Fluorescence Imaging as a Tool in Preclinical Evaluation of Polymer-Based Nano-DDS Systems Intended for Cancer Treatment




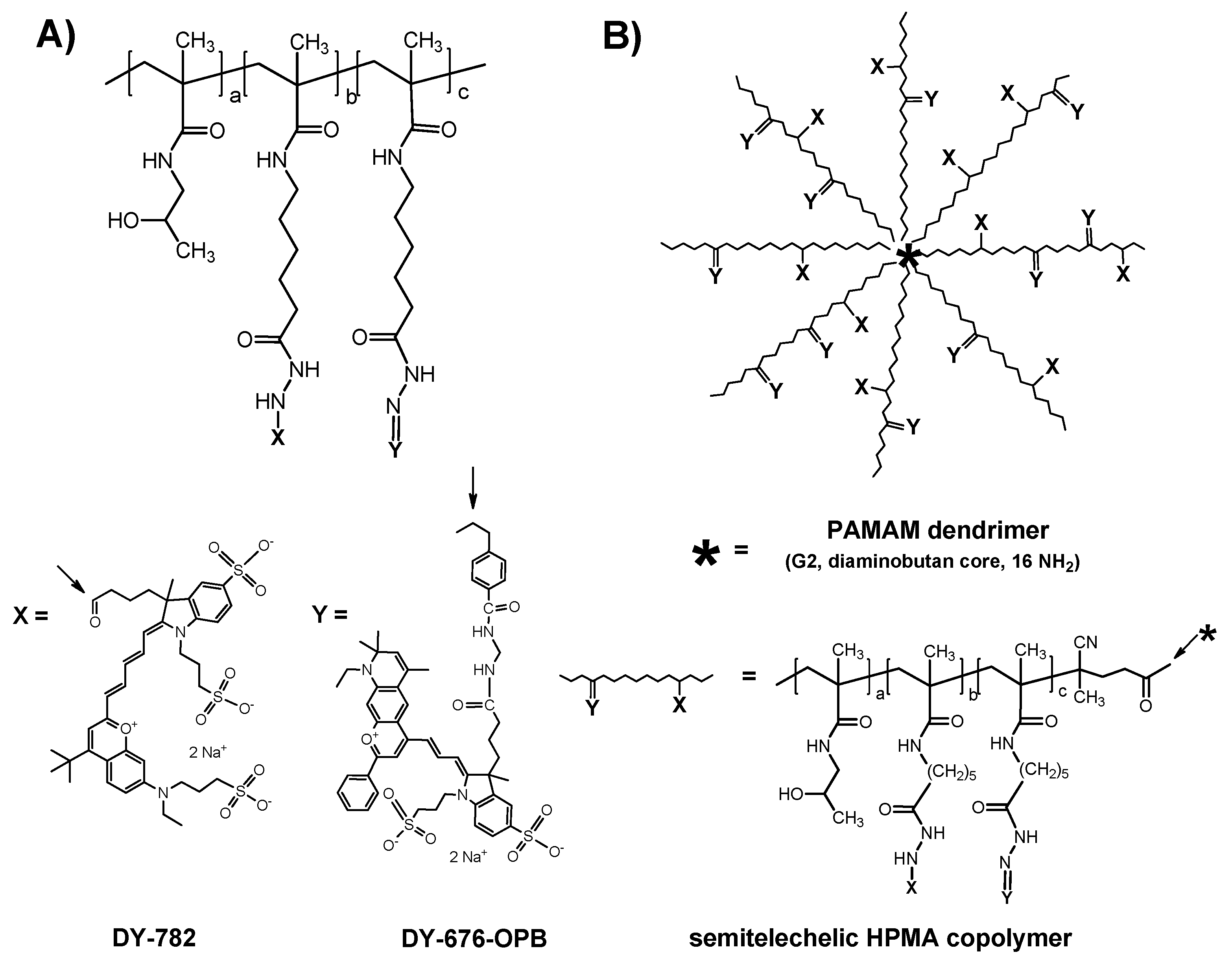{kind=link}
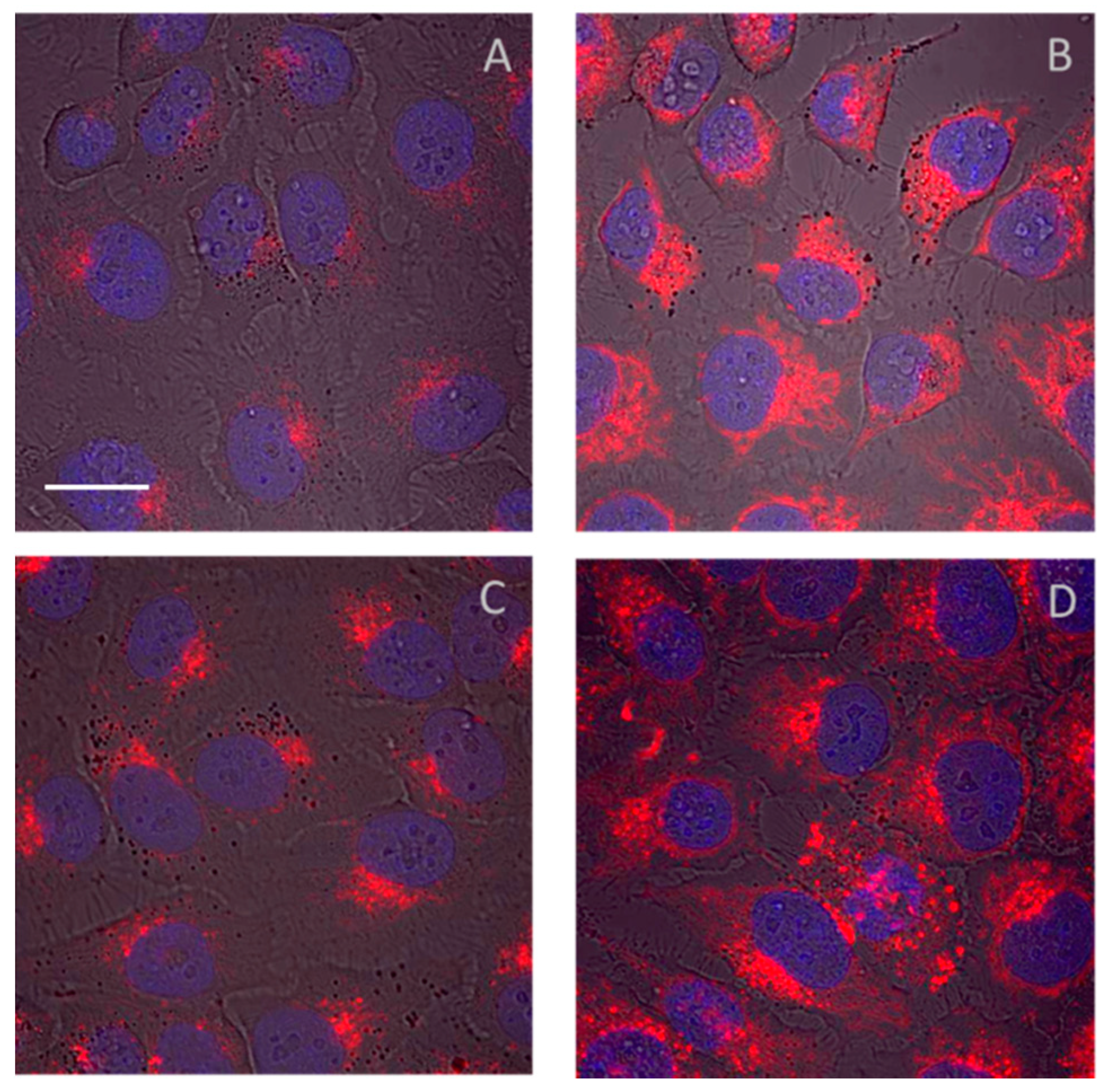{kind=link}
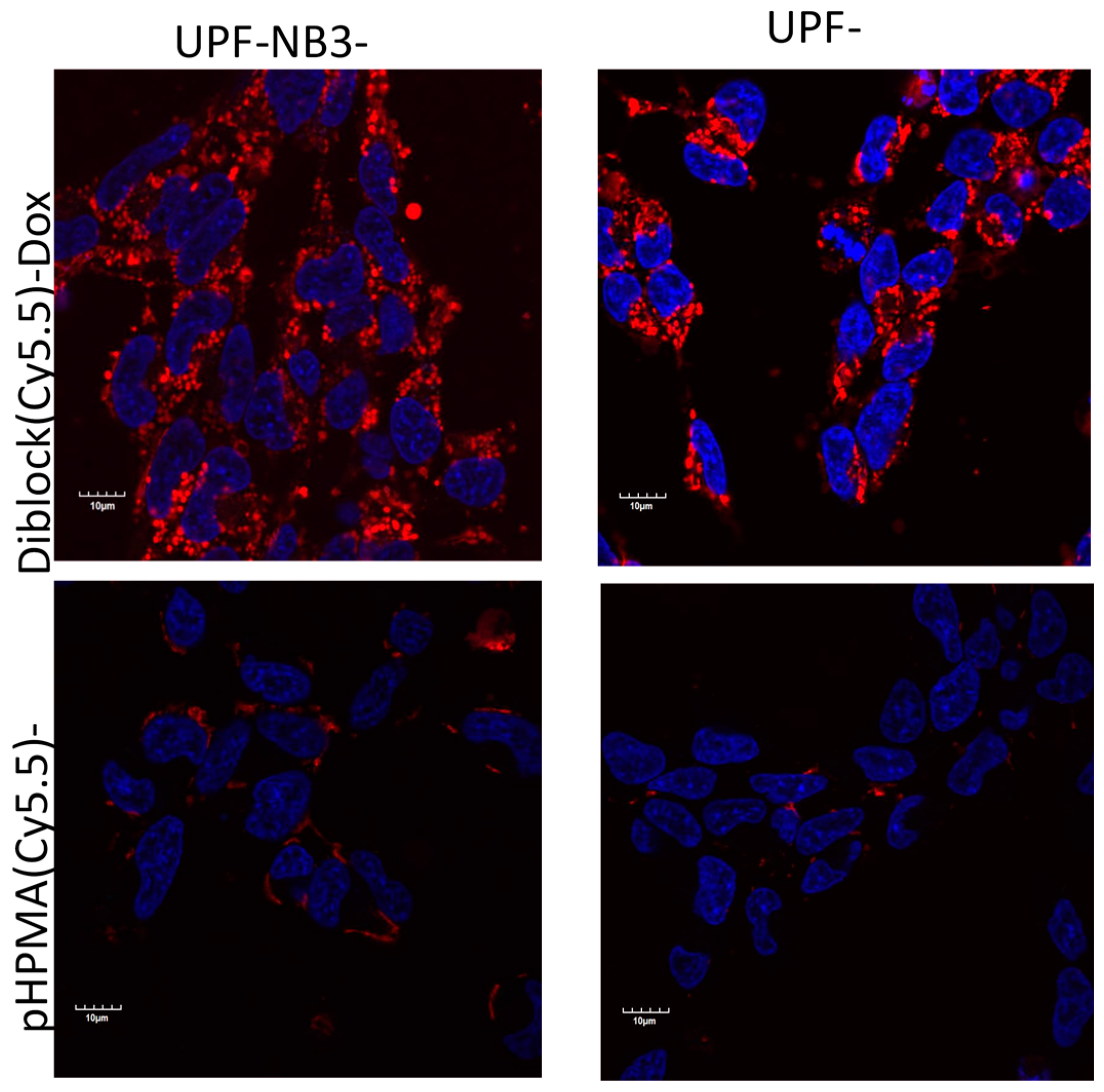{kind=link}
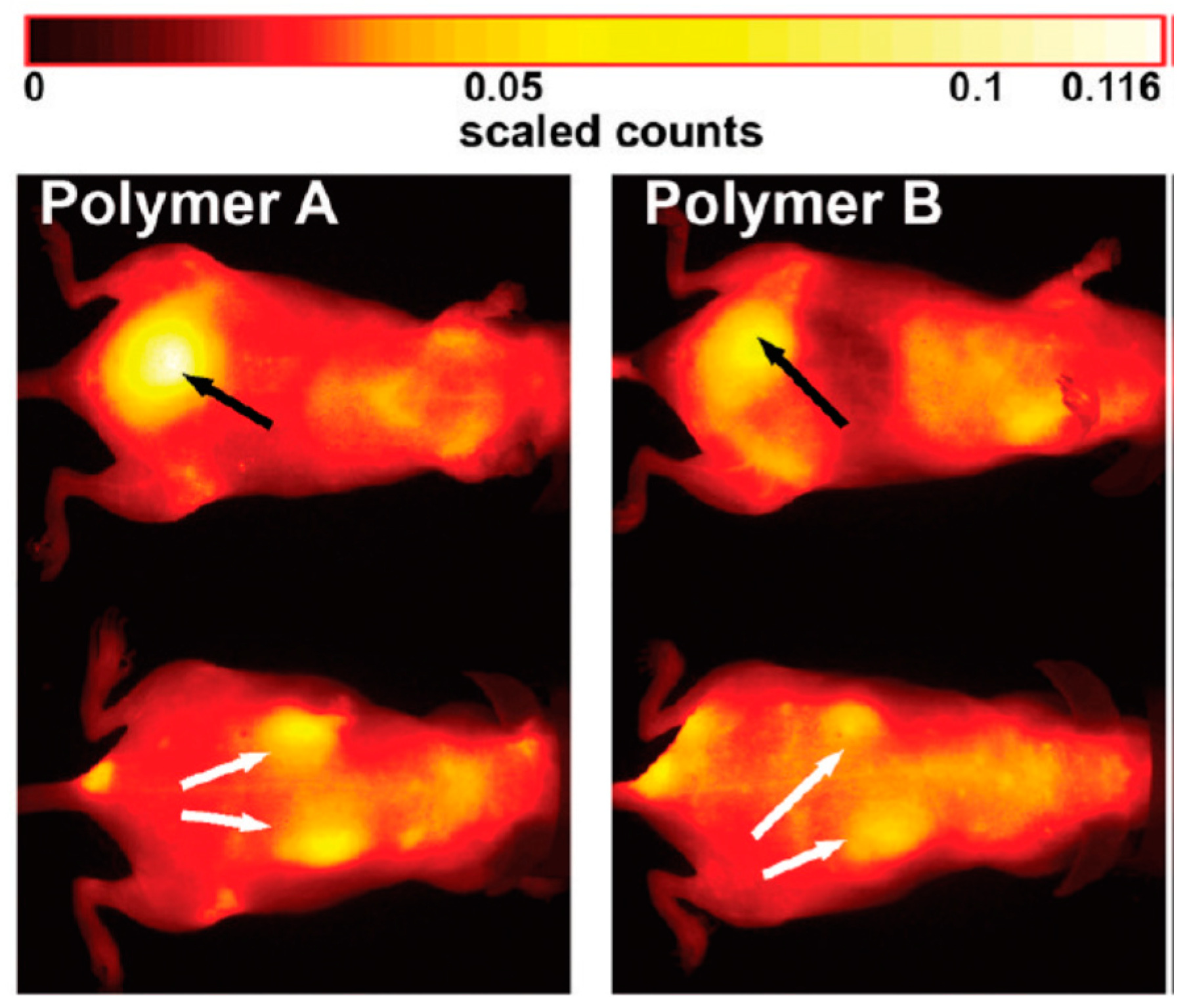{kind=link}
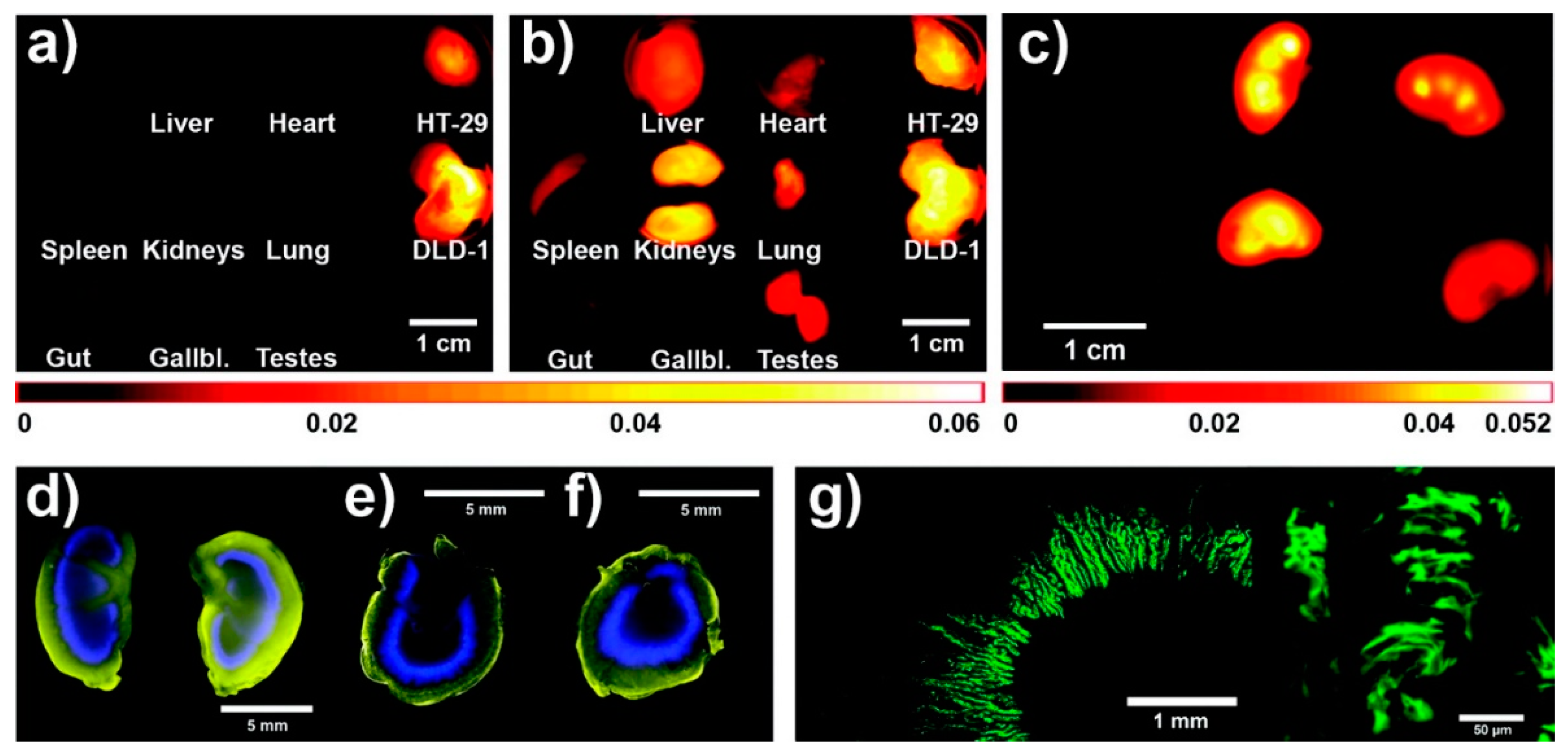{kind=link}
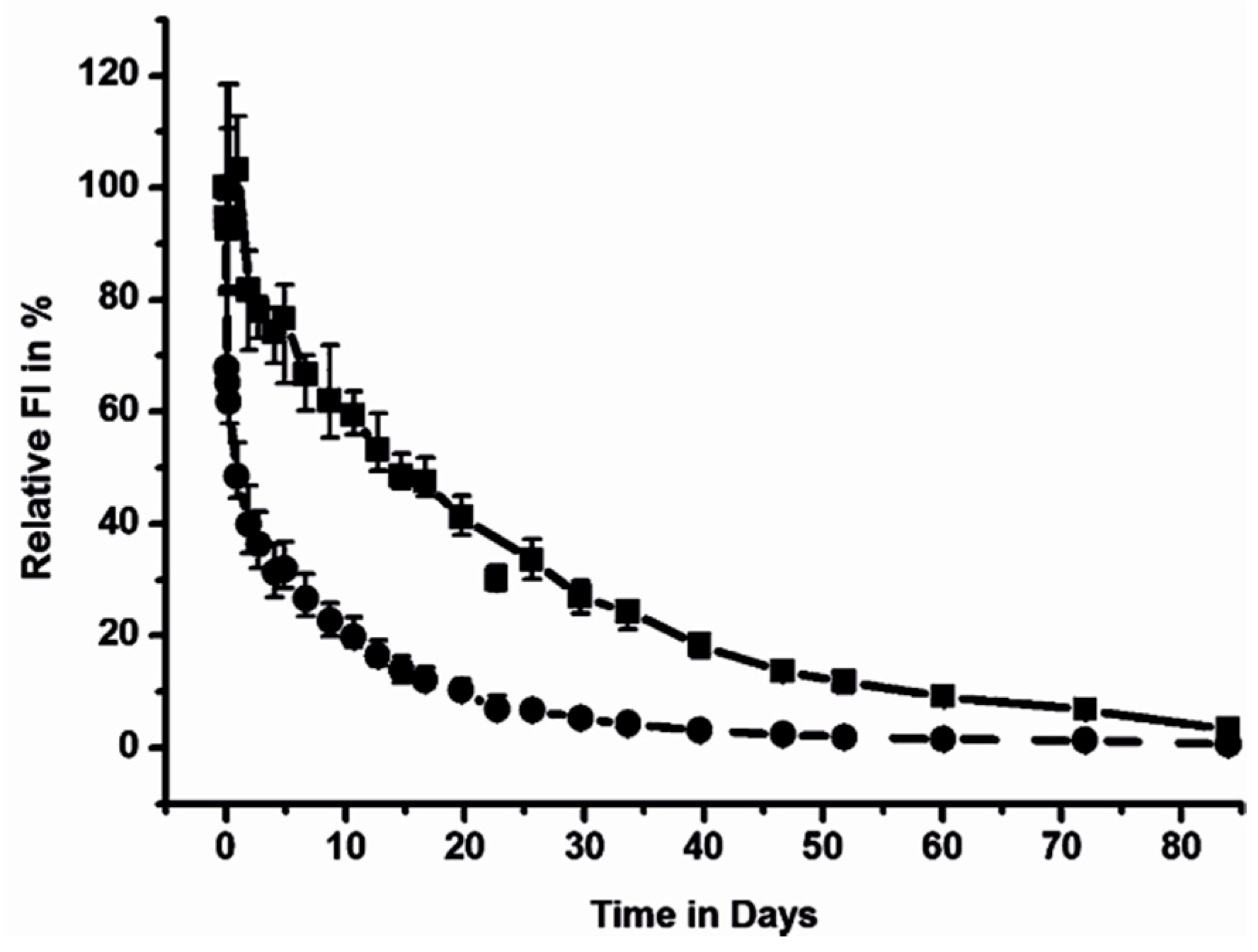{kind=link}
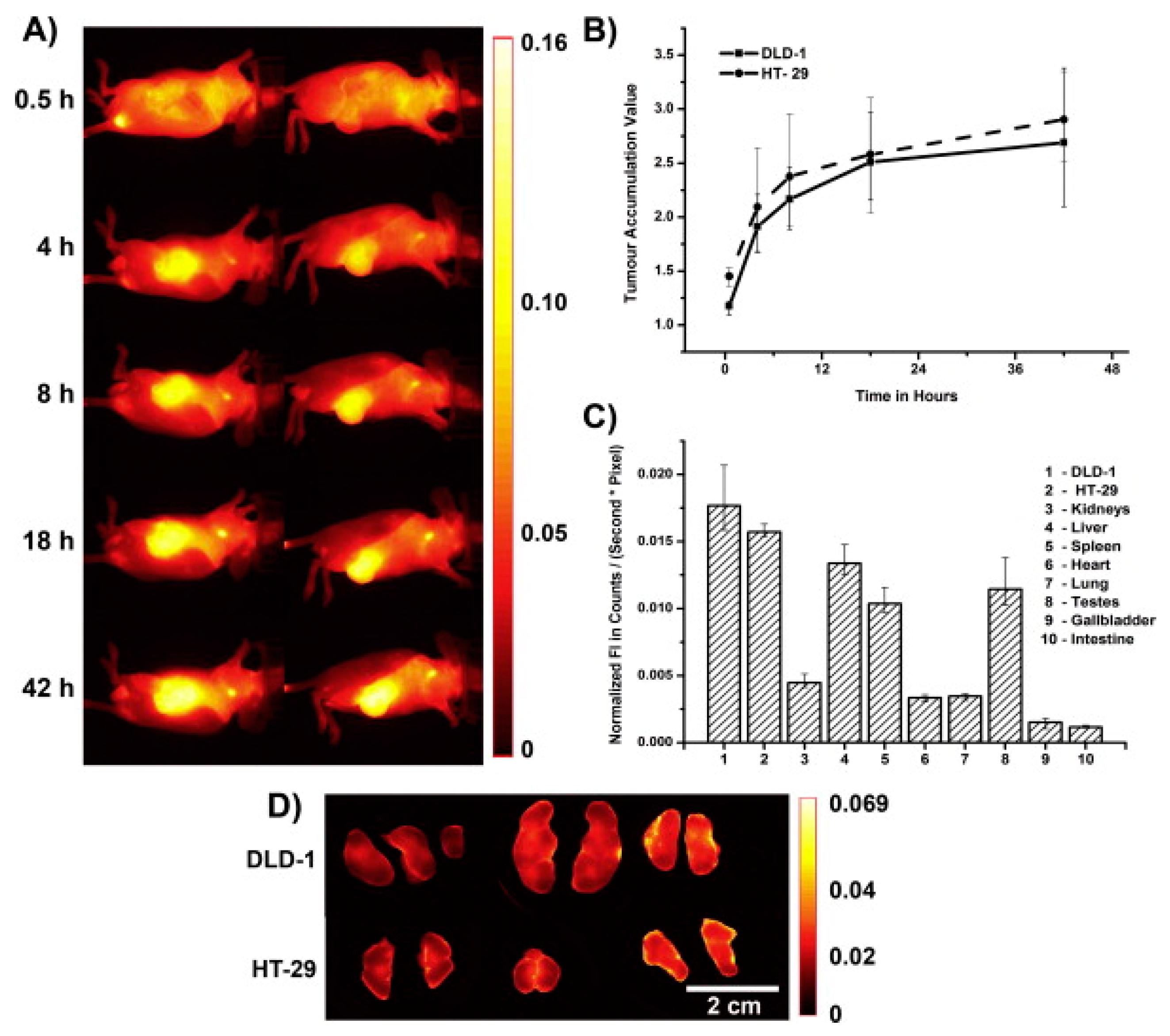{kind=link}
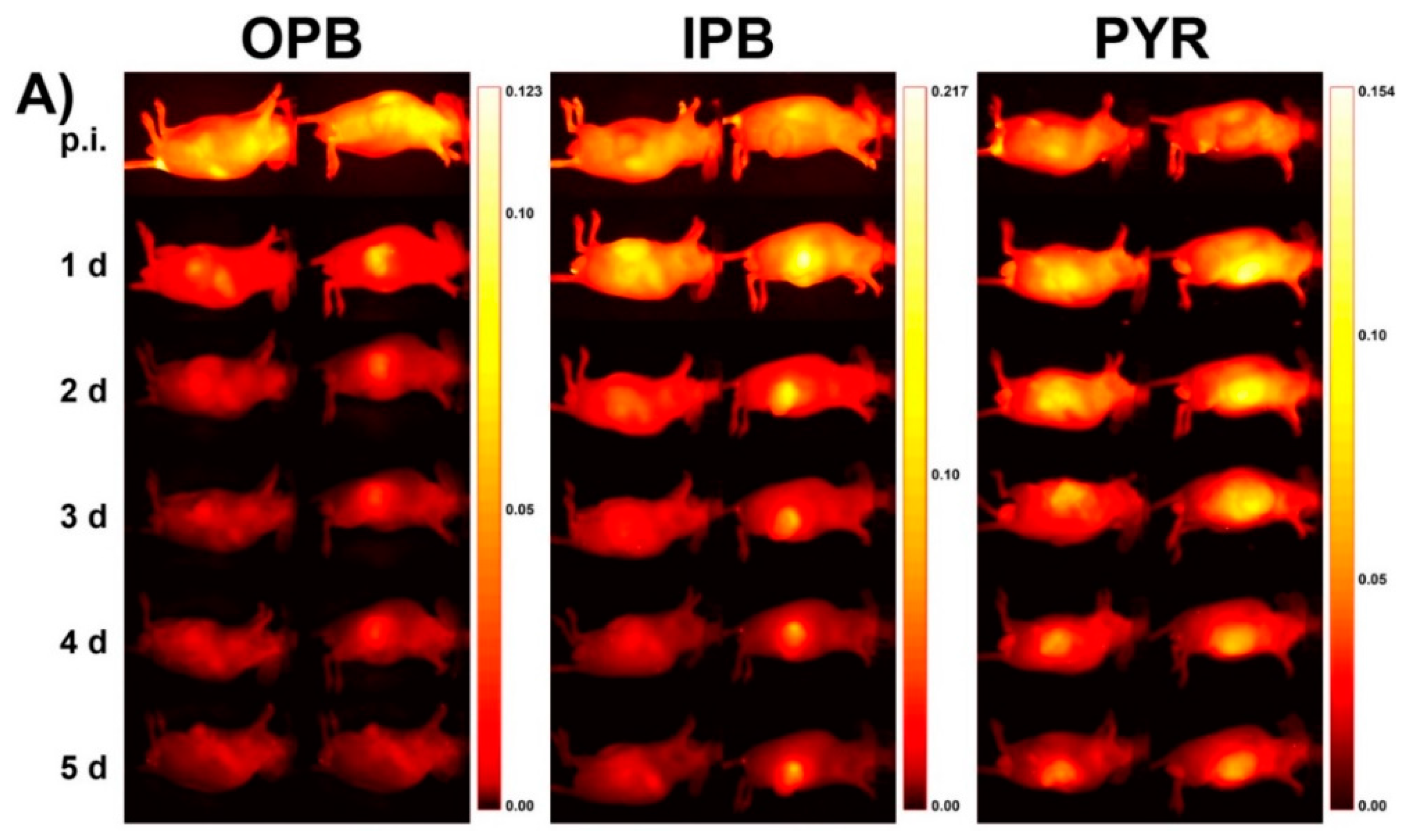{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Imaging at the Cellular Level
3. In Vivo Imaging
4. Fluorescence-Based Tomography and Future Prospects
5. Conclusions
Funding
Conflicts of Interest
References
- Ulbrich, K.; Holá, K.; Šubr, V.; Bakandritsos, A.; Tuček, J.; Zbořil, R. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chem. Rev. 2016, 116, 5338–5431. [Google Scholar] [CrossRef] [PubMed]
- Boas, D.A.; Brooks, D.H.; Miller, E.L.; DiMarzio, C.A.; Kilmer, M.; Gaudette, R.J.; Zhang, Q. Imaging the body with diffuse optical tomography. IEEE Signal. Proc. Mag. 2001, 18, 57–75. [Google Scholar] [CrossRef]
- Gibson, A.P.; Hebden, J.C.; Arridge, S.R. Recent advances in diffuse optical imaging. Phys. Med. Biol. 2005, 50, R1–R43. [Google Scholar] [CrossRef] [PubMed]
- Leblond, F.; Davis, S.C.; Valdes, P.A.; Pogue, B.W. Pre-clinical whole-body fluorescence imaging: Review of instruments, methods and applications. J. Photochem. Photobiol. B 2010, 98, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Etrych, T.; Lucas, H.; Janoušková, O.; Chytil, P.; Mueller, T.; Mäder, K. Fluorescence optical imaging in anticancer drug delivery. J. Control. Release 2016, 226, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.d.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release 2008, 126, 187–204. [Google Scholar] [CrossRef]
- Dozono, H.; Yanazume, S.; Nakamura, H.; Etrych, T.; Chytil, P.; Ulbrich, K.; Fang, J.; Arimura, T.; Douchi, T.; Kobayashi, H.; et al. HPMA Copolymer-Conjugated Pirarubicin in Multimodal Treatment of a Patient with Stage IV Prostate Cancer and Extensive Lung and Bone Metastases. Target. Oncol. 2016, 11, 101–106. [Google Scholar] [CrossRef]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the Microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer-Chemotherapy - Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Hoffman, A.S. The origins and evolution of “controlled” drug delivery systems. J. Control. Release 2008, 132, 153–163. [Google Scholar] [CrossRef]
- Mulder, W.J.M.; Strijkers, G.J.; Van Tilborg, G.A.F.; Cormode, D.P.; Fayad, Z.A.; Nicolay, K. Nanoparticulate Assemblies of Amphiphiles and Diagnostically Active Materials for Multimodality Imaging. Acc. Chem. Res. 2009, 42, 904–914. [Google Scholar] [CrossRef]
- Ulbrich, K.; Šubr, V. Structural and chemical aspects of HPMA copolymers as drug carriers. Adv. Drug Deliv. Rev. 2010, 62, 150–166. [Google Scholar] [CrossRef]
- Venditto, V.J.; Szoka, F.C. Cancer nanomedicines: So many papers and so few drugs! Adv. Drug Deliv. Rev. 2013, 65, 80–88. [Google Scholar] [CrossRef]
- Kunjachan, S.; Jayapaul, J.; Mertens, M.E.; Storm, G.; Kiessling, F.; Lammers, T. Theranostic Systems and Strategies for Monitoring Nanomedicine-Mediated Drug Targeting. Curr. Pharm. Biotechnol. 2012, 13, 609–622. [Google Scholar] [CrossRef]
- Lammers, T.; Aime, S.; Hennink, W.E.; Storm, G.; Kiessling, F. Theranostic Nanomedicine. Acc. Chem. Res. 2011, 44, 1029–1038. [Google Scholar] [CrossRef]
- Phillips, M.A.; Gran, M.L.; Peppas, N.A. Targeted nanodelivery of drugs and diagnostics. Nano. Today 2010, 5, 143–159. [Google Scholar] [CrossRef]
- Allmeroth, M.; Moderegger, D.; Biesalski, B.; Koynov, K.; Rosch, F.; Thews, O.; Zentel, R. Modifying the Body Distribution of HPMA-Based Copolymers by Molecular Weight and Aggregate Formation. Biomacromolecules 2011, 12, 2841–2849. [Google Scholar] [CrossRef]
- Lammers, T.; Kuhnlein, R.; Kissel, M.; Šubr, V.; Etrych, T.; Pola, R.; Pechar, M.; Ulbrich, K.; Storm, G.; Huber, P.; et al. Effect of physicochemical modification on the biodistribution and tumor accumulation of HPMA copolymers. J. Control. Release 2005, 110, 103–118. [Google Scholar] [CrossRef]
- Lu, Z.R. Molecular imaging of HPMA copolymers: Visualizing drug delivery in cell, mouse and man. Adv. Drug Deliv. Rev. 2010, 62, 246–257. [Google Scholar] [CrossRef]
- Licha, K.; Olbrich, C. Optical imaging in drug discovery and diagnostic applications. Adv. Drug Deliv. Rev. 2005, 57, 1087–1108. [Google Scholar] [CrossRef]
- Ntziachristos, V. Fluorescence molecular imaging. Annu. Rev. Biomed. Eng. 2006, 8, 1–33. [Google Scholar] [CrossRef]
- Ke, S.; Wen, X.X.; Gurfinkel, M.; Charnsangavej, C.; Wallace, S.; Sevick-Muraca, E.M.; Li, C. Near-infrared optical imaging of epidermal growth factor receptor in breast cancer xenografts. Cancer Res. 2003, 63, 7870–7875. [Google Scholar]
- Wunder, A.; Tung, C.H.; Muller-Ladner, U.; Weissleder, R.; Mahmood, U. In vivo imaging of protease activity in arthritis—A novel approach for monitoring treatment response. Arthritis Rheum. 2004, 50, 2459–2465. [Google Scholar] [CrossRef]
- Zaheer, A.; Lenkinski, R.E.; Mahmood, A.; Jones, A.G.; Cantley, L.C.; Frangioni, J.V. In vivo near-infrared fluorescence imaging of osteoblastic activity. Nat. Biotechnol. 2001, 19, 1148–1154. [Google Scholar] [CrossRef]
- Ntziachristos, V.; Ripoll, J.; Wang, L.H.V.; Weissleder, R. Looking and listening to light: The evolution of whole-body photonic imaging. Nat. Biotechnol. 2005, 23, 313–320. [Google Scholar] [CrossRef]
- Hebden, J.C.; Arridge, S.R.; Delpy, D.T. Optical imaging in medicine.1. Experimental techniques. Phys. Med. Biol. 1997, 42, 825–840. [Google Scholar] [CrossRef]
- Mahmood, U.; Weissleder, R. Near-infrared optical imaging of proteases in cancer. Mol. Cancer Ther. 2003, 2, 489–496. [Google Scholar]
- Hoffmann, S.; Vystrčilová, L.; Ulbrich, K.; Etrych, T.; Caysa, H.; Mueller, T.; Mäder, K. Dual Fluorescent HPMA Copolymers for Passive Tumor Targeting with pH-Sensitive Drug Release: Synthesis and Characterization of Distribution and Tumor Accumulation in Mice by Noninvasive Multispectral Optical Imaging. Biomacromolecules 2012, 13, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Chytil, P.; Hoffmann, S.; Schindler, L.; Kostka, L.; Ulbrich, K.; Caysa, H.; Mueller, T.; Mäder, K.; Etrych, T. Dual fluorescent HPMA copolymers for passive tumor targeting with pH- sensitive drug release II: Impact of release rate on biodistribution. J. Control. Release 2013, 172, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Tang, R.; Xue, J.; Wang, W.B.; Xu, B.; Achilefu, S. Synthesis of dye conjugates to visualize the cancer cells using fluorescence microscopy. Appl. Opt. 2014, 53, 2345–2351. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, H.; Acebrón, M.; Iborra, F.J.; Arias-Gonzalez, J.R.; Juárez, B.H. Photoluminescence Activation of Organic Dyes via Optically Trapped Quantum Dots. ACS Nano 2019, 13, 7223–7230. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Cao, X.; Yang, S.; Mo, Z.; Wang, W.; Zeng, W. Fluorescent Probes for Detection of Protein: From Bench to Bed. Protein Pept. Lett. 2018, 25, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Richards-Kortum, R. Optical molecular imaging agents for cancer diagnostics and therapeutics. Nanomedicine-Uk 2006, 1, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Freidus, L.G.; Pradeep, P.; Kumar, P.; Choonara, Y.E.; Pillay, V. Alternative fluorophores designed for advanced molecular imaging. Drug Discov. Today 2018, 23, 115–133. [Google Scholar] [CrossRef]
- Gao, X.H.; Nie, S.M. Molecular profiling of single cells and tissue specimens with quantum dots. Trends Biotechnol. 2003, 21, 371–373. [Google Scholar] [CrossRef]
- Xue, J.P.; Shan, L.L.; Chen, H.Y.; Li, Y.; Zhu, H.Y.; Deng, D.W.; Qian, Z.Y.; Achilefu, S.; Gu, Y.Q. Visual detection of STAT5B gene expression in living cell using the hairpin DNA modified gold nanoparticle beacon. Biosens. Bioelectron. 2013, 41, 71–77. [Google Scholar] [CrossRef]
- Hoffman, R.M. Application of GFP imaging in cancer. Lab. Investig. 2015, 95, 432–452. [Google Scholar] [CrossRef]
- McCann, T.; Kosaka, N.; Choyke, P.; Kobayashi, H. The Use of Fluorescent Proteins for Developing Cancer-Specific Target Imaging Probes. In In Vivo Cellular Imaging Using Fluorescent Proteins; Hoffman, R.M., Ed.; Humana Press: Totowa, NJ, USA, 2012; Volume 872, pp. 191–204. [Google Scholar]
- Karasev, M.M.; Stepanenko, O.V.; Rumyantsev, K.A.; Turoverov, K.K.; Verkhusha, V.V. Near-Infrared Fluorescent Proteins and Their Applications. Biochem-Moscow 2019, 84, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, A.K.; Lucas, H.; Schindler, L.; Chytil, P.; Etrych, T.; Mäder, K.; Mueller, T. Improved Tumor-Specific Drug Accumulation by Polymer Therapeutics with pH-Sensitive Drug Release Overcomes Chemotherapy Resistance. Mol. Cancer Ther. 2016, 15, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Dolloff, N.G.; Ma, X.H.; Dicker, D.T.; Humphreys, R.C.; Li, L.Z.; El-Deiry, W.S. Spectral imaging-based methods for quantifying autophagy and apoptosis. Cancer Biol. Ther. 2011, 12, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Galateanu, B.; Hudita, A.; Negrei, C.; Ion, R.M.; Costache, M.; Stan, M.; Nikitovic, D.; Hayes, A.W.; Spandidos, D.A.; Tsatsakis, A.M.; et al. Impact of multicellular tumor spheroids as an in vivo-like tumor model on anticancer drug response. Int. J. Oncol. 2016, 48, 2295–2302. [Google Scholar] [CrossRef] [PubMed]
- Ballou, B.; Fisher, G.W.; Hakala, T.R.; Farkas, D.L. Tumor detection and visualization using cyanine fluorochrome-labeled antibodies. Biotechnol. Progr. 1997, 13, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Lidický, O.; Janoušková, O.; Strohalm, J.; Alam, M.; Klener, P.; Etrych, T. Anti-Lymphoma Efficacy Comparison of Anti-Cd20 Monoclonal Antibody-Targeted and Non-Targeted Star-Shaped Polymer-Prodrug Conjugates. Molecules 2015, 20, 19849–19864. [Google Scholar] [CrossRef] [PubMed]
- Folli, S.; Westermann, P.; Braichotte, D.; Pelegrin, A.; Wagnieres, G.; van den Bergh, H.; Mach, J.P. Antibody-indocyanin conjugates for immunophotodetection of human squamous cell carcinoma in nude mice. Cancer Res. 1994, 54, 2643–2649. [Google Scholar]
- Pechar, M.; Pola, R.; Janoušková, O.; Sieglová, I.; Král, V.; Fábry, M.; Tomalová, B.; Kovář, M. Polymer Cancerostatics Targeted with an Antibody Fragment Bound via a Coiled Coil Motif: In Vivo Therapeutic Efficacy against Murine BCL1 Leukemia. Macromol. Biosci. 2018, 18, 1700173. [Google Scholar] [CrossRef]
- Pola, R.; Studenovsky, M.; Pechar, M.; Ulbrich, K.; Hovorka, O.; Vetvicka, D.; Rihova, B. HPMA-copolymer conjugates targeted to tumor endothelium using synthetic oligopeptides. J. Drug Target. 2009, 17, 763–776. [Google Scholar] [CrossRef]
- Studenovsky, M.; Pola, R.; Pechar, M.; Etrych, T.; Ulbrich, K.; Kovar, L.; Kabesova, M.; Rihova, B. Polymer carriers for anticancer drugs targeted to EGF receptor. Macromol. Biosci. 2012, 12, 1714–1720. [Google Scholar] [CrossRef]
- Song, Y.; Zhu, Z.; An, Y.; Zhang, W.; Zhang, H.; Liu, D.; Yu, C.; Duan, W.; Yang, C.J. Selection of DNA aptamers against epithelial cell adhesion molecule for cancer cell imaging and circulating tumor cell capture. Anal. Chem. 2013, 85, 4141–4149. [Google Scholar] [CrossRef]
- Tung, C.H. Fluorescent peptide probes for in vivo diagnostic imaging. Biopolymers 2004, 76, 391–403. [Google Scholar] [CrossRef]
- Weissleder, R. Molecular imaging: Exploring the next frontier. Radiology 1999, 212, 609–614. [Google Scholar] [CrossRef]
- Shi, H.; Lei, Y.; Ge, J.; He, X.; Cui, W.; Ye, X.; Liu, J.; Wang, K. A Simple, pH-Activatable Fluorescent Aptamer Probe with Ultralow Background for Bispecific Tumor Imaging. Anal. Chem. 2019, 91, 9154–9160. [Google Scholar] [CrossRef]
- Muller-Taubenberger, A.; Anderson, K.I. Recent advances using green and red fluorescent protein variants. Appl. Microbiol. Biotechnol. 2007, 77, 1–12. [Google Scholar] [CrossRef]
- Hoffman, R.M. The multiple uses of fluorescent proteins to visualize cancer in vivo. Nat. Rev. Cancer 2005, 5, 796–806. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef]
- Choy, G.; Choyke, P.; Libutti, S.K. Current Advances in Molecular Imaging: Noninvasive in Vivo Bioluminescent and Fluorescent Optical Imaging in Cancer Research. Mol. Imaging 2003, 2, 15353500200303142. [Google Scholar] [CrossRef]
- Barua, S.; Yoo, J.W.; Kolhar, P.; Wakankar, A.; Gokarn, Y.R.; Mitragotri, S. Particle shape enhances specificity of antibody-displaying nanoparticles. PNAS 2013, 110, 3270–3275. [Google Scholar] [CrossRef]
- Gratton, S.E.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. PNAS 2008, 105, 11613–11618. [Google Scholar] [CrossRef]
- Huang, X.; Teng, X.; Chen, D.; Tang, F.; He, J. The effect of the shape of mesoporous silica nanoparticles on cellular uptake and cell function. Biomaterials 2010, 31, 438–448. [Google Scholar] [CrossRef]
- Shi, J.; Choi, J.L.; Chou, B.; Johnson, R.N.; Schellinger, J.G.; Pun, S.H. Effect of polyplex morphology on cellular uptake, intracellular trafficking, and transgene expression. ACS nano 2013, 7, 10612–10620. [Google Scholar] [CrossRef]
- Koziolová, E.; Goel, S.; Chytil, P.; Janoušková, O.; Barnhart, T.E.; Cai, W.; Etrych, T. A tumor-targeted polymer theranostics platform for positron emission tomography and fluorescence imaging. Nanoscale 2017, 9, 10906–10918. [Google Scholar] [CrossRef]
- Pola, R.; Laga, R.; Ulbrich, K.; Sieglová, I.; Král, V.; Fábry, M.; Kabešová, M.; Kovář, M.; Pechar, M. Polymer Therapeutics with a Coiled Coil Motif Targeted against Murine BCL1 Leukemia. Biomacromolecules 2013, 14, 881–889. [Google Scholar] [CrossRef]
- Pola, R.; Král, V.; Filippov, S.K.; Kaberov, L.; Etrych, T.; Sieglová, I.; Sedláček, J.; Fábry, M.; Pechar, M. Polymer Cancerostatics Targeted by Recombinant Antibody Fragments to GD2-Positive Tumor Cells. Biomacromolecules 2019, 20, 412–421. [Google Scholar] [CrossRef]
- Jiang, S.; Gnanasammandhan, M.K.; Zhang, Y. Optical imaging-guided cancer therapy with fluorescent nanoparticles. J. R. Soc. Interface 2010, 7, 3–18. [Google Scholar] [CrossRef]
- Koziolová, E.; Machová, D.; Pola, R.; Janoušková, O.; Chytil, P.; Laga, R.; Filippov, S.K.; Šubr, V.; Etrych, T.; Pechar, M. Micelle-forming HPMA copolymer conjugates of ritonavir bound via a pH-sensitive spacer with improved cellular uptake designed for enhanced tumor accumulation. J. Mater. Chem. B 2016, 4, 7620–7629. [Google Scholar] [CrossRef]
- Hovorka, O.; Etrych, T.; Šubr, V.; Strohalm, J.; Ulbrich, K.; Říhová, B. HPMA based macromolecular therapeutics: Internalization, intracellular pathway and cell death depend on the character of covalent bond between the drug and the peptidic spacer and also on spacer composition. J. Drug Target. 2006, 14, 391–403. [Google Scholar] [CrossRef]
- Machová, D.; Koziolová, E.; Chytil, P.; Venclíková, K.; Etrych, T.; Janoušková, O. Nanotherapeutics with suitable properties for advanced anticancer therapy based on HPMA copolymer-bound ritonavir via pH-sensitive spacers. Eur. J. Pharm. Biopharm. 2018, 131, 141–150. [Google Scholar] [CrossRef]
- Chen, Y.; Walsh, R.J.; Arriaga, E.A. Selective determination of the doxorubicin content of individual acidic organelles in impure subcellular fractions. Anal. Chem. 2005, 77, 2281–2287. [Google Scholar] [CrossRef]
- Shen, F.; Chu, S.; Bence, A.K.; Bailey, B.; Xue, X.; Erickson, P.A.; Montrose, M.H.; Beck, W.T.; Erickson, L.C. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J. Pharmacol. Exp. Ther. 2008, 324, 95–102. [Google Scholar] [CrossRef]
- Priem, B.; Tian, C.; Tang, J.; Zhao, Y.; Mulder, W.J.M. Fluorescent nanoparticles for the accurate detection of drug delivery. Expert Opin. Drug Deliv. 2015, 12, 1881–1894. [Google Scholar] [CrossRef]
- Nori, A.; Kopecek, J. Intracellular targeting of polymer-bound drugs for cancer chemotherapy. Adv. Drug Deliv. Rev. 2005, 57, 609–636. [Google Scholar] [CrossRef]
- Chytil, P.; Koziolová, E.; Janoušková, O.; Kostka, L.; Ulbrich, K.; Etrych, T. Synthesis and Properties of Star HPMA Copolymer Nanocarriers Synthesised by RAFT Polymerisation Designed for Selective Anticancer Drug Delivery and Imaging. Macromol. Biosci. 2015, 15, 839–850. [Google Scholar] [CrossRef]
- Laga, R.; Janoušková, O.; Ulbrich, K.; Pola, R.; Blažková, J.; Filippov, S.K.; Etrych, T.; Pechar, M. Thermoresponsive Polymer Micelles as Potential Nanosized Cancerostatics. Biomacromolecules 2015, 16, 2493–2505. [Google Scholar] [CrossRef]
- Braunová, A.; Kostka, L.; Sivák, L.; Cuchalová, L.; Hvězdová, Z.; Laga, R.; Filippov, S.; Černoch, P.; Pechar, M.; Janoušková, O.; et al. Tumor-targeted micelle-forming block copolymers for overcoming of multidrug resistance. J. Control. Release 2017, 245, 41–51. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, J.; Radford, D.C.; Fang, Y.; Kopeček, J. FRET Imaging of Enzyme-Responsive HPMA Copolymer Conjugate. Macromol. Biosci. 2017, 17, 1600125. [Google Scholar] [CrossRef]
- Yang, J.Y.; Zhang, R.; Radford, D.C.; Kopecek, J. FRET-trackable biodegradable HPMA copolymer-epirubicin conjugates for ovarian carcinoma therapy. J. Control. Release 2015, 218, 36–44. [Google Scholar] [CrossRef]
- Fan, W.; Shi, W.; Zhang, W.; Jia, Y.; Zhou, Z.; Brusnahan, S.K.; Garrison, J.C. Cathepsin S-cleavable, multi-block HPMA copolymers for improved SPECT/CT imaging of pancreatic cancer. Biomaterials 2016, 103, 101–115. [Google Scholar] [CrossRef]
- Bhuckory, S.; Kays, J.C.; Dennis, A.M. In Vivo Biosensing Using Resonance Energy Transfer. Biosensors 2019, 9, 76. [Google Scholar] [CrossRef]
- Basuki, J.S.; Duong, H.T.T.; Macmillan, A.; Erlich, R.B.; Esser, L.; Akerfeldt, M.C.; Whan, R.M.; Kavallaris, M.; Boyer, C.; Davis, T.P. Using Fluorescence Lifetime Imaging Microscopy to Monitor Theranostic Nanoparticle Uptake and Intracellular Doxorubicin Release. ACS Nano 2013, 7, 10175–10189. [Google Scholar] [CrossRef]
- Dai, X.W.; Yue, Z.L.; Eccleston, M.E.; Swartling, J.; Slater, N.K.H.; Kaminski, C.F. Fluorescence intensity and lifetime imaging of free and micellar-encapsulated doxorubicin in living cells. Nanomed-Nanotechnol. 2008, 4, 49–56. [Google Scholar] [CrossRef]
- Mansfield, J.R.; Gossage, K.W.; Hoyt, C.C.; Levenson, R.M. Autofluorescence removal, multiplexing, and automated analysis methods for in-vivo fluorescence imaging. J. Biomed. Opt. 2005, 10, 041207. [Google Scholar] [CrossRef]
- Weissleder, R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 19, 316–317. [Google Scholar] [CrossRef]
- Hoffmann, S.; Caysa, H.; Kuntsche, J.; Kreideweiss, P.; Leimert, A.; Mueller, T.; Mäder, K. Carbohydrate plasma expanders for passive tumor targeting: In vitro and in vivo studies. Carbohyd. Polym. 2013, 95, 404–413. [Google Scholar] [CrossRef]
- Han, Y.-H.; Kankala, R.K.; Wang, S.-B.; Chen, A.-Z. Leveraging Engineering of Indocyanine Green-Encapsulated Polymeric Nanocomposites for Biomedical Applications. Nanomaterials 2018, 8, 360. [Google Scholar] [CrossRef]
- Kolitz-Domb, M.; Grinberg, I.; Corem-Salkmon, E.; Margel, S. Engineering of near infrared fluorescent proteinoid-poly(L-lactic acid) particles for in vivo colon cancer detection. J. Nanobiotechnol. 2014, 12, 30. [Google Scholar] [CrossRef]
- Hirsjarvi, S.; Sancey, L.; Dufort, S.; Belloche, C.; Vanpouille-Box, C.; Garcion, E.; Coll, J.L.; Hindre, F.; Benoit, J.P. Effect of particle size on the biodistribution of lipid nanocapsules: Comparison between nuclear and fluorescence imaging and counting. Int. J. Pharm. 2013, 453, 594–600. [Google Scholar] [CrossRef]
- Studenovský, M.; Heinrich, A.-K.; Lucas, H.; Mueller, T.; Mäder, K.; Etrych, T. Dual fluorescent N-(2-hydroxypropyl)methacrylamide-based conjugates for passive tumor targeting with reduction-sensitive drug release: Proof of the concept, tumor accumulation, and biodistribution. J. Bioact. Compat. Pol. 2016. [Google Scholar] [CrossRef]
- Pola, R.; Heinrich, A.K.; Mueller, T.; Kostka, L.; Mäder, K.; Pechar, M.; Etrych, T. Passive Tumor Targeting of Polymer Therapeutics: In Vivo Imaging of Both the Polymer Carrier and the Enzymatically Cleavable Drug Model. Macromol. Biosci. 2016, 16, 1577–1582. [Google Scholar] [CrossRef]
- Cho, H.; Kwon, G.S. Polymeric Micelles for Neoadjuvant Cancer Therapy and Tumor-Primed Optical Imaging. ACS Nano 2011, 5, 8721–8729. [Google Scholar] [CrossRef]
- Pola, R.; Parnica, J.; Zuska, K.; Böhmová, E.; Filipová, M.; Pechar, M.; Pankrác, J.; Mucksová, J.; Kalina, J.; Trefil, P.; et al. Oligopeptide-targeted polymer nanoprobes for fluorescence-guided endoscopic surgery. Multifunct. Mater. 2019, 2, 024004. [Google Scholar] [CrossRef]
- Ko, J.Y.; Park, S.; Lee, H.; Koo, H.; Kim, M.S.; Choi, K.; Kwon, I.C.; Jeong, S.Y.; Kim, K.; Lee, D.S. pH-Sensitive Nanoflash for Tumoral Acidic pH Imaging in Live Animals. Small 2010, 6, 2539–2544. [Google Scholar] [CrossRef]
- Gao, G.H.; Li, Y.; Lee, D.S. Environmental pH-sensitive polymeric micelles for cancer diagnosis and targeted therapy. J. Control. release 2013, 169, 180–184. [Google Scholar] [CrossRef]
- Etrych, T.; Daumová, L.; Pokorná, E.; Tušková, D.; Lidický, O.; Kolářová, V.; Pankrác, J.; Šefc, L.; Chytil, P.; Klener, P. Effective doxorubicin-based nano-therapeutics for simultaneous malignant lymphoma treatment and lymphoma growth imaging. J. Control. Release 2018, 289, 44–55. [Google Scholar] [CrossRef]
- Berg, K.; Selbo, P.K.; Weyergang, A.; Dietze, A.; Prasmickaite, L.; Bonsted, A.; Engesaeter, B.O.; Angell-Petersen, E.; Warloe, T.; Frandsen, N.; et al. Porphyrin-related photosensitizers for cancer imaging and therapeutic applications. J. Microsc-Oxford 2005, 218, 133–147. [Google Scholar] [CrossRef]
- Nakamura, H.; Liao, L.; Hitaka, Y.; Tsukigawa, K.; Šubr, V.; Fang, J.; Ulbrich, K.; Maeda, H. Micelles of zinc protoporphyrin conjugated to N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer for imaging and light-induced antitumor effects in vivo. J. Control. Release 2013, 165, 191–198. [Google Scholar] [CrossRef]
- Hackbarth, S.; Islam, W.; Fang, J.; Šubr, V.; Röder, B.; Etrych, T.; Maeda, H. Singlet oxygen phosphorescence detection in vivo identifies PDT-induced anoxia in solid tumors. Photochem. Photobiol. Sci. 2019, 18, 1304–1314. [Google Scholar] [CrossRef]
- Niedre, M.J.; de Kleine, R.H.; Aikawa, E.; Kirsch, D.G.; Weissleder, R.; Ntziachristos, V. Early photon tomography allows fluorescence detection of lung carcinomas and disease progression in mice in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 19126–19131. [Google Scholar] [CrossRef]
- Hall, D.; Ma, G.B.; Lesage, F.; Yong, W. Simple time-domain optical method for estimating the depth and concentration of a fluorescent inclusion in a turbid medium. Opt. Lett. 2004, 29, 2258–2260. [Google Scholar] [CrossRef]
- Swartling, J.; Svensson, J.; Bengtsson, D.; Terike, K.; Andersson-Engels, S. Fluorescence spectra provide information on the depth of fluorescent lesions in tissue. Appl. Opt. 2005, 44, 1934–1941. [Google Scholar] [CrossRef]
- Shi, J.W.; Liu, F.; Pu, H.S.; Zuo, S.M.; Luo, J.W.; Bai, J. An adaptive support driven reweighted L1-regularization algorithm for fluorescence molecular tomography. Biomed. Opt. Express 2014, 5, 4039–4052. [Google Scholar] [CrossRef]
- Favicchio, R.; Psycharakis, S.; Schonig, K.; Bartsch, D.; Mamalaki, C.; Papamatheakis, J.; Ripoll, J.; Zacharakis, G. Quantitative performance characterization of three-dimensional noncontact fluorescence molecular tomography. J. Biomed. Opt. 2016, 21, 026009. [Google Scholar] [CrossRef]
- Pian, Q.; Yao, R.Y.; Zhao, L.L.; Intes, X. Hyperspectral time-resolved wide-field fluorescence molecular tomography based on structured light and single-pixel detection. Opt. Lett. 2015, 40, 431–434. [Google Scholar] [CrossRef]
- An, Y.; Liu, J.; Zhang, G.L.; Ye, J.Z.; Du, Y.; Mao, Y.; Chi, C.W.; Tian, J. A Novel Region Reconstruction Method for Fluorescence Molecular Tomography. IEEE Trans. Biomed. Eng. 2015, 62, 1818–1826. [Google Scholar] [CrossRef]
- Chi, C.; Du, Y.; Ye, J.; Kou, D.; Qiu, J.; Wang, J.; Tian, J.; Chen, X. Intraoperative Imaging-Guided Cancer Surgery: From Current Fluorescence Molecular Imaging Methods to Future Multi-Modality Imaging Technology. Theranostics 2014, 4, 1072–1084. [Google Scholar] [CrossRef]
- Kelly, K.; Alencar, H.; Funovics, M.; Mahmood, U.; Weissleder, R. Detection of invasive colon cancer using a novel, targeted, library-derived fluorescent peptide. Cancer Res. 2004, 64, 6247–6251. [Google Scholar] [CrossRef]
- Heffer, E.; Pera, V.; Schutz, O.; Siebold, H.; Fantini, S. Near-infrared imaging of the human breast: Complementing hemoglobin concentration maps with oxygenation images. J. Biomed. Opt. 2004, 9, 1152–1160. [Google Scholar] [CrossRef][Green Version]
- Choe, R.; Corlu, A.; Lee, K.; Durduran, T.; Konecky, S.D.; Grosicka-Koptyra, M.; Arridge, S.R.; Czerniecki, B.J.; Fraker, D.L.; DeMichele, A.; et al. Diffuse optical tomography of breast cancer during neoadjuvant chemotherapy: A case study with comparison to MRI. Medical. Phys. 2005, 32, 1128–1139. [Google Scholar] [CrossRef]
- Taroni, P.; Danesini, G.; Torricelli, A.; Pifferi, A.; Spinelli, L.; Cubeddu, R. Clinical trial of time-resolved scanning optical mammography at 4 wavelengths between 683 and 975 nm. J. Biomed. Opt. 2004, 9, 464–473. [Google Scholar] [CrossRef]
- Corlu, A.; Choe, R.; Durduran, T.; Rosen, M.A.; Schweiger, M.; Arridge, S.R.; Schnall, M.D.; Yodh, A.G. Three-dimensional in vivo fluorescence diffuse optical tomography of breast cancer in humans. Opt. Express 2007, 15, 6696–6716. [Google Scholar] [CrossRef]
- Intes, X.; Ripoll, J.; Chen, Y.; Nioka, S.; Yodh, A.G.; Chance, B. In vivo continuous-wave optical breast imaging enhanced with Indocyanine Green. Med. Phys. 2003, 30, 1039–1047. [Google Scholar] [CrossRef]
- Graves, E.E.; Ripoll, J.; Weissleder, R.; Ntziachristos, V. A submillimeter resolution fluorescence molecular imaging system for small animal imaging. Med. Phys. 2003, 30, 901–911. [Google Scholar] [CrossRef]
- Ntziachristos, V.; Weissleder, R. Experimental three-dimensional fluorescence reconstruction of diffuse media by use of a normalized Born approximation. Opt. Lett. 2001, 26, 893–895. [Google Scholar] [CrossRef]
- Ale, A.; Ermolayev, V.; Herzog, E.; Cohrs, C.; de Angelis, M.H.; Ntziachristos, V. FMT-XCT: In vivo animal studies with hybrid fluorescence molecular tomography-X-ray computed tomography. Nat. Methods 2012, 9, 615–620. [Google Scholar] [CrossRef]
- Panizzi, P.; Nahrendorf, M.; Figueiredo, J.L.; Panizzi, J.; Marinelli, B.; Iwamoto, Y.; Keliher, E.; Maddur, A.A.; Waterman, P.; Kroh, H.K.; et al. In vivo detection of Staphylococcus aureus endocarditis by targeting pathogen-specific prothrombin activation. Nat. Med. 2011, 17, 1142–1146. [Google Scholar] [CrossRef]
- Vonwil, D.; Christensen, J.; Fischer, S.; Ronneberger, O.; Shastri, V.P. Validation of Fluorescence Molecular Tomography/Micro-CT Multimodal Imaging In Vivo in Rats. Mol. Imaging Biol. 2014, 16, 350–361. [Google Scholar] [CrossRef]
- Schulz, R.B.; Ale, A.; Sarantopoulos, A.; Freyer, M.; Soehngen, E.; Zientkowska, M.; Ntziachristos, V. Hybrid System for Simultaneous Fluorescence and X-Ray Computed Tomography. IEEE Trans. Med. Imaging 2010, 29, 465–473. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Keliher, E.; Marinelli, B.; Waterman, P.; Feruglio, P.F.; Fexon, L.; Pivovarov, M.; Swirski, F.K.; Pittet, M.J.; Vinegoni, C.; et al. Hybrid PET-optical imaging using targeted probes. Proc. Natl. Acad. Sci. USA 2010, 107, 7910–7915. [Google Scholar] [CrossRef]
- Ma, X.; Phi Van, V.; Kimm, M.A.; Prakash, J.; Kessler, H.; Kosanke, K.; Feuchtinger, A.; Aichler, M.; Gupta, A.; Rummeny, E.J.; et al. Integrin-Targeted Hybrid Fluorescence Molecular Tomography/X-ray Computed Tomography for Imaging Tumor Progression and Early Response in Non-Small Cell Lung Cancer. Neoplasia 2017, 19, 8–16. [Google Scholar] [CrossRef]
- Kunjachan, S.; Gremse, F.; Theek, B.; Koczera, P.; Pola, R.; Pechar, M.; Etrych, T.; Ulbrich, K.; Storm, G.; Kiessling, F.; et al. Noninvasive Optical Imaging of Nanomedicine Biodistribution. ACS Nano 2013, 7, 252–262. [Google Scholar] [CrossRef]
- Kunjachan, S.; Pola, R.; Gremse, F.; Theek, B.; Ehling, J.; Moeckel, D.; Hermanns-Sachweh, B.; Pechar, M.; Ulbrich, K.; Hennink, W.E.; et al. Passive versus Active Tumor Targeting Using RGD- and NGR-Modified Polymeric Nanomedicines. Nano. Lett. 2014, 14, 972–981. [Google Scholar] [CrossRef]
- Giddabasappa, A.; Gupta, V.R.; Norberg, R.; Gupta, P.; Spilker, M.E.; Wentland, J.; Rago, B.; Eswaraka, J.; Leal, M.; Sapra, P. Biodistribution and Targeting of Anti-5T4 Antibody-Drug Conjugate Using Fluorescence Molecular Tomography. Mol. Cancer Ther. 2016, 15, 2530–2540. [Google Scholar] [CrossRef]
- Theek, B.; Baues, M.; Gremse, F.; Pola, R.; Pechar, M.; Negwer, I.; Koynov, K.; Weber, B.; Barz, M.; Jahnen-Dechent, W.; et al. Histidine-rich glycoprotein-induced vascular normalization improves EPR-mediated drug targeting to and into tumors. J. Control. Release 2018, 282, 25–34. [Google Scholar] [CrossRef]
- Lee, H.; Lytton-Jean, A.K.R.; Chen, Y.; Love, K.T.; Park, A.I.; Karagiannis, E.D.; Sehgal, A.; Querbes, W.; Zurenko, C.S.; Jayaraman, M.; et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 2012, 7, 389–393. [Google Scholar] [CrossRef]
- Novobrantseva, T.I.; Borodovsky, A.; Wong, J.; Klebanov, B.; Zafari, M.; Yucius, K.; Querbes, W.; Ge, P.; Ruda, V.M.; Milstein, S.; et al. Systemic RNAi-mediated Gene Silencing in Nonhuman Primate and Rodent Myeloid Cells. Mol. Ther-Nucl Acids 2012, 1, e4. [Google Scholar] [CrossRef]
- Al Rawashdeh, W.; Zuo, S.; Melle, A.; Appold, L.; Koletnik, S.; Tsvetkova, Y.; Beztsinna, N.; Pich, A.; Lammers, T.; Kiessling, F.; et al. Noninvasive Assessment of Elimination and Retention using CT-FMT and Kinetic Whole-body Modeling. Theranostics 2017, 7, 1499–1510. [Google Scholar] [CrossRef]
- Li, B.Q.; Maafi, F.; Berti, R.; Pouliot, P.; Rheaume, E.; Tardif, J.C.; Lesage, F. Hybrid FMT-MRI applied to in vivo atherosclerosis imaging. Biomed. Opt. Express 2014, 5, 1664–1676. [Google Scholar] [CrossRef]
- Sosnovik, D.E.; Nahrendorf, M.; Deliolanis, N.; Novikov, M.; Aikawa, E.; Josephson, L.; Rosenzweig, A.; Weissleder, R.; Ntziachristos, V. Fluorescence tomography and magnetic resonance imaging of myocardial macrophage infiltration in infarcted myocardium in vivo. Circulation 2007, 115, 1384–1391. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, B.; Liu, F.; Luo, J.W.; Bai, J. In vivo tomographic imaging with fluorescence and MRI using tumor-targeted dual-labeled nanoparticles. Int. J. Nanomed. 2014, 9, 33–41. [Google Scholar] [CrossRef]
- Gaedicke, S.; Braun, F.; Prasad, S.; Machein, M.; Firat, E.; Hettich, M.; Gudihal, R.; Zhu, X.; Klingner, K.; Schüler, J.; et al. Noninvasive positron emission tomography and fluorescence imaging of CD133+ tumor stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E692–E701. [Google Scholar] [CrossRef] [PubMed]
- Boutet, J.; Herve, L.; Debourdeau, M.; Guyon, L.; Peltie, P.; Dinten, J.M.; Saroul, L.; Duboeuf, F.; Vray, D. Bimodal ultrasound and fluorescence approach for prostate cancer diagnosis. J. Biomed. Opt. 2009, 14, 064001. [Google Scholar] [CrossRef] [PubMed]
- Laidevant, A.; Herve, L.; Debourdeau, M.; Boutet, J.; Grenier, N.; Dinten, J.M. Fluorescence time-resolved imaging system embedded in an ultrasound prostate probe. Biomed. Opt. Express 2011, 2, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Theek, B.; Gremse, F.; Kunjachan, S.; Fokong, S.; Pola, R.; Pechar, M.; Deckers, R.; Storm, G.; Ehling, J.; Kiessling, F.; et al. Characterizing EPR-mediated passive drug targeting using contrast-enhanced functional ultrasound imaging. J. Control. Release 2014, 182, 83–89. [Google Scholar] [CrossRef] [PubMed]
- McCann, C.M.; Waterman, P.; Figueiredo, J.L.; Aikawa, E.; Weissleder, R.; Chen, J.W. Combined magnetic resonance and fluorescence imaging of the living mouse brain reveals glioma response to chemotherapy. Neuroimage 2009, 45, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Penet, M.F.; Mikhaylova, M.; Li, C.; Krishnamachary, B.; Glunde, K.; Pathak, A.P.; Bhujwalla, Z.M. Applications of molecular MRI and optical imaging in cancer. Future Med. Chem. 2010, 2, 975–988. [Google Scholar] [CrossRef]
- Mikhaylova, M.; Stasinopoulos, I.; Kato, Y.; Artemov, D.; Bhujwalla, Z.M. Imaging of cationic multifunctional liposome-mediated delivery of COX-2 siRNA. Cancer Gene Ther. 2009, 16, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Medarova, Z.; Pham, W.; Farrar, C.; Petkova, V.; Moore, A. In vivo imaging of siRNA delivery and silencing in tumors. Nat. Med. 2007, 13, 372–377. [Google Scholar] [CrossRef]














© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etrych, T.; Janoušková, O.; Chytil, P. Fluorescence Imaging as a Tool in Preclinical Evaluation of Polymer-Based Nano-DDS Systems Intended for Cancer Treatment. Pharmaceutics 2019, 11, 471. https://doi.org/10.3390/pharmaceutics11090471
Etrych T, Janoušková O, Chytil P. Fluorescence Imaging as a Tool in Preclinical Evaluation of Polymer-Based Nano-DDS Systems Intended for Cancer Treatment. Pharmaceutics. 2019; 11(9):471. https://doi.org/10.3390/pharmaceutics11090471
Chicago/Turabian StyleEtrych, Tomáš, Olga Janoušková, and Petr Chytil. 2019. "Fluorescence Imaging as a Tool in Preclinical Evaluation of Polymer-Based Nano-DDS Systems Intended for Cancer Treatment" Pharmaceutics 11, no. 9: 471. https://doi.org/10.3390/pharmaceutics11090471
APA StyleEtrych, T., Janoušková, O., & Chytil, P. (2019). Fluorescence Imaging as a Tool in Preclinical Evaluation of Polymer-Based Nano-DDS Systems Intended for Cancer Treatment. Pharmaceutics, 11(9), 471. https://doi.org/10.3390/pharmaceutics11090471