CrossRef]
- Syvänen, S.; Eriksson, J. Advances in PET imaging of P-glycoprotein function at the blood-brain barrier. ACS Chem. Neurosci. 2013, 4, 225–237. [Google Scholar] [CrossRef]
- Traxl, A.; Wanek, T.; Mairinger, S.; Stanek, J.; Filip, T.; Sauberer, M.; Muller, M.; Kuntner, C.; Langer, O. Breast Cancer Resistance Protein and P-Glycoprotein Influence In Vivo Disposition of 11C-Erlotinib. J. Nucl. Med. 2015, 56, 1930–1936. [Google Scholar] [CrossRef]
- Tournier, N.; Goutal, S.; Auvity, S.; Traxl, A.; Mairinger, S.; Wanek, T.; Helal, O.; Buvat, I.; Soussan, M.; Caillé, F.; et al. Strategies to Inhibit ABCB1- and ABCG2-Mediated Efflux Transport of Erlotinib at the Blood–Brain Barrier: A PET Study on Nonhuman Primates. J. Nucl. Med. 2017, 58, 117–122. [Google Scholar] [CrossRef]
- Bauer, M.; Karch, R.; Wulkersdorfer, B.; Philippe, C.; Nics, L.; Klebermass, E.; Weber, M.; Poschner, S.; Haslacher, H.; Jäger, W.; et al. A Proof-of-Concept Study to Inhibit ABCG2- and ABCB1-Mediated Efflux Transport at the Human Blood–Brain Barrier. J. Nucl. Med. 2019, 60, 486–491. [Google Scholar] [CrossRef]
- Kawamura, K.; Konno, F.; Yamasaki, T.; Yui, J.; Hatori, A.; Yanamoto, K.; Irie, T.; Fukumura, T.; Suzuki, K.; Kanno, I.; et al. Synthesis and evaluation of C-11 labeled dual modulator for P-gp and BCRP as a PET probe. J. Label. Compd. Radiopharm. 2009, 52, S370. [Google Scholar]
- Wanek, T.; Kuntner, C.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Sauberer, M.; Mairinger, S.; Strommer, S.; Wacheck, V.; Löscher, W.; et al. A comparative small-animal PET evaluation of [11C]tariquidar, [11C]elacridar and (R)-[11C]verapamil for detection of P-glycoprotein-expressing murine breast cancer. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 149–159. [Google Scholar] [CrossRef][Green Version]
- Bauer, M.; Karch, R.; Zeitlinger, M.; Stanek, J.; Philippe, C.; Wadsak, W.; Mitterhauser, M.; Jager, W.; Haslacher, H.; Muller, M.; et al. Interaction of 11C-Tariquidar and 11C-Elacridar with P-Glycoprotein and Breast Cancer Resistance Protein at the Human Blood-Brain Barrier. J. Nucl. Med. 2013, 54, 1181–1187. [Google Scholar] [CrossRef]
- Dörner, B.; Kuntner, C.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Wanek, T.; Stundner, G.; Mairinger, S.; Löscher, W.; Müller, M.; et al. Synthesis and Small-Animal Positron Emission Tomography Evaluation of [11C]-Elacridar As a Radiotracer to Assess the Distribution of P-Glycoprotein at the Blood−Brain Barrier. J. Med. Chem. 2009, 52, 6073–6082. [Google Scholar] [CrossRef]
- Wanek, T.; Kuntner, C.; Bankstahl, J.P.; Mairinger, S.; Bankstahl, M.; Stanek, J.; Sauberer, M.; Filip, T.; Erker, T.; Müller, M.; et al. A Novel PET Protocol for Visualization of Breast Cancer Resistance Protein Function at the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 2002–2011. [Google Scholar] [CrossRef]
- Bauer, F.; Kuntner, C.; Bankstahl, J.P.; Wanek, T.; Bankstahl, M.; Stanek, J.; Mairinger, S.; Dörner, B.; Löscher, W.; Müller, M.; et al. Synthesis and in vivo evaluation of [11C]tariquidar, a positron emission tomography radiotracer based on a third-generation P-glycoprotein inhibitor. Bioorg. Med. Chem. 2010, 18, 5489–5497. [Google Scholar] [CrossRef]
- Saleem, A.; Brown, G.D.; Brady, F.; Aboagye, E.O.; Osman, S.; Luthra, S.K.; Ranicar, A.S.O.; Brock, C.S.; Stevens, M.F.G.; Newlands, E.; et al. Metabolic activation of temozolomide measured in vivo using positron emission tomography. Cancer Res. 2003, 63, 2409–2415. [Google Scholar]
- Römermann, K.; Wanek, T.; Bankstahl, M.; Bankstahl, J.P.; Fedrowitz, M.; Müller, M.; Löscher, W.; Kuntner, C.; Langer, O. (R)-[11C]verapamil is selectively transported by murine and human P-glycoprotein at the blood–brain barrier, and not by MRP1 and BCRP. Nucl. Med. Biol. 2013, 40, 873–878. [Google Scholar] [CrossRef]
- Luurtsema, G.; Molthoff, C.F.; Windhorst, A.; Smit, J.; Keizer, H.; Boellaard, R.; Lammertsma, A.; Franssen, E.J. (R)- and (S)-[11C]verapamil as PET-tracers for measuring P-glycoprotein function: In vitro and in vivo evaluation. Nucl. Med. Biol. 2003, 30, 747–751. [Google Scholar] [CrossRef]
- Hendrikse, N.H.; De Vries, E.G.E.; Eriks-Fluks, L.; Van Der Graaf, W.T.A.; Hospers, G.A.P.; Willemsen, A.T.M.; Vaalburg, W.; Franssen, E.J.F. A new in vivo method to study P-glycoprotein transport in tumors and the blood-brain barrier. Cancer Res. 1999, 59, 2411–2416. [Google Scholar]
- Sasongko, L.; Link, J.; Muzi, M.; Mankoff, D.; Yang, X.; Collier, A.; Shoner, S.; Unadkat, J. Imaging P-glycoprotein transport activity at the human blood-brain barrier with positron emission tomography. Clin. Pharmacol. Ther. 2005, 77, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Kannan, P.; Brimacombe, K.R.; Zoghbi, S.S.; Liow, J.-S.; Morse, C.; Taku, A.K.; Pike, V.W.; Halldin, C.; Innis, R.B.; Gottesman, M.M.; et al. N-desmethyl -Loperamide Is Selective for P-Glycoprotein among Three ATP-Binding Cassette Transporters at the Blood-Brain Barrier. Drug Metab. Dispos. 2010, 38, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Damont, A.; Goutal, S.; Auvity, S.; Valette, H.; Kuhnast, B.; Saba, W.; Tournier, N. Imaging the impact of cyclosporin A and dipyridamole on P-glycoprotein (ABCB1) function at the blood-brain barrier: A [11C]-N-desmethyl-loperamide PET study in nonhuman primates. Eur. J. Pharm. Sci. 2016, 91, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, S.S.; Liow, J.-S.; Yasuno, F.; Hong, J.; Tuan, E.; Lazarova, N.; Gladding, R.L.; Pike, V.W.; Innis, R.B. 11C-Loperamide and Its N-Desmethyl Radiometabolite Are Avid Substrates for Brain Permeability-Glycoprotein Efflux. J. Nucl. Med. 2008, 49, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Hosten, B.; Boisgard, R.; Jacob, A.; Goutal, S.; Saubaméa, B.; Dollé, F.; Scherrmann, J.-M.; Cisternino, S.; Tournier, N. [11C]befloxatone brain kinetics is not influenced by Bcrp function at the blood–brain barrier: A PET study using Bcrp TGEM knockout rats. Eur. J. Pharm. Sci. 2013, 50, 520–525. [Google Scholar] [CrossRef]
- Mairinger, S.; Langer, O.; Kuntner, C.; Wanek, T.; Bankstahl, J.P.; Bankstahl, M.; Stanek, J.; Dörner, B.; Bauer, F.; Baumgartner, C.; et al. Synthesis and in vivo evaluation of the putative breast cancer resistance protein inhibitor [11C]methyl 4-((4-(2-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)ethyl)phenyl)amino-carbonyl)-2-(quinoline-2-carbonylamino)benzoate. Nucl. Med. Biol. 2010, 37, 637–644. [Google Scholar] [CrossRef][Green Version]
- Sivapackiam, J.; Harpstrite, S.E.; Prior, J.L.; Mattingly, S.; Sharma, V. 67/68Galmydar: A metalloprobe for monitoring breast cancer resistance protein (BCRP)-mediated functional transport activity. Nucl. Med. Biol. 2016, 43, 191–197. [Google Scholar] [CrossRef]
- Okamura, T.; Kikuchi, T.; Okada, M.; Toramatsu, C.; Fukushi, K.; Takei, M.; Irie, T. Noninvasive and Quantitative Assessment of the Function of Multidrug Resistance-Associated Protein 1 in the Living Brain. J. Cereb. Blood Flow Metab. 2009, 29, 504–511. [Google Scholar] [CrossRef]
- Krohn, M.; Wanek, T.; Menet, M.-C.; Noack, A.; Declèves, X.; Langer, O.; Löscher, W.; Pahnke, J. Humanization of the blood–brain barrier transporter ABCB1 in mice disrupts genomic locus — lessons from three unsuccessful approaches. Eur. J. Microbiol. Immunol. 2018, 8, 78–86. [Google Scholar] [CrossRef]
- Bart, J.; Willemsen, A.T.M.; Groen, H.J.M.; van der Graaf, W.T.; Wegman, T.D.; Vaalburg, W.; de Vries, E.G.; Hendrikse, N.H. Quantitative assessment of P-glycoprotein function in the rat blood–brain barrier by distribution volume of [11C]verapamil measured with PET. Neuroimage 2003, 20, 1775–1782. [Google Scholar] [CrossRef]
- Bauer, M.; Tournier, N.; Langer, O. Imaging P-Glycoprotein Function at the Blood–Brain Barrier as a Determinant of the Variability in Response to Central Nervous System Drugs. Clin. Pharmacol. Ther. 2019, 105, 1061–1064. [Google Scholar] [CrossRef]
- Goutal, S.; Gerstenmayer, M.; Auvity, S.; Caillé, F.; Mériaux, S.; Buvat, I.; Larrat, B.; Tournier, N. Physical blood-brain barrier disruption induced by focused ultrasound does not overcome the transporter-mediated efflux of erlotinib. J. Control. Release 2018, 292, 210–220. [Google Scholar] [CrossRef]
- Prideaux, B.; Stoeckli, M. Mass spectrometry imaging for drug distribution studies. J. Proteomics 2012, 75, 4999–5013. [Google Scholar] [CrossRef]
- Nilsson, A.; Goodwin, R.J.A.; Shariatgorji, M.; Vallianatou, T.; Webborn, P.J.H.; Andrén, P.E. Mass Spectrometry Imaging in Drug Development. Anal. Chem. 2015, 87, 1437–1455. [Google Scholar] [CrossRef]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef]
- Dilillo, M.; Ait-Belkacem, R.; Esteve, C.; Pellegrini, D.; Nicolardi, S.; Costa, M.; Vannini, E.; de Graaf, E.L.; Caleo, M.; McDonnell, L.A. Ultra-High Mass Resolution MALDI Imaging Mass Spectrometry of Proteins and Metabolites in a Mouse Model of Glioblastoma. Sci. Rep. 2017, 7, 603. [Google Scholar] [CrossRef]
- Jarmusch, A.K.; Alfaro, C.M.; Pirro, V.; Hattab, E.M.; Cohen-Gadol, A.A.; Cooks, R.G. Differential Lipid Profiles of Normal Human Brain Matter and Gliomas by Positive and Negative Mode Desorption Electrospray Ionization—Mass Spectrometry Imaging. PLoS ONE 2016, 11, e0163180. [Google Scholar] [CrossRef]
- Liu, X.; Ide, J.L.; Norton, I.; Marchionni, M.A.; Ebling, M.C.; Wang, L.Y.; Davis, E.; Sauvageot, C.M.; Kesari, S.; Kellersberger, K.A.; et al. Molecular imaging of drug transit through the blood-brain barrier with MALDI mass spectrometry imaging. Sci. Rep. 2013, 3, 2859. [Google Scholar] [CrossRef]
- Jove, M.; Spencer, J.; Clench, M.; Loadman, P.M.; Twelves, C. Precision pharmacology: Mass spectrometry imaging and pharmacokinetic drug resistance. Crit. Rev. Oncol. Hematol. 2019, 141, 153–162. [Google Scholar] [CrossRef]
- Eberlin, L.S.; Norton, I.; Orringer, D.; Dunn, I.F.; Liu, X.; Ide, J.L.; Jarmusch, A.K.; Ligon, K.L.; Jolesz, F.A.; Golby, A.J.; et al. Ambient mass spectrometry for the intraoperative molecular diagnosis of human brain tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 1611–1616. [Google Scholar] [CrossRef]
- Eberlin, L.S.; Norton, I.; Dill, A.L.; Golby, A.J.; Ligon, K.L.; Santagata, S.; Cooks, R.G.; Agar, N.Y.R. Classifying Human Brain Tumors by Lipid Imaging with Mass Spectrometry. Cancer Res. 2012, 72, 645–654. [Google Scholar] [CrossRef]
- Clark, A.R.; Calligaris, D.; Regan, M.S.; Pomeranz Krummel, D.; Agar, J.N.; Kallay, L.; MacDonald, T.; Schniederjan, M.; Santagata, S.; Pomeroy, S.L.; et al. Rapid discrimination of pediatric brain tumors by mass spectrometry imaging. J. Neurooncol. 2018, 140, 269–279. [Google Scholar] [CrossRef]
- Le Rhun, E.; Duhamel, M.; Wisztorski, M.; Gimeno, J.-P.; Zairi, F.; Escande, F.; Reyns, N.; Kobeissy, F.; Maurage, C.-A.; Salzet, M.; et al. Evaluation of non-supervised MALDI mass spectrometry imaging combined with microproteomics for glioma grade III classification. Biochim. Biophys. Acta - Proteins Proteomics 2017, 1865, 875–890. [Google Scholar] [CrossRef]
- de Lange, E.C.M.; van den Brink, W.; Yamamoto, Y.; de Witte, W.E.A.; Wong, Y.C. Novel CNS drug discovery and development approach: Model-based integration to predict neuro-pharmacokinetics and pharmacodynamics. Expert Opin. Drug Discov. 2017, 12, 1207–1218. [Google Scholar] [CrossRef]
- Bendels, S.; Kansy, M.; Wagner, B.; Huwyler, J. In silico prediction of brain and CSF permeation of small molecules using PLS regression models. Eur. J. Med. Chem. 2008, 43, 1581–1592. [Google Scholar] [CrossRef]
- Loryan, I.; Sinha, V.; Mackie, C.; Van Peer, A.; Drinkenburg, W.H.; Vermeulen, A.; Heald, D.; Hammarlund-Udenaes, M.; Wassvik, C.M. Molecular Properties Determining Unbound Intracellular and Extracellular Brain Exposure of CNS Drug Candidates. Mol. Pharm. 2015, 12, 520–532. [Google Scholar] [CrossRef]
- Fleming, N. How artificial intelligence is changing drug discovery. Nature 2018, 557, S55–S57. [Google Scholar] [CrossRef]
- Chen, H.; Winiwarter, S.; Fridén, M.; Antonsson, M.; Engkvist, O. In silico prediction of unbound brain-to-plasma concentration ratio using machine learning algorithms. J. Mol. Graph. Model. 2011, 29, 985–995. [Google Scholar] [CrossRef]
- Plisson, F.; Piggott, A. Predicting Blood–Brain Barrier Permeability of Marine-Derived Kinase Inhibitors Using Ensemble Classifiers Reveals Potential Hits for Neurodegenerative Disorders. Mar. Drugs 2019, 17, 81. [Google Scholar] [CrossRef]
- de Lange, E.C.M. PBPK Modeling Approach for Predictions of Human CNS Drug Brain Distribution. In Blood-Brain Barrier in Drug Discovery; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 296–323. [Google Scholar]
- Bouzom, F.; Ball, K.; Perdaems, N.; Walther, B. Physiologically based pharmacokinetic (PBPK) modelling tools: How to fit with our needs? Biopharm. Drug Dispos. 2012, 33, 55–71. [Google Scholar] [CrossRef]
- Ball, K.; Bouzom, F.; Scherrmann, J.-M.; Walther, B.; Declèves, X. Development of a Physiologically Based Pharmacokinetic Model for the Rat Central Nervous System and Determination of an In Vitro–In Vivo Scaling Methodology for the Blood–Brain Barrier Permeability of Two Transporter Substrates, Morphine and Oxycodone. J. Pharm. Sci. 2012, 101, 4277–4292. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Välitalo, P.A.; Wong, Y.C.; Huntjens, D.R.; Proost, J.H.; Vermeulen, A.; Krauwinkel, W.; Beukers, M.W.; Kokki, H.; Kokki, M.; et al. Prediction of human CNS pharmacokinetics using a physiologically-based pharmacokinetic modeling approach. Eur. J. Pharm. Sci. 2018, 112, 168–179. [Google Scholar] [CrossRef]
- Uchida, Y.; Wakayama, K.; Ohtsuki, S.; Chiba, M.; Ohe, T.; Ishii, Y.; Terasaki, T. Blood-Brain Barrier Pharmacoproteomics-Based Reconstruction of the In Vivo Brain Distribution of P-Glycoprotein Substrates in Cynomolgus Monkeys. J. Pharmacol. Exp. Ther. 2014, 350, 578–588. [Google Scholar] [CrossRef]
- Trapa, P.E.; Troutman, M.D.; Lau, T.Y.; Wager, T.T.; Maurer, T.S.; Patel, N.C.; West, M.A.; Umland, J.P.; Carlo, A.A.; Feng, B.; et al. In Vitro–In Vivo Extrapolation of Key Transporter Activity at the Blood–Brain Barrier. Drug Metab. Dispos. 2019, 47, 405–411. [Google Scholar] [CrossRef]
- Al Feteisi, H.; Al-Majdoub, Z.M.; Achour, B.; Couto, N.; Rostami-Hodjegan, A.; Barber, J. Identification and quantification of blood–brain barrier transporters in isolated rat brain microvessels. J. Neurochem. 2018, 146, 670–685. [Google Scholar] [CrossRef]
- Chun, H.B.; Scott, M.; Niessen, S.; Hoover, H.; Baird, A.; Yates, J.; Torbett, B.E.; Eliceiri, B.P. The proteome of mouse brain microvessel membranes and basal lamina. J. Cereb. Blood Flow Metab. 2011, 31, 2267–2281. [Google Scholar] [CrossRef]
- Chasseigneaux, S.; Moraca, Y.; Cochois-Guégan, V.; Boulay, A.-C.; Gilbert, A.; Le Crom, S.; Blugeon, C.; Firmo, C.; Cisternino, S.; Laplanche, J.-L.; et al. Isolation and differential transcriptome of vascular smooth muscle cells and mid-capillary pericytes from the rat brain. Sci. Rep. 2018, 8, 12272. [Google Scholar] [CrossRef]
- Porte, B.; Chatelain, C.; Hardouin, J.; Derambure, C.; Zerdoumi, Y.; Hauchecorne, M.; Dupré, N.; Bekri, S.; Gonzalez, B.; Marret, S.; et al. Proteomic and transcriptomic study of brain microvessels in neonatal and adult mice. PLoS ONE 2017, 12, e0171048. [Google Scholar] [CrossRef]
- Karamanos, Y.; Gosselet, F.; Dehouck, M.-P.; Cecchelli, R. Blood–Brain Barrier Proteomics: Towards the Understanding of Neurodegenerative Diseases. Arch. Med. Res. 2014, 45, 730–737. [Google Scholar] [CrossRef]
- Uchida, Y.; Tachikawa, M.; Obuchi, W.; Hoshi, Y.; Tomioka, Y.; Ohtsuki, S.; Terasaki, T. A study protocol for quantitative targeted absolute proteomics (QTAP) by LC-MS/MS: Application for inter-strain differences in protein expression levels of transporters, receptors, claudin-5, and marker proteins at the blood--brain barrier in ddY, FVB, an. Fluids Barriers CNS 2013, 10, 21. [Google Scholar] [CrossRef]
- Gomez-Zepeda, D.; Taghi, M.; Smirnova, M.; Sergent, P.; Liu, W.; Chhuon, C.; Vidal, M.; Picard, M.; Thioulouse, E.; Broutin, I.; et al. LC–MS/MS-based quantification of efflux transporter proteins at the BBB. J. Pharm. Biomed. Anal. 2019, 164, 496–508. [Google Scholar] [CrossRef]
- Gerber, S.A.; Rush, J.; Stemman, O.; Kirschner, M.W.; Gygi, S.P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. USA 2003, 100, 6940–6945. [Google Scholar] [CrossRef]
- Billington, S.; Salphati, L.; Hop, C.E.C.A.; Chu, X.; Evers, R.; Burdette, D.; Rowbottom, C.; Lai, Y.; Xiao, G.; Humphreys, W.G.; et al. Interindividual and Regional Variability in Drug Transporter Abundance at the Human Blood–Brain Barrier Measured by Quantitative Targeted Proteomics. Clin. Pharmacol. Ther. 2019, 106, 228–237. [Google Scholar] [CrossRef]
- Prasad, B.; Achour, B.; Artursson, P.; Hop, C.E.C.A.; Lai, Y.; Smith, P.C.; Barber, J.; Wisniewski, J.R.; Spellman, D.; Uchida, Y.; et al. Toward a Consensus on Applying Quantitative Liquid Chromatography-Tandem Mass Spectrometry Proteomics in Translational Pharmacology Research: A White Paper. Clin. Pharmacol. Ther. 2019, 106, 525–543. [Google Scholar] [CrossRef]
- Plate, K.H.; Scholz, A.; Dumont, D.J. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012, 124, 763–775. [Google Scholar] [CrossRef]
- Ohgaki, H. Genetic Pathways to Glioblastoma: A Population-Based Study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef]
- Thompson, C.B. Metabolic Enzymes as Oncogenes or Tumor Suppressors. N. Engl. J. Med. 2009, 360, 813–815. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef]
- Sun, H.; Yin, L.; Li, S.; Han, S.; Song, G.; Liu, N.; Yan, C. Prognostic significance of IDH mutation in adult low-grade gliomas: A meta-analysis. J. Neurooncol. 2013, 113, 277–284. [Google Scholar] [CrossRef]
- Choi, C.; Ganji, S.K.; DeBerardinis, R.J.; Hatanpaa, K.J.; Rakheja, D.; Kovacs, Z.; Yang, X.-L.; Mashimo, T.; Raisanen, J.M.; Marin-Valencia, I.; et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat. Med. 2012, 18, 624–629. [Google Scholar] [CrossRef]
- Schumacher, T.; Bunse, L.; Pusch, S.; Sahm, F.; Wiestler, B.; Quandt, J.; Menn, O.; Osswald, M.; Oezen, I.; Ott, M.; et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014, 512, 324–327. [Google Scholar] [CrossRef]
- Forman, D.; Bray, F.; Brewster, D.H.; Gombe Mbalawa, C.; Kohler, B.; Piñeros, M.; Steliarova-Foucher, E.; Swaminathan, R.; Ferlay, J. (Eds.) Cancer Incidence in Five Continents Volume X; IARC Scientific Publication: Lyon, France, 2014; Volume 164, ISBN 978-92-832-2165-4. [Google Scholar]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A state of the science review. Neuro. Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Bauchet, L.; Mathieu-Daude, H.; Fabbro-Peray, P.; Rigau, V.; Fabbro, M.; Chinot, O.; Pallusseau, L.; Carnin, C.; Laine, K.; Schlama, A.; et al. Oncological patterns of care and outcome for 952 patients with newly diagnosed glioblastoma in 2004. Neuro. Oncol. 2010, 12, 725–735. [Google Scholar] [CrossRef]
- Dropcho, E.J.; Soong, S.J. The prognostic impact of prior low grade histology in patients with anaplastic gliomas: A case-control study. Neurology 1996, 47, 684–690. [Google Scholar] [CrossRef]
- Walsh, K.M.; Ohgaki, H.; Wrensch, M.R. Epidemiology. In Handbook of Clinical Neurology; Berger, M., Weller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 134, pp. 3–18. ISBN 9780128029978. [Google Scholar]
- Bredel, M.; Zentner, J. Brain-tumour drug resistance: The bare essentials. Lancet Oncol. 2002, 3, 397–406. [Google Scholar] [CrossRef]
- Shen, D.W.; Goldenberg, S.; Pastan, I.; Gottesman, M.M. Decreased accumulation of [14C]carboplatin in human cisplatin-resistant cells results from reduced energy-dependent uptake. J. Cell. Physiol. 2000, 183, 108–116. [Google Scholar] [CrossRef]
- Korashy, H.M.; Abuohashish, H.M.; Maayah, Z.H. The role of aryl hydrocarbon receptor-regulated cytochrome P450 enzymes in glioma. Curr. Pharm. Des. 2013, 19, 7155–7166. [Google Scholar] [CrossRef]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993, 74, 957–967. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001, 15, 719–730. [Google Scholar] [CrossRef]
- Ramirez, Y.; Weatherbee, J.; Wheelhouse, R.; Ross, A. Glioblastoma Multiforme Therapy and Mechanisms of Resistance. Pharmaceuticals 2013, 6, 1475–1506. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R.; Chen, Z.-S.; Ashby, C.R., Jr.; Chen, Z.-S. The modulation of ABC transporter-mediated multidrug resistance in cancer: A review of the past decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef]
- Machein, M.R.; Kullmer, J.; Fiebich, B.L.; Plate, K.H.; Warnke, P.C. Vascular endothelial growth factor expression, vascular volume, and, capillary permeability in human brain tumors. Neurosurgery 1999, 44, 732–740. [Google Scholar] [CrossRef]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef]
- Dhermain, F.G.; Hau, P.; Lanfermann, H.; Jacobs, A.H.; van den Bent, M.J.; van den Bent, M.; Vogelbaum, M.; Wen, P.; Macdonald, D.; Chang, S.; et al. Advanced MRI and PET imaging for assessment of treatment response in patients with gliomas. Lancet. Neurol. 2010, 9, 906–920. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro. Oncol. 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Agarwal, S.; Manchanda, P.; Vogelbaum, M.A.; Ohlfest, J.R.; Elmquist, W.F. Function of the Blood-Brain Barrier and Restriction of Drug Delivery to Invasive Glioma Cells: Findings in an Orthotopic Rat Xenograft Model of Glioma. Drug Metab. Dispos. 2013, 41, 33–39. [Google Scholar] [CrossRef]
- Parrish, K.E.; Cen, L.; Murray, J.; Calligaris, D.; Kizilbash, S.; Mittapalli, R.K.; Carlson, B.L.; Schroeder, M.A.; Sludden, J.; Boddy, A.V.; et al. Efficacy of PARP Inhibitor Rucaparib in Orthotopic Glioblastoma Xenografts Is Limited by Ineffective Drug Penetration into the Central Nervous System. Mol. Cancer Ther. 2015, 14, 2735–2743. [Google Scholar] [CrossRef]
- Parrish, K.E.; Pokorny, J.; Mittapalli, R.K.; Bakken, K.; Sarkaria, J.N.; Elmquist, W.F. Efflux Transporters at the Blood-Brain Barrier Limit Delivery and Efficacy of Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib (PD-0332991) in an Orthotopic Brain Tumor Model. J. Pharmacol. Exp. Ther. 2015, 355, 264–271. [Google Scholar] [CrossRef]
- Mittapalli, R.K.; Chung, A.H.; Parrish, K.E.; Crabtree, D.; Halvorson, K.G.; Hu, G.; Elmquist, W.F.; Becher, O.J. ABCG2 and ABCB1 Limit the Efficacy of Dasatinib in a PDGF-B-Driven Brainstem Glioma Model. Mol. Cancer Ther. 2016, 15, 819–829. [Google Scholar] [CrossRef]
- Randall, E.C.; Emdal, K.B.; Laramy, J.K.; Kim, M.; Roos, A.; Calligaris, D.; Regan, M.S.; Gupta, S.K.; Mladek, A.C.; Carlson, B.L.; et al. Integrated mapping of pharmacokinetics and pharmacodynamics in a patient-derived xenograft model of glioblastoma. Nat. Commun. 2018, 9, 4904. [Google Scholar] [CrossRef]
- Declèves, X.; Bihorel, S.; Debray, M.; Yousif, S.; Camenisch, G.; Scherrmann, J.M. ABC transporters and the accumulation of imatinib and its active metabolite CGP74588 in rat C6 glioma cells. Pharmacol. Res. 2008, 57, 214–222. [Google Scholar] [CrossRef]
- Declèves, X.; Fajac, A.; Lehmann-Che, J.; Tardy, M.; Mercier, C.; Hurbain, I.; Laplanche, J.-L.; Bernaudin, J.-F.; Scherrmann, J.-M. Molecular and functional MDR1-Pgp and MRPs expression in human glioblastoma multiforme cell lines. Int. J. Cancer 2002, 98, 173–180. [Google Scholar] [CrossRef]
- Breedveld, P. The Effect of Bcrp1 (Abcg2) on the In vivo Pharmacokinetics and Brain Penetration of Imatinib Mesylate (Gleevec): Implications for the Use of Breast Cancer Resistance Protein and P-Glycoprotein Inhibitors to Enable the Brain Penetration of Imatinib in Pat. Cancer Res. 2005, 65, 2577–2582. [Google Scholar] [CrossRef]
- Oberoi, R.K.; Mittapalli, R.K.; Elmquist, W.F. Pharmacokinetic Assessment of Efflux Transport in Sunitinib Distribution to the Brain. J. Pharmacol. Exp. Ther. 2013, 347, 755–764. [Google Scholar] [CrossRef]
- Balaji, S.A.; Udupa, N.; Chamallamudi, M.R.; Gupta, V.; Rangarajan, A. Role of the drug transporter ABCC3 in breast cancer chemoresistance. PLoS ONE 2016, 11, 1–22. [Google Scholar] [CrossRef]
- Calcagno, A.M.; Fostel, J.M.; To, K.K.W.; Salcido, C.D.; Martin, S.E.; Chewning, K.J.; Wu, C.-P.; Varticovski, L.; Bates, S.E.; Caplen, N.J.; et al. Single-step doxorubicin-selected cancer cells overexpress the ABCG2 drug transporter through epigenetic changes. Br. J. Cancer 2008, 98, 1515–1524. [Google Scholar] [CrossRef]
- Loging, W.T.; Lal, A.; Siu, I.M.; Loney, T.L.; Wikstrand, C.J.; Marra, M.A.; Prange, C.; Bigner, D.D.; Strausberg, R.L.; Riggins, G.J. Identifying potential tumor markers and antigens by database mining and rapid expression screening. Genome Res. 2000, 10, 1393–1402. [Google Scholar] [CrossRef]
- Haga, S.; Hinoshita, E.; Ikezaki, K.; Fukui, M.; Scheffer, G.L.; Uchiumi, T.; Kuwano, M. Involvement of the multidrug resistance protein 3 in drug sensitivity and its expression in human glioma. Jpn. J. Cancer Res. 2001, 92, 211–219. [Google Scholar] [CrossRef]
- Kuan, C.-T.; Wakiya, K.; Herndon, J.E.; Lipp, E.S.; Pegram, C.N.; Riggins, G.J.; Rasheed, A.; Szafranski, S.E.; McLendon, R.E.; Wikstrand, C.J.; et al. MRP3: A molecular target for human glioblastoma multiforme immunotherapy. BMC Cancer 2010, 10, 468. [Google Scholar] [CrossRef]
- Wang, F. Identification of CD44 and ABCC3 as chemotherapy resistant markers for Glioblastoma. Neuro. Oncol. 2016, 18, 54–55. [Google Scholar] [CrossRef][Green Version]
- Demeule, M.; Shedid, D.; Beaulieu, E.; Del Maestro, R.F.; Moghrabi, A.; Ghosn, P.B.; Moumdjian, R.; Berthelet, F.; Béliveau, R. Expression of multidrug-resistance P-glycoprotein (MDR1) in human brain tumors. Int. J. cancer 2001, 93, 62–66. [Google Scholar] [CrossRef]
- Benyahia, B.; Huguet, S.; Declèves, X.; Mokhtari, K.; Crinière, E.; Bernaudin, J.F.; Scherrmann, J.M.; Delattre, J.Y. Multidrug resistance-associated protein MRP1 expression in human gliomas: Chemosensitization to vincristine and etoposide by indomethacin in human glioma cell lines overexpressing MRP1. J. Neurooncol. 2004, 66, 65–70. [Google Scholar] [CrossRef]
- Kirches, E.; Oda, Y.; Von Bossanyi, P.; Diete, S.; Schneider, T.; Warich-Kirches, M.; Dietzmann, K. Mdr1 mRNA expression differs between grade III astrocytomas and glioblastomas. Clin. Neuropathol. 1997, 16, 34–36. [Google Scholar]
- Liebner, S.; Fischmann, A.; Rascher, G.; Duffner, F.; Grote, E.-H.; Kalbacher, H.; Wolburg, H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000, 100, 323–331. [Google Scholar] [CrossRef]
- Anfuso, C.D.; Motta, C.; Giurdanella, G.; Arena, V.; Alberghina, M.; Lupo, G. Endothelial PKCα-MAPK/ERK-phospholipase A2 pathway activation as a response of glioma in a triple culture model. A new role for pericytes? Biochimie 2014, 99, 77–87. [Google Scholar] [CrossRef]
- Walter, F.R.; Veszelka, S.; Pásztói, M.; Péterfi, Z.A.; Tóth, A.; Rákhely, G.; Cervenak, L.; Ábrahám, C.S.; Deli, M.A. Tesmilifene modifies brain endothelial functions and opens the blood-brain/blood-glioma barrier. J. Neurochem. 2015, 134, 1040–1054. [Google Scholar] [CrossRef]
- Raub, T.J.; Kuentzel, S.L.; Sawada, G.A. Permeability of bovine brain microvessel endothelial cells in vitro: Barrier tightening by a factor released from astroglioma cells. Exp. Cell Res. 1992, 199, 330–340. [Google Scholar] [CrossRef]
- DeBault, L.E.; Cancilla, P.A. γ-glutamyl transpeptidase in isolated brain endothelial cells: Induction by glial cells in vitro. Science 1980, 207, 653–655. [Google Scholar] [CrossRef]
- Terrell-Hall, T.B.; Ammer, A.G.; Griffith, J.I.G.; Lockman, P.R. Permeability across a novel microfluidic blood-tumor barrier model. Fluids Barriers CNS 2017, 14, 1–10. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.; Yang, J.; Hagan, J.P.; Li, M. Vertebrate animal models of glioma: Understanding the mechanisms and developing new therapies. Biochim. Biophys. Acta - Rev. Cancer 2013, 1836, 158–165. [Google Scholar] [CrossRef]
- Becher, O.J.; Holland, E.C. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006, 66, 3355–3358. [Google Scholar] [CrossRef]
- Reilly, K.M.; Loisel, D.A.; Bronson, R.T.; McLaughlin, M.E.; Jacks, T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat. Genet. 2000, 26, 109–113. [Google Scholar] [CrossRef]
- Hu, X.; Pandolfi, P.P.; Li, Y.; Koutcher, J.A.; Rosenblum, M.; Holland, E.C. mTOR Promotes Survival and Astrocytic Characteristics Induced by Pten/Akt Signaling in Glioblastoma. Neoplasia 2005, 7, 356–368. [Google Scholar] [CrossRef][Green Version]
- Dai, C.; Celestino, J.C.; Okada, Y.; Louis, D.N.; Fuller, G.N.; Holland, E.C. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001, 15, 1913–1925. [Google Scholar] [CrossRef]
- Xiao, A.; Wu, H.; Pandolfi, P.P.; Louis, D.N.; Van Dyke, T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell 2002, 1, 157–168. [Google Scholar] [CrossRef][Green Version]
- Chen, P.-Y.; Ozawa, T.; Drummond, D.C.; Kalra, A.; Fitzgerald, J.B.; Kirpotin, D.B.; Wei, K.-C.; Butowski, N.; Prados, M.D.; Berger, M.S.; et al. Comparing routes of delivery for nanoliposomal irinotecan shows superior anti-tumor activity of local administration in treating intracranial glioblastoma xenografts. Neuro. Oncol. 2013, 15, 189–197. [Google Scholar] [CrossRef]
- Yamashita, Y.; Krauze, M.T.; Kawaguchi, T.; Noble, C.O.; Drummond, D.C.; Park, J.W.; Bankiewicz, K.S. Convection-enhanced delivery of a topoisomerase I inhibitor (nanoliposomal topotecan) and a topoisomerase II inhibitor (pegylated liposomal doxorubicin) in intracranial brain tumor xenografts1. Neuro. Oncol. 2007, 9, 20–28. [Google Scholar] [CrossRef]
- Takamiya, Y.; Abe, Y.; Tanaka, Y.; Tsugu, A.; Kazuno, M.; Oshika, Y.; Maruo, K.; Ohnishi, Y.; Sato, O.; Yamazaki, H.; et al. Murine P-glycoprotein on stromal vessels mediates multidrug resistance in intracerebral human glioma xenografts. Br. J. Cancer 1997, 76, 445–450. [Google Scholar] [CrossRef]
- Schlageter, K.E.; Molnar, P.; Lapin, G.D.; Groothuis, D.R. Microvessel Organization and Structure in Experimental Brain Tumors: Microvessel Populations with Distinctive Structural and Functional Properties. Microvasc. Res. 1999, 58, 312–328. [Google Scholar] [CrossRef]
- Tanaka, Y.; Abe, Y.; Tsugu, A.; Takamiya, Y.; Akatsuka, A.; Tsuruo, T.; Yamazaki, H.; Ueyama, Y.; Sato, O.; Tamaoki, N. Ultrastructural localization of P-glycoprotein on capillary endothelial cells in human gliomas. Virchows Arch. 1994, 425, 133–138. [Google Scholar] [CrossRef]
- Pardridge, W.M. CSF, blood-brain barrier, and brain drug delivery. Expert Opin. Drug Deliv. 2016, 13, 963–975. [Google Scholar] [CrossRef]
- Parrish, K.; Sarkaria, J.; Elmquist, W. Improving drug delivery to primary and metastatic brain tumors: Strategies to overcome the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 336–346. [Google Scholar] [CrossRef]
- Bush, N.A.O.; Chang, S.M.; Berger, M.S. Current and future strategies for treatment of glioma. Neurosurg. Rev. 2017, 40, 1–14. [Google Scholar] [CrossRef]
- Oberoi, R.K.; Parrish, K.E.; Sio, T.T.; Mittapalli, R.K.; Elmquist, W.F.; Sarkaria, J.N. Strategies to improve delivery of anticancer drugs across the blood-brain barrier to treat glioblastoma. Neuro. Oncol. 2016, 18, 27–36. [Google Scholar] [CrossRef]
- Pennock, G.D.; Dalton, W.S.; Roeske, W.R.; Appleton, C.P.; Mosley, K.; Plezia, P.; Miller, T.P.; Salmon, S.E. Systemic Toxic Effects Associated With High-Dose Verapamil Infusion and Chemotherapy Administration. JNCI J. Natl. Cancer Inst. 1991, 83, 105–110. [Google Scholar] [CrossRef]
- Ferry, D.R.; Traunecker, H.; Kerr, D.J. Clinical trials of p-glycoprotein reversal in solid tumours. Eur. J. Cancer 1996, 32, 1070–1081. [Google Scholar] [CrossRef]
- de Bruin, M.; Miyake, K.; Litman, T.; Robey, R.; Bates, S.E. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 1999, 146, 117–126. [Google Scholar] [CrossRef]
- Tsvankin, V.; Hashizume, R.; Katagi, H.; Herndon, J.E.; Lascola, C.; Venkatraman, T.N.; Picard, D.; Burrus, B.; Becher, O.J.; Thompson, E.M. ABC Transporter Inhibition Plus Dexamethasone Enhances the Efficacy of Convection Enhanced Delivery in H3.3K27M Mutant Diffuse Intrinsic Pontine Glioma. Neurosurgery 2019, 92, nyz212. [Google Scholar] [CrossRef]
- Da Silva, C.G.; Peters, G.J.; Ossendorp, F.; Cruz, L.J. The potential of multi-compound nanoparticles to bypass drug resistance in cancer. Cancer Chemother. Pharmacol. 2017, 80, 881–894. [Google Scholar] [CrossRef]
- Choo, E.F.; Kurnik, D.; Muszkat, M.; Ohkubo, T.; Shay, S.D.; Higginbotham, J.N.; Glaeser, H.; Kim, R.B.; Wood, A.J.J.; Wilkinson, G.R. Differential in vivo sensitivity to inhibition of P-glycoprotein located in lymphocytes, testes, and the blood-brain barrier. J. Pharmacol. Exp. Ther. 2006, 317, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Rabindran, S.K.; Ross, D.D.; Doyle, L.A.; Yang, W.; Greenberger, L.M. Fumitremorgin C reverses multidrug resistance in cells transfected with the breast cancer resistance protein. Cancer Res. 2000, 60, 47–50. [Google Scholar] [PubMed]
- Allen, J.D.; van Loevezijn, A.; Lakhai, J.M.; van der Valk, M.; van Tellingen, O.; Reid, G.; Schellens, J.H.M.; Koomen, G.-J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar]
- Shukla, S.; Sauna, Z.E.; Ambudkar, S. V Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2008, 22, 445–447. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J. Short Communication Nucleoside, Nucleotide, and Non-Nucleoside Reverse ABSTRACT. Drug Metab. Dispos. 2007, 35, 340–344. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar]
- Li, K.; Lai, H. TanshinoneIIA enhances the chemosensitivity of breast cancer cells to doxorubicin through down-regulating the expression of MDR-related ABC transporters. Biomed. Pharmacother. 2017, 96, 371–377. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Zhou, W.; Yang, M.; Ding, Y.; Wang, Q.; Hu, R. Fasudil increases temozolomide sensitivity and suppresses temozolomide-resistant glioma growth via inhibiting ROCK2/ABCG2. Cell Death Dis. 2018, 9, 190. [Google Scholar] [CrossRef]
- Chowdhury, E.H. Strategies for tumor-directed delivery of siRNA. Expert Opin. Drug Deliv. 2011, 8, 389–401. [Google Scholar] [CrossRef]
- Wu, H.; Hait, W.N.; Yang, J.-M. Small interfering RNA-induced suppression of MDR1 (P-glycoprotein) restores sensitivity to multidrug-resistant cancer cells. Cancer Res. 2003, 63, 1515–1519. [Google Scholar]
- Ee, P.L.R.; He, X.; Ross, D.D.; Beck, W.T. Modulation of breast cancer resistance protein (BCRP/ABCG2) gene expression using RNA interference. Mol. Cancer Ther. 2004, 3, 1577–1583. [Google Scholar]
- Li, Y.T.; Chua, M.J.; Kunnath, A.P.; Chowdhury, E.H. Reversing multidrug resistance in breast cancer cells by silencing ABC transporter genes with nanoparticle-facilitated delivery of target siRNAs. Int. J. Nanomed. 2012, 7, 2473–2481. [Google Scholar] [CrossRef][Green Version]
- Michalak, K.; Wesolowska, O. Polyphenols Counteract Tumor Cell Chemoresistance Conferred by Multidrug Resistance Proteins. Anticancer. Agents Med. Chem. 2012, 12, 880–890. [Google Scholar] [CrossRef]
- Huang, X.-C.; Xiao, X.; Zhang, Y.-K.; Talele, T.; Salim, A.; Chen, Z.-S.; Capon, R. Lamellarin O, a Pyrrole Alkaloid from an Australian Marine Sponge, Ianthella sp., Reverses BCRP Mediated Drug Resistance in Cancer Cells. Mar. Drugs 2014, 12, 3818–3837. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; Martinez-Luis, S. Marine Natural Products with P-Glycoprotein Inhibitor Properties. Mar. Drugs 2014, 12, 525–546. [Google Scholar] [CrossRef]
- Pan, M.; Cui, J.; Jiao, L.; Ghaleb, H.; Liao, C.; Zhou, J.; Kairuki, M.; Lin, H.; Huang, W.; Qian, H. Synthesis and biological evaluation of JL-A7 derivatives as potent ABCB1 inhibitors. Bioorganic Med. Chem. 2017. [Google Scholar] [CrossRef]
- Gampa, G.; Vaidhyanathan, S.; Sarkaria, J.N.; Elmquist, W.F. Drug delivery to melanoma brain metastases: Can current challenges lead to new opportunities? Pharmacol. Res. 2017, 123, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Salphati, L.; Heffron, T.P.; Alicke, B.; Nishimura, M.; Barck, K.; Carano, R.A.; Cheong, J.; Edgar, K.A.; Greve, J.; Kharbanda, S.; et al. Targeting the PI3K Pathway in the Brain--Efficacy of a PI3K Inhibitor Optimized to Cross the Blood-Brain Barrier. Clin. Cancer Res. 2012, 18, 6239–6248. [Google Scholar] [CrossRef] [PubMed]
- Salphati, L.; Alicke, B.; Heffron, T.P.; Shahidi-Latham, S.; Nishimura, M.; Cao, T.; Carano, R.A.; Cheong, J.; Greve, J.; Koeppen, H.; et al. Brain distribution and efficacy of the brain penetrant PI3K inhibitor GDC-0084 in orthotopic mouse models of human glioblastoma. Drug Metab. Dispos. 2016, 44, 1881–1889. [Google Scholar] [CrossRef]
- Salphati, L.; Shahidi-Latham, S.; Quiason, C.; Barck, K.; Nishimura, M.; Alicke, B.; Pang, J.; Carano, R.A.; Olivero, A.G.; Phillips, H.S. Distribution of the phosphatidylinositol 3-kinase inhibitors pictilisib (GDC-0941) and GNE-317 in U87 and GS2 intracranial glioblastoma models - Assessment by matrix-assisted laser desorption ionization imaging. Drug Metab. Dispos. 2014, 42, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.M.; Oberoi, R.K.; McFarren, S.J.; Muldoon, D.M.; Pafundi, D.H.; Pokorny, J.L.; Brinkmann, D.H.; Ohlfest, J.R.; Sarkaria, J.N.; Largaespada, D.A.; et al. Decreased affinity for efflux transporters increases brain penetrance and molecular targeting of a PI3K/mTOR inhibitor in a mouse model of glioblastoma. Neuro. Oncol. 2015, 17, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- NIH ClinicalTrials.gov. Safety, Pharmacokinetics and Efficacy of GDC-0084 in Newly-Diagnosed Glioblastoma Multiforme. Available online: https://clinicaltrials.gov/ct2/show/NCT03522298 (accessed on 12 November 2019).
- Wait, S.D.; Prabhu, R.S.; Burri, S.H.; Atkins, T.G.; Asher, A.L. Polymeric drug delivery for the treatment of glioblastoma. Neuro. Oncol. 2015, 17, ii9–ii23. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Hilt, D.C.; Bortey, E.; Delavault, P.; Olivares, R.; Warnke, P.C.; Whittle, I.R.; Jääskeläinen, J.; Ram, Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro. Oncol. 2003, 5, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Brem, H.; Piantadosi, S.; Burger, P.; Walker, M.; Selker, R.; Vick, N.; Black, K.; Sisti, M.; Brem, S.; Mohr, G.; et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. Lancet 1995, 345, 1008–1012. [Google Scholar] [CrossRef]
- Valtonen, S.; la Timonen, U.; Toivanen, P.; Kalimo, H.; Kivipelto, L.; Heiskanen, O.; Unsgaard, G.; Kuurne, T. Interstitial Chemotherapy with Carmustine-loaded Polymers for High-grade Gliomas: A Randomized Double-blind Study. Neurosurgery 1997, 41, 44–49. [Google Scholar] [CrossRef]
- Bregy, A.; Shah, A.H.; Diaz, M.V.; Pierce, H.E.; Ames, P.L.; Diaz, D.; Komotar, R.J. The role of Gliadel wafers in the treatment of high-grade gliomas. Expert Rev. Anticancer Ther. 2013, 13, 1453–1461. [Google Scholar] [CrossRef]
- Morrison, P.F.; Laske, D.W.; Bobo, H.; Oldfield, E.H.; Dedrick, R.L. High-flow microinfusion: Tissue penetration and pharmacodynamics. Am. J. Physiol. Integr. Comp. Physiol. 1994, 266, R292–R305. [Google Scholar] [CrossRef]
- Bobo, R.H.; Laske, D.W.; Akbasak, A.; Morrison, P.F.; Dedrick, R.L.; Oldfield, E.H. Convection-enhanced delivery of macromolecules in the brain. Proc. Natl. Acad. Sci. USA 1994, 91, 2076–2080. [Google Scholar] [CrossRef]
- Lonser, R.R.; Sarntinoranont, M.; Morrison, P.F.; Oldfield, E.H. Convection-enhanced delivery to the central nervous system. J. Neurosurg. 2015, 122, 697–706. [Google Scholar] [CrossRef]
- Shi, M.; Sanche, L. Convection-Enhanced Delivery in Malignant Gliomas: A Review of Toxicity and Efficacy. J. Oncol. 2019, 2019, 1–13. [Google Scholar] [CrossRef]
- Sampson, J.H.; Archer, G.; Pedain, C.; Wembacher-Schröder, E.; Westphal, M.; Kunwar, S.; Vogelbaum, M.A.; Coan, A.; Herndon, J.E.; Raghavan, R.; et al. Poor drug distribution as a possible explanation for the results of the PRECISE trial. J. Neurosurg. 2010, 113, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Asthagiri, A.R.; Walbridge, S.; Heiss, J.D.; Lonser, R.R. Effect of concentration on the accuracy of convective imaging distribution of a gadolinium-based surrogate tracer. J. Neurosurg. 2011, 115, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Chittiboina, P.; Heiss, J.D.; Warren, K.E.; Lonser, R.R. Magnetic resonance imaging properties of convective delivery in diffuse intrinsic pontine gliomas. J. Neurosurg. Pediatr. 2014, 13, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro. Oncol. 2010, 12, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sivakumar, W.; Torres, S.; Jhaveri, N.; Vaikari, V.P.; Gong, A.; Howard, A.; Golden, E.B.; Louie, S.G.; Schönthal, A.H.; et al. Effects of convection-enhanced delivery of bevacizumab on survival of glioma-bearing animals. Neurosurg. Focus 2015, 38, E8. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.S.; Neira, J.A.; Yun, J.; Alexiades, N.G.; Banu, M.; Englander, Z.K.; Kennedy, B.C.; Ung, T.H.; Rothrock, R.J.; Romanov, A.; et al. Validation of an effective implantable pump-infusion system for chronic convection-enhanced delivery of intracerebral topotecan in a large animal model. J. Neurosurg. 2019, 2, 1–10. [Google Scholar] [CrossRef]
- Neuwelt, E.A.; Maravilla, K.R.; Frenkel, E.P.; Rapaport, S.I.; Hill, S.A.; Barnett, P.A. Osmotic blood-brain barrier disruption. Computerized tomographic monitoring of chemotherapeutic agent delivery. J. Clin. Invest. 1979, 64, 684–688. [Google Scholar] [CrossRef]
- Angelov, L.; Doolittle, N.D.; Kraemer, D.F.; Siegal, T.; Barnett, G.H.; Peereboom, D.M.; Stevens, G.; McGregor, J.; Jahnke, K.; Lacy, C.A.; et al. Blood-Brain Barrier Disruption and Intra-Arterial Methotrexate-Based Therapy for Newly Diagnosed Primary CNS Lymphoma: A Multi-Institutional Experience. J. Clin. Oncol. 2009, 27, 3503–3509. [Google Scholar] [CrossRef]
- Chakraborty, S.; Filippi, C.G.; Wong, T.; Ray, A.; Fralin, S.; Tsiouris, A.J.; Praminick, B.; Demopoulos, A.; McCrea, H.J.; Bodhinayake, I.; et al. Superselective intraarterial cerebral infusion of cetuximab after osmotic blood/brain barrier disruption for recurrent malignant glioma: Phase I study. J. Neurooncol. 2016, 128, 405–415. [Google Scholar] [CrossRef]
- Burkhardt, J.-K.; Riina, H.; Shin, B.J.; Christos, P.; Kesavabhotla, K.; Hofstetter, C.P.; Tsiouris, A.J.; Boockvar, J.A. Intra-Arterial Delivery of Bevacizumab after Blood-Brain Barrier Disruption for the Treatment of Recurrent Glioblastoma: Progression-Free Survival and Overall Survival. World Neurosurg. 2012, 77, 130–134. [Google Scholar] [CrossRef]
- NIH ClinicalTrials.gov. Super-Selective Intraarterial Cerebral Infusion of Cetuximab (Erbitux) for Treatment of Relapsed/Refractory GBM and AA. Available online: https://clinicaltrials.gov/ct2/show/NCT01238237 (accessed on 12 November 2019).
- Rodriguez, A.; Tatter, S.; Debinski, W. Neurosurgical Techniques for Disruption of the Blood–Brain Barrier for Glioblastoma Treatment. Pharmaceutics 2015, 7, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Raymond, S.; Weissleder, R.; Jolesz, F.A.; Sheikov, N. Focal disruption of the blood–brain barrier due to 260-kHz ultrasound bursts: A method for molecular imaging and targeted drug delivery. J. Neurosurg. 2006, 105, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Etame, A.B.; Diaz, R.J.; Smith, C.A.; Mainprize, T.G.; Hynynen, K.; Rutka, J.T. Focused ultrasound disruption of the blood-brain barrier: A new frontier for therapeutic delivery in molecular neurooncology. Neurosurg. Focus 2012, 32, E3. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.-H.; Liu, H.-L.; Huang, C.-Y.; Ma, Y.-J.; Yen, T.-C.; Yeh, C.-K. Detection of Intracerebral Hemorrhage and Transient Blood-Supply Shortage in Focused-Ultrasound-Induced Blood–Brain Barrier Disruption by Ultrasound Imaging. Ultrasound Med. Biol. 2012, 38, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.-Y.; Fan, C.-H.; Liu, H.-L.; Huang, C.-Y.; Hsieh, H.-Y.; Yen, T.-C.; Wei, K.-C.; Yeh, C.-K. Concurrent blood–brain barrier opening and local drug delivery using drug-carrying microbubbles and focused ultrasound for brain glioma treatment. Biomaterials 2012, 33, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-K.; Santos, M.A.; Marcus, S.L.; Hynynen, K. MR-guided Focused Ultrasound Facilitates Sonodynamic Therapy with 5-Aminolevulinic Acid in a Rat Glioma Model. Sci. Rep. 2019, 9, 10465. [Google Scholar] [CrossRef]
- Horodyckid, C.; Canney, M.; Vignot, A.; Boisgard, R.; Drier, A.; Huberfeld, G.; François, C.; Prigent, A.; Santin, M.D.; Adam, C.; et al. Safe long-term repeated disruption of the blood-brain barrier using an implantable ultrasound device: A multiparametric study in a primate model. J. Neurosurg. 2017, 126, 1351–1361. [Google Scholar] [CrossRef]
- Beccaria, K.; Canney, M.; Goldwirt, L.; Fernandez, C.; Piquet, J.; Perier, M.; Lafon, C.; Chapelon, J.; Carpentier, A. Ultrasound-induced opening of the blood-brain barrier to enhance temozolomide and irinotecan delivery: An experimental study in rabbits. J. Neurosurg. 2016, 124, 1602–1610. [Google Scholar] [CrossRef]
- NIH ClinicalTrials.gov. Safety of BBB Opening With the SonoCloud (SONOCLOUD). Available online: https://clinicaltrials.gov/ct2/show/NCT02253212 (accessed on 12 November 2019).
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood–Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef]
- Teleanu, D.M.; Chircov, C.; Grumezescu, A.M.; Teleanu, R.I. Neuronanomedicine: An up-to-date overview. Pharmaceutics 2019, 11, 101. [Google Scholar] [CrossRef]
- Jiang, Y.; Lv, L.; Shi, H.; Hua, Y.; Lv, W.; Wang, X.; Xin, H.; Xu, Q. PEGylated Polyamidoamine dendrimer conjugated with tumor homing peptide as a potential targeted delivery system for glioma. Colloids Surfaces B Biointerfaces 2016, 147, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.K.; Gajbhiye, V.; Kesharwani, P.; Jain, N.K. Ligand anchored poly(propyleneimine) dendrimers for brain targeting: Comparative in vitro and in vivo assessment. J. Colloid Interface Sci. 2016, 482, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.K.; Adamo, B.; Karginova, O.; Deal, A.M.; Rawal, S.; Darr, D.; Schorzman, A.; Santos, C.; Bash, R.; Kafri, T.; et al. Pharmacokinetics and Efficacy of PEGylated Liposomal Doxorubicin in an Intracranial Model of Breast Cancer. PLoS ONE 2013, 8, e61359. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, D.; Kerklaan, B.M.; Diéras, V.; Altintas, S.; Anders, C.K.; Ballester, M.A.; Gelderblom, H.; Soetekouw, P.M.M.B.; Gladdines, W.; Lonnqvist, F.; et al. 472 Phase 1/2 a Study of Glutathione Pegylated Liposomal Doxorubicin (2B3-101) in Patients with Brain Metastases (BM) from Solid Tumors or Recurrent High Grade Gliomas (HGG). Ann. Oncol. 2014, 25, iv157–iv158. [Google Scholar] [CrossRef]
- Tian, C.; Asghar, S.; Xu, Y.; Chen, Z.; Zhang, J.; Ping, Q.; Xiao, Y. Tween 80-modified hyaluronic acid-ss-curcumin micelles for targeting glioma: Synthesis, characterization and their in vitro evaluation. Int. J. Biol. Macromol. 2018, 120, 2579–2588. [Google Scholar] [CrossRef]
- Kafa, H.; Wang, J.T.-W.; Rubio, N.; Klippstein, R.; Costa, P.M.; Hassan, H.A.F.M.; Sosabowski, J.K.; Bansal, S.S.; Preston, J.E.; Abbott, N.J.; et al. Translocation of LRP1 targeted carbon nanotubes of different diameters across the blood–brain barrier in vitro and in vivo. J. Control. Release 2016, 225, 217–229. [Google Scholar] [CrossRef]
- Duan, J.; Mansour, H.M.; Zhang, Y.; Deng, X.; Chen, Y.; Wang, J.; Pan, Y.; Zhao, J. Reversion of multidrug resistance by co-encapsulation of doxorubicin and curcumin in chitosan/poly(butyl cyanoacrylate) nanoparticles. Int. J. Pharm. 2012, 426, 193–201. [Google Scholar] [CrossRef]
- Pramanik, D.; Campbell, N.R.; Das, S.; Gupta, S.; Chenna, V.; Bisht, S.; Sysa-shah, P.; Bedja, D.; Karikari, C.; Steenbergen, C.; et al. A composite polymer nanoparticle overcomes multidrug resistance and ameliorates doxorubicin-associated cardiomyopathy. Oncotarget 2012, 3, 640–650. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.-L.; Qi, G.-B.; Chen, X.-G.; Wang, H. Dynamic disordering of liposomal cocktails and the spatio-temporal favorable release of cargoes to circumvent drug resistance. Biomaterials 2014, 35, 3406–3415. [Google Scholar] [CrossRef]





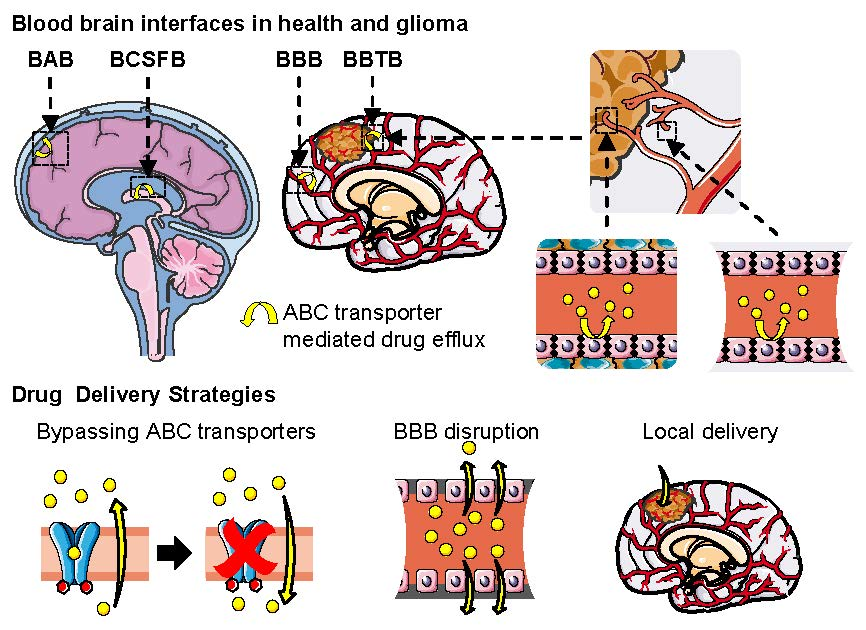{kind=link}
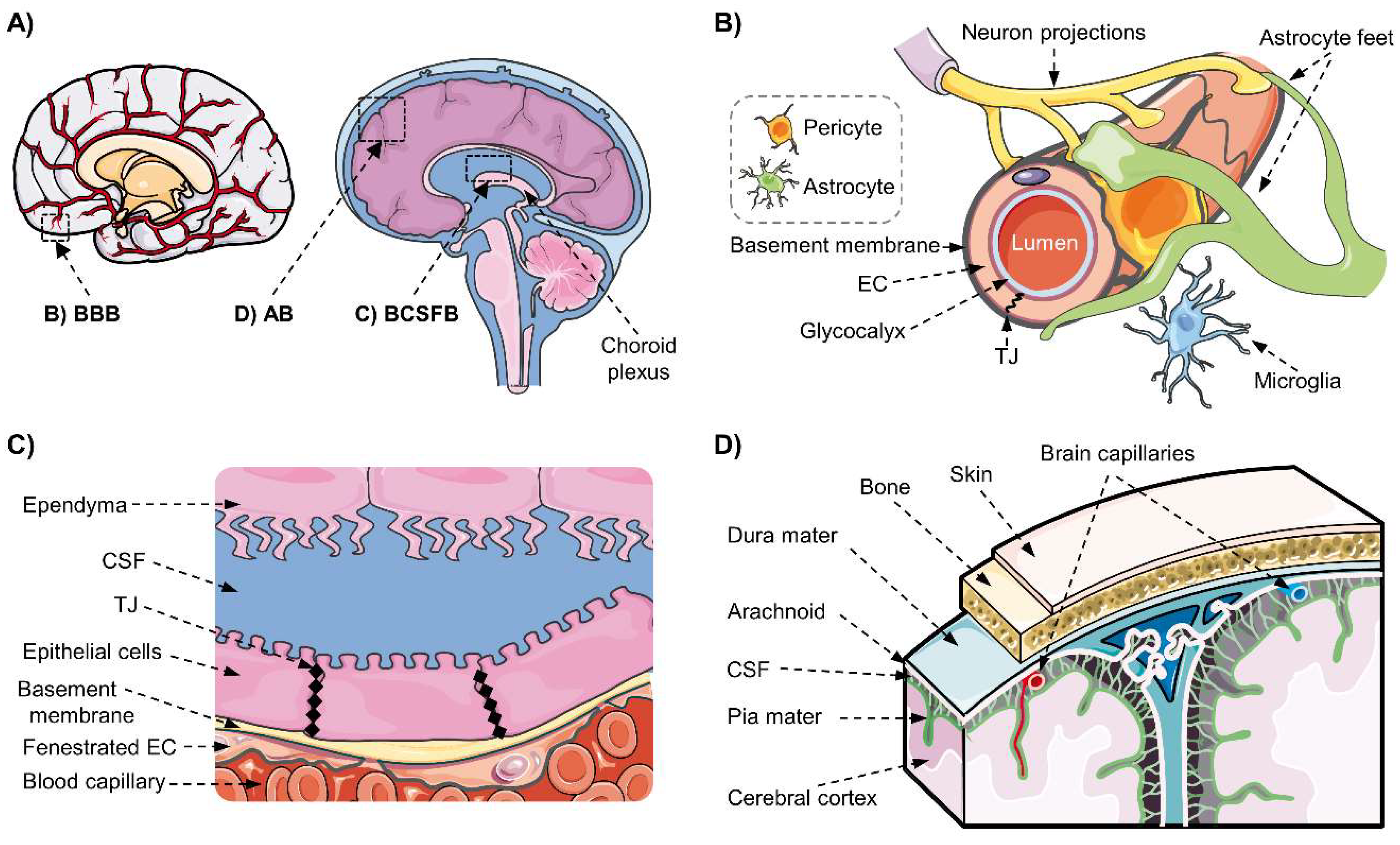{kind=link}
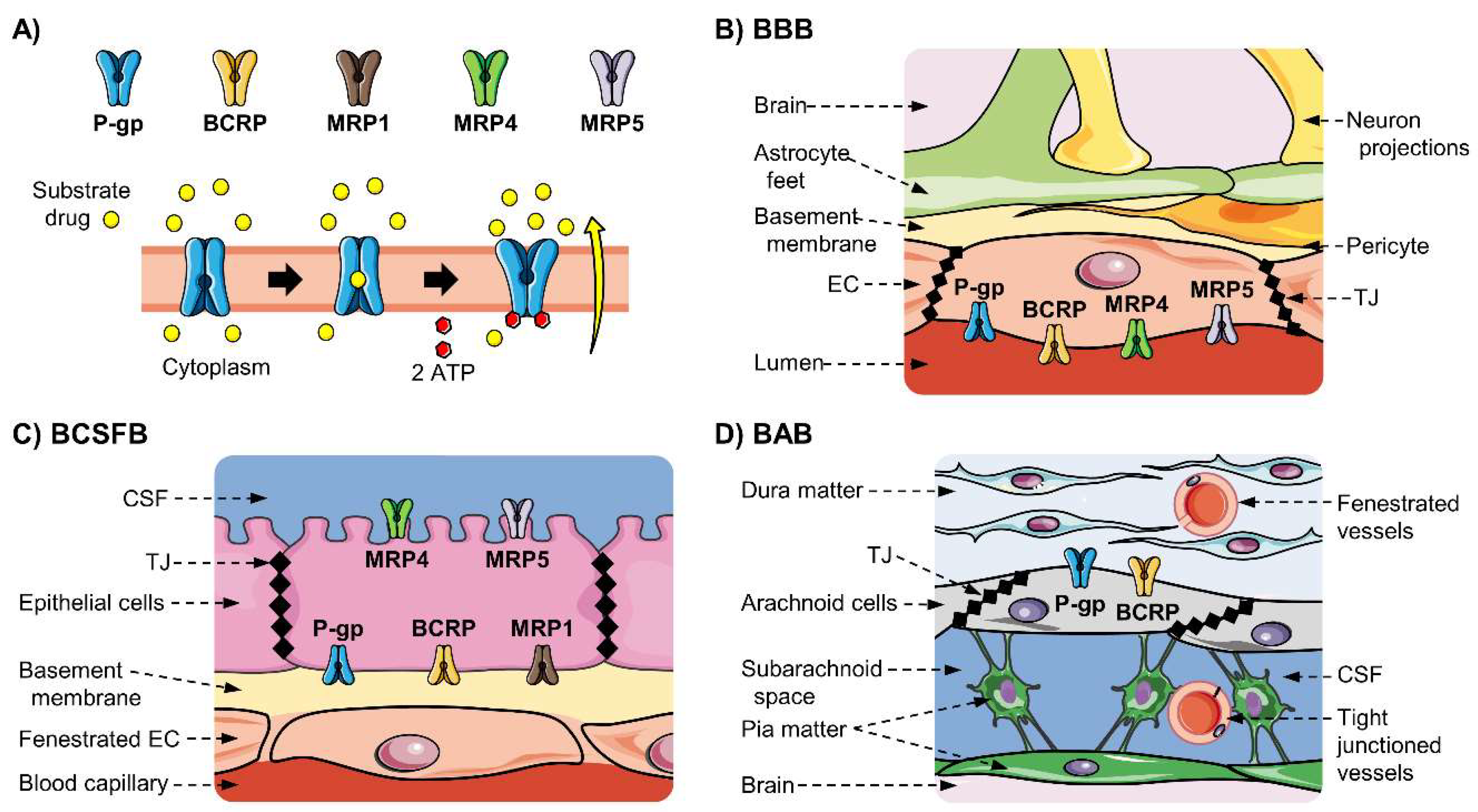{kind=link}
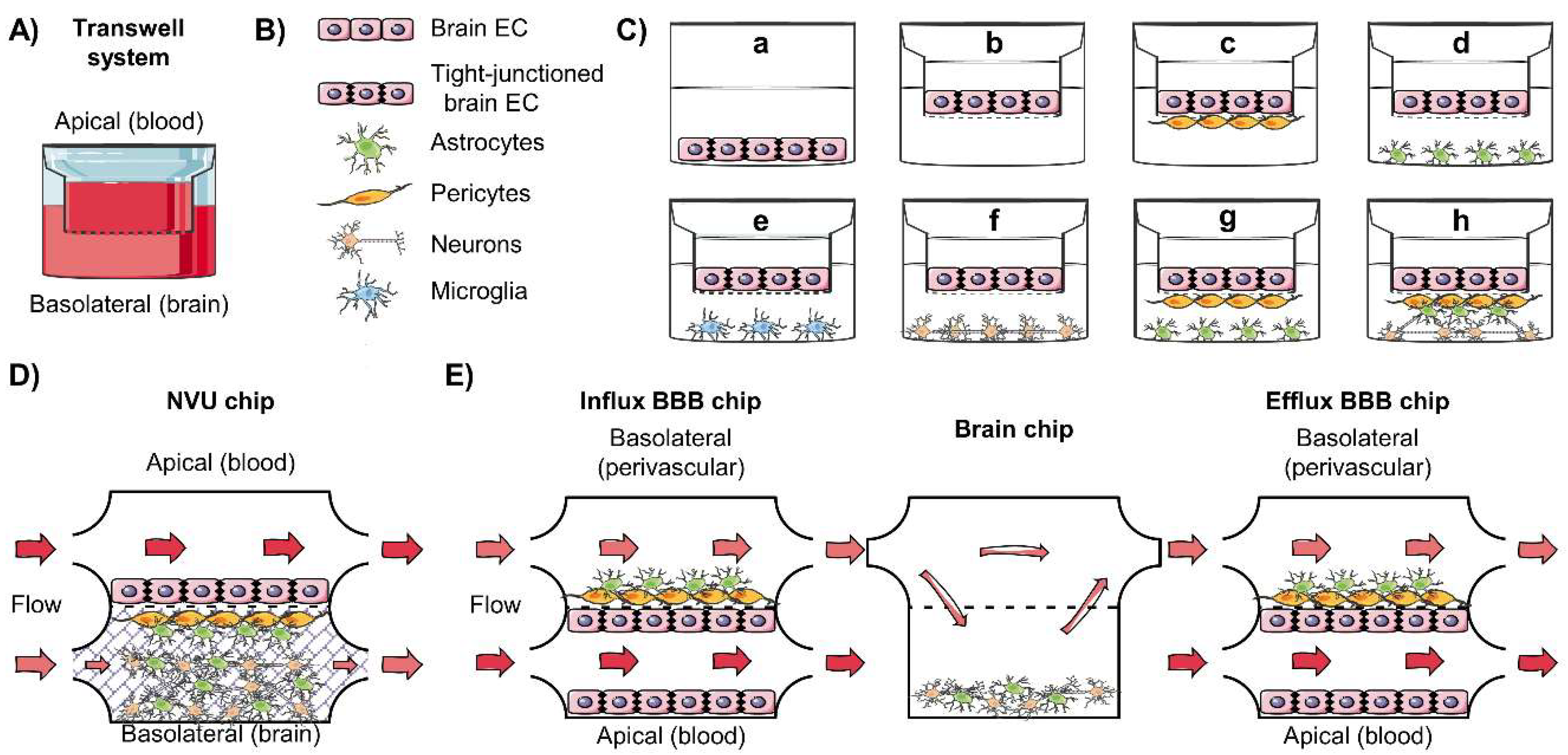{kind=link}
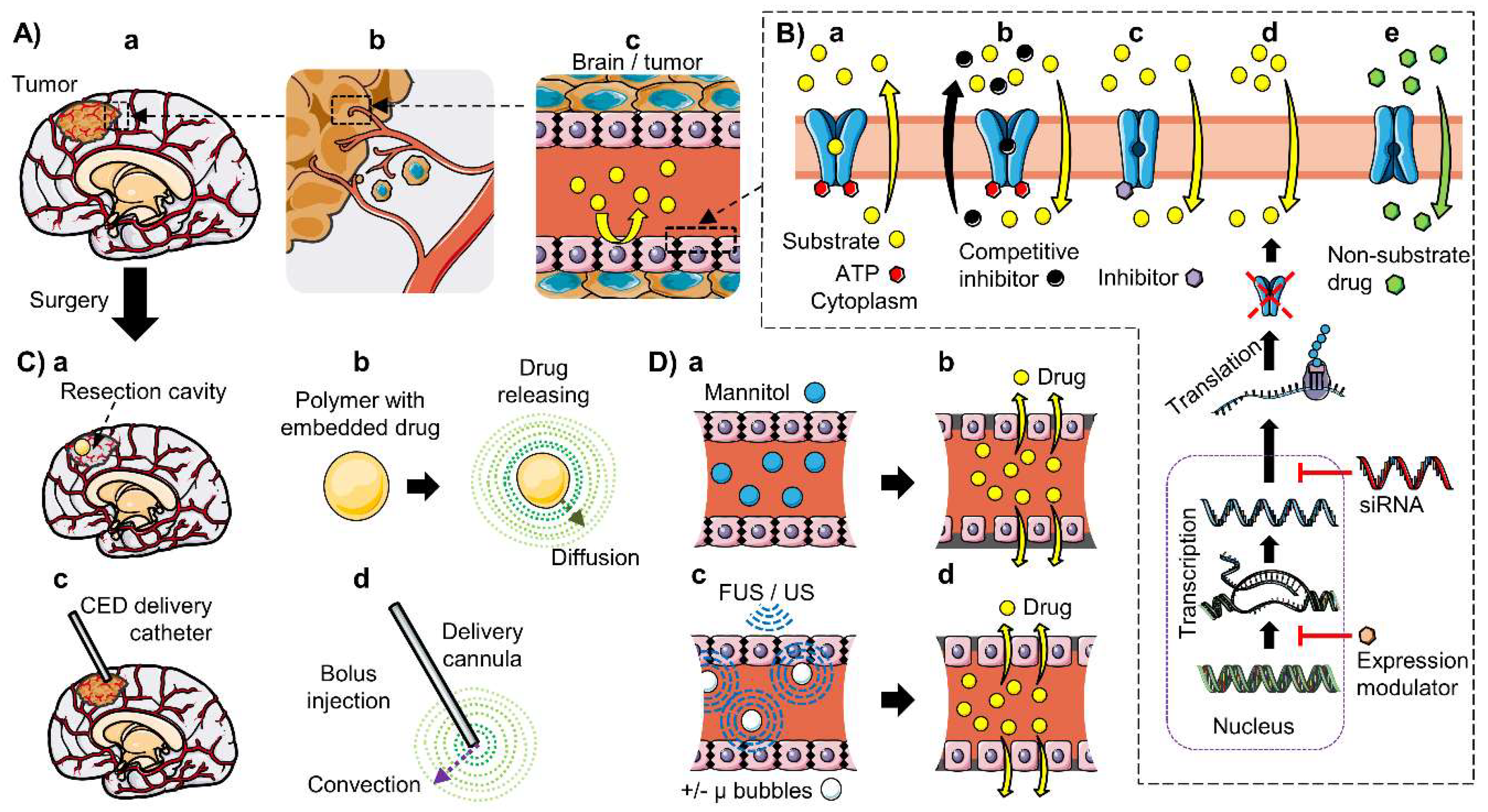{kind=link}
| Gene; Protein | Substrates Classes | Examples of Chemotherapeutics Substrates |
|---|---|---|
| ABCB1; P-gp MDR1 [16,42,50,51] | Amphipathic cations, organic molecules. No structure–activity relationship has been identified | Alkylating agents: temozolomide (TMZ) *, procarbazine *, carmustin * Topoisomerase inhibitors: etoposide *, topotecan *, irinotecan *, teniposide, doxorubicin, daunorubicin, carboplatin *, mitoxantrone Tyrosine kinase inhibitors: erlotinib *, dasatinib *, sunitinib *, sorafenib *, imatinib mesylate, gefitinib Anti-microtubule taxanes: paclitaxel *, docetaxel Dihydrofolate reductase inhibitor: methotrexate Vinca alkaloids: vinblastine *, vincristine * PARP1/2 inhibitor: veliparib (ABT-888) * MGMT inhibitor: lomeguatrib (O6Benzylguanine/O6BG) * |
| ABCG2; BCRP [16,42,52,53,54] | Partial overlap with P-gp substrates | Alkylating agents: temozolomide (TMZ) * Topoisomerase inhibitors: etoposide *, topotecan *, mitoxantrone, irinotecan *, SN-38, 9-aminocamptothecin, doxorubicin Tyrosine kinase inhibitors (TKI): erlotinib *, dasatinib *, sunitinib *, sorafenib *, imatinib, gefitinib, nilotinib PARP1/2 inhibitor: veliparib (ABT-888) * MGMT inhibitor: lomeguatrib (O6Benzylguanine/O6BG) * |
| ABBC1; MRP1 [16,55,56,57] | Organic anions, glutathione conjugates. Glutathione (GSH)-dependent | Alkylating agents: cyclophosphamide Topoisomerase inhibitors: doxorubicin, etoposide *, campathecin, camptothecin, irinotecan * (CPT-11) Anti-microtubule taxanes: paclitaxel * Dihydrofolate reductase inhibitor: methotrexate Vinca alkaloids: vinblastine *, vincristine * |
| ABCC2; MRP2 [16,55,56,57] | Organic anions, glutathione conjugates. Allosteric stimulation by bile acids, sulfinpyranzone, penicillin G, and indomethacin; but not GSH | Alkylating agents: chlorambucil, cyclophosphamide, cisplatin *, oxaliplatin Topoisomerase inhibitors: doxorubicin, etoposide *, epirubicin mitoxantrone, irinotecan *, glucuronidated SN-38 Vinca alkaloids: vinblastine *, vincristine * Antineoplastic, dihydrofolate reductase inhibitor: methotrexate Antineoplastic, angiotensin inhibitors: valsartan, olmesartan |
| ABCC3; MRP3 [16,55,56,57] | Organic anions, glutathione conjugates. Not stimulated by GSH nor bile acids | Alkylating agents: cisplatin * Antineoplastic, dihydrofolate reductase inhibitor: methotrexate Topoisomerase inhibitors: etoposide *, teniposide, doxorubicin Vinca alkaloids: vincristine * Conjugates: dinitrophenyl S-glutathione, acetaminophen glucuronide |
| ABBC4; MRP4 [16,55,56,57] | Organic anions, glutathione conjugates, cyclic nucleotides. GSH requirement depending on substrate; but not for cAMP or cGMP | Antineoplastic, dihydrofolate reductase inhibitor: methotrexate Topoisomerase inhibitors: topotecan * Nucleotide analogues: 6-mercaptopurine, 6-thioguanine |
| ABBC5; MRP5 [16,55,56,57] | Organic anions, glutathione conjugates, cyclic nucleotides. GSH requirement not exactly established, depending on substrate; but not for cAMP or cGMP | Antineoplastic, dihydrofolate reductase inhibitors: methotrexate Platinum-based drugs: cisplatin * Nucleotide analogues: 6-mercaptopurine, 6-thioguanine Conjugates: dinitrophenyl S-glutathione Heavy metals: cadmium chloride, potassium antimonyl tartrate |
| ABCC6; MRP6 [16,55,56,57] | Organic anions, glutathione conjugates. GSH requirement not stablished | Alkylating agents: cisplatin * Topoisomerase inhibitors: etoposide *, doxorubicin, daunorubicin |
| Gene; Protein | BBB | Parenchymal Cells | BCSFB | AB |
|---|---|---|---|---|
| ABCB1; P-gp/MDR1 Abcb1a and Abc1b Mdr1a and Mdr1b (r, m) | Luminal: h, r (Mdr1a), m (Mdr1a) mRNA and protein: h, r (Abcb1a), m (Abcb1a) [22,58,59,60,61,62,63] | Not detected in healthy tissue (h, r, m) [64,65,66,67,68,69] | Apical: h, r, m mRNA and protein: h, r (Abcb1a, Abcb1b), m (Abcb1a) [70,71,72,73] | Apical: h, r, m mRNA and protein: h, r, m [33,34,74,75,76,77] |
| ABCG2; BCRP | Luminal: h, r, m mRNA and protein: h, r, m, p [22,63,78,79,80,81,82,83] | Unclear mRNA and protein: Neuropil (h); cultured astrocytes (h, r) mRNA: Microglia (h, m) [65,68,84,85] | Apical: h, m mRNA and protein: h, r, m [20,33,70,81] | Apical: h, r, m mRNA and protein: h, r, m [33,34,74,75,76,77] |
| ABBC1; MRP1 | Luminal: h * protein: h * mRNA: h *, r, m, c (low) [46,86] | Not detected [64] | Basolateral: h, r, m Protein and mRNA: h, m, r [70,71,73,83,87,88,89] | mRNA: h, r, m Protein: r [33,34] |
| ABCC2; MRP2 | Luminal: r, m protein: r, m mRNA: r (low), m, c (low) [46,86,90,91] | mRNA and protein: neuropil, glial and neuronal cells (h) [64] | mRNA: h, r [87,88] | Not detected (h, r, m) [33,34] |
| ABCC3; MRP3 | mRNA: h * (low), r (low), m [46,86] | Not detected [64] | mRNA: h, r [87,88] | Not detected (h, r, m) [33,34] |
| ABBC4; MRP4 | Luminal: h, r, m Protein: h, r, m mRNA: h, r, m [22,46,86,92,93,94,95] | Not detected [64] | Basolateral: h, r, m Protein & mRNA: h, r, m [83,87,88,96] | mRNA: h, r, m Protein: r [34] |
| ABBC5; MRP5 | Luminal: h, r, m mRNA: h, r, m [46,86] | mRNA & protein: Neuropil (h) [64] | Basolateral: r mRNA & protein: h, r [83,87,88] | Not detected (h, r, m) [33,34] |
| ABCC6; MRP6 | mRNA: h *, r, m [46,86] | Not analyzed [64] | mRNA: h, r [87,88] | mRNA & protein: r [34] |
| Gene; Protein | Location in Human BRAIN Tumors |
|---|---|
| ABCB1; P-gp/MDR1 | Tumor capillaries; schwannomas, gangliogliomas, meningiomas, low-grade gliomas (astrocytomas, pilocytic astrocytomas) and high-grade gliomas (glioblastoma multiforme (GBM), anaplastic astrocytomas and anaplastic oligodendrogliomas) |
| ABBC1; MRP1 | Tumor capillaries, glioma cells, neuronal components of gangliosomas |
| ABCC2; MRP2 | ND |
| ABCC3; MRP3 | Anaplastic astrocytomas (grade III), GBM; cultured cancer and ECs from GBM |
| ABBC4; MRP4 | Tumor capillaries; astrocytic tumors; and astrocytic portions of oligoastrocytomas |
| ABBC5; MRP5 | Tumor capillaries; astrocytic tumors; and astrocytic portions of oligoastrocytomas |
| ABCC6; MRP6 | NAn |
| ABCG2; BCRP | Tumor capillaries; ND in glioma cells in situ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Zepeda, D.; Taghi, M.; Scherrmann, J.-M.; Decleves, X.; Menet, M.-C. ABC Transporters at the Blood–Brain Interfaces, Their Study Models, and Drug Delivery Implications in Gliomas. Pharmaceutics 2020, 12, 20. https://doi.org/10.3390/pharmaceutics12010020
Gomez-Zepeda D, Taghi M, Scherrmann J-M, Decleves X, Menet M-C. ABC Transporters at the Blood–Brain Interfaces, Their Study Models, and Drug Delivery Implications in Gliomas. Pharmaceutics. 2020; 12(1):20. https://doi.org/10.3390/pharmaceutics12010020
Chicago/Turabian StyleGomez-Zepeda, David, Méryam Taghi, Jean-Michel Scherrmann, Xavier Decleves, and Marie-Claude Menet. 2020. "ABC Transporters at the Blood–Brain Interfaces, Their Study Models, and Drug Delivery Implications in Gliomas" Pharmaceutics 12, no. 1: 20. https://doi.org/10.3390/pharmaceutics12010020
APA StyleGomez-Zepeda, D., Taghi, M., Scherrmann, J.-M., Decleves, X., & Menet, M.-C. (2020). ABC Transporters at the Blood–Brain Interfaces, Their Study Models, and Drug Delivery Implications in Gliomas. Pharmaceutics, 12(1), 20. https://doi.org/10.3390/pharmaceutics12010020