Development and Characterization of the Solvent-Assisted Active Loading Technology (SALT) for Liposomal Loading of Poorly Water-Soluble Compounds


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Challenges in Delivery of Poorly Water-Soluble Drugs
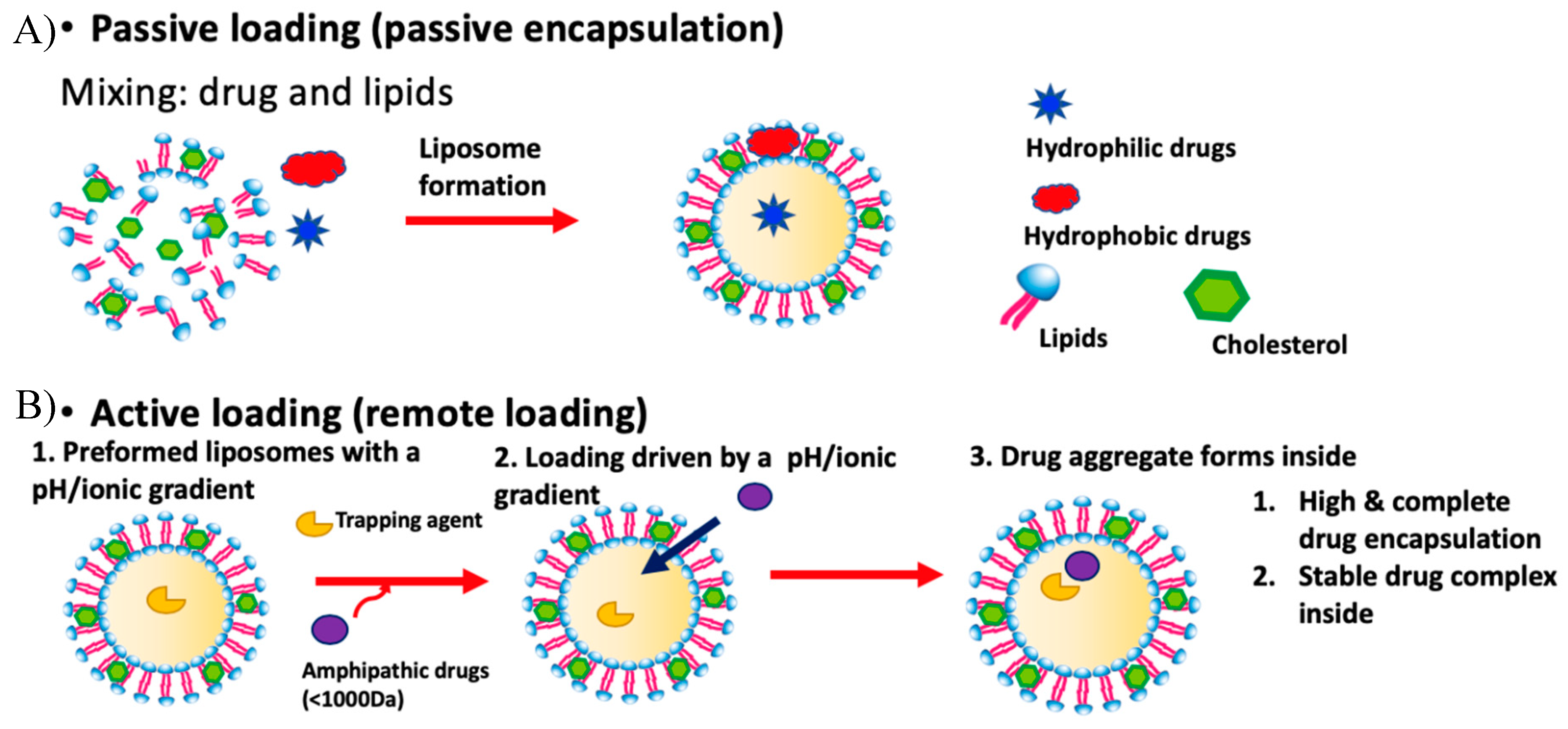
1.1.1. Liposomes and Drug Loading
1.1.2. Passive Loading
1.1.3. Limitations of Passive Loading
1.1.4. Active Loading
1.1.5. Limitations of Standard Active Loading
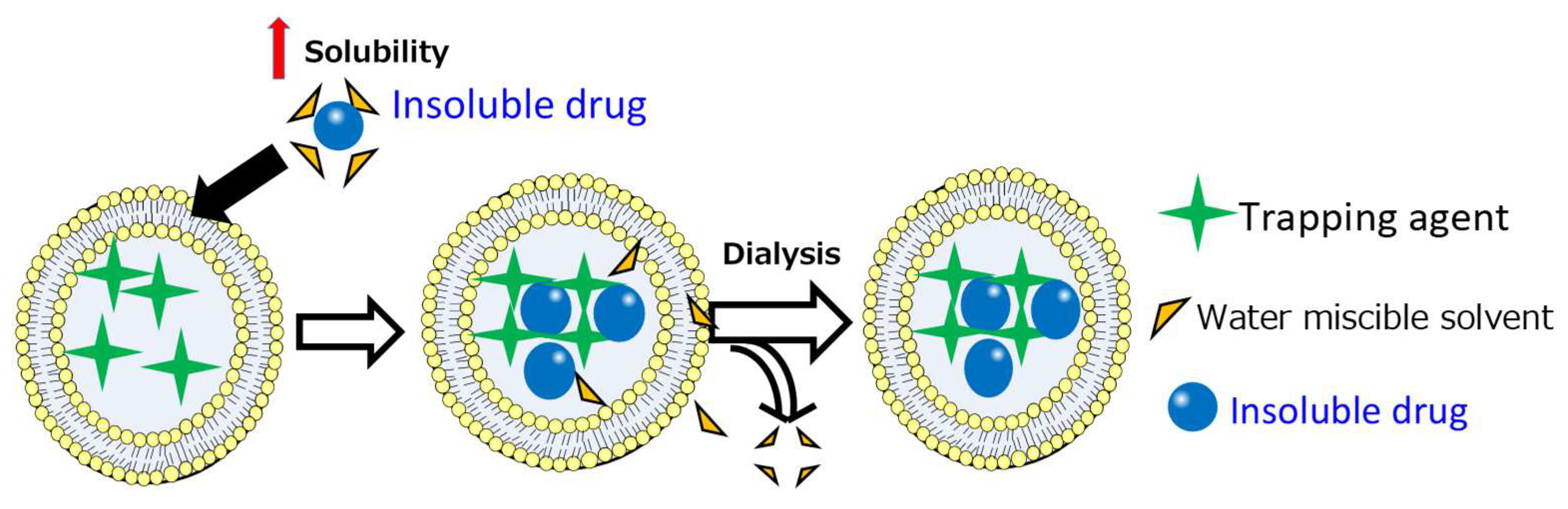
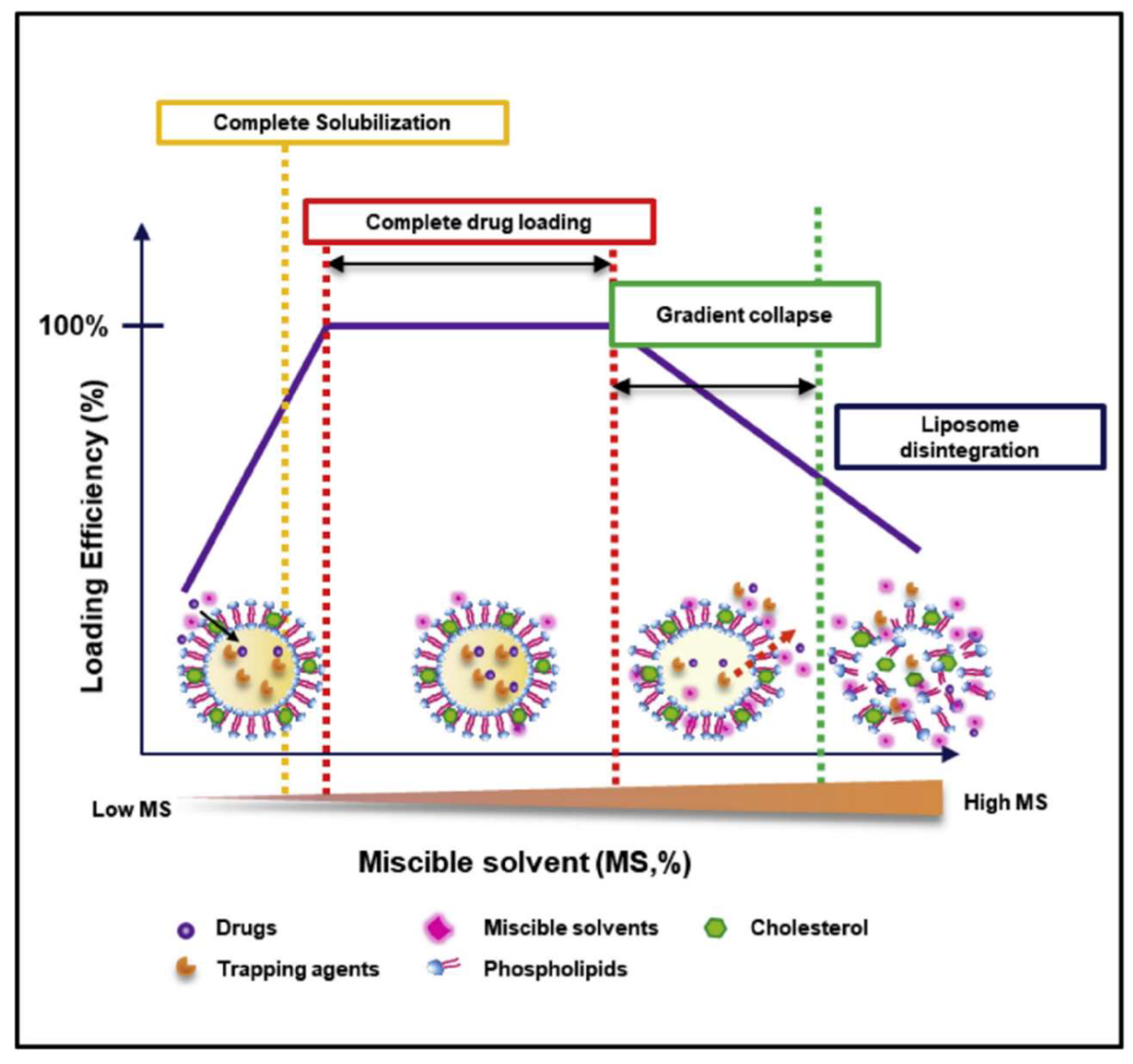
2. Solvent-Assisted Active Loading Technology (SALT)
2.1. Introduction
2.2. Applications and Mechanism
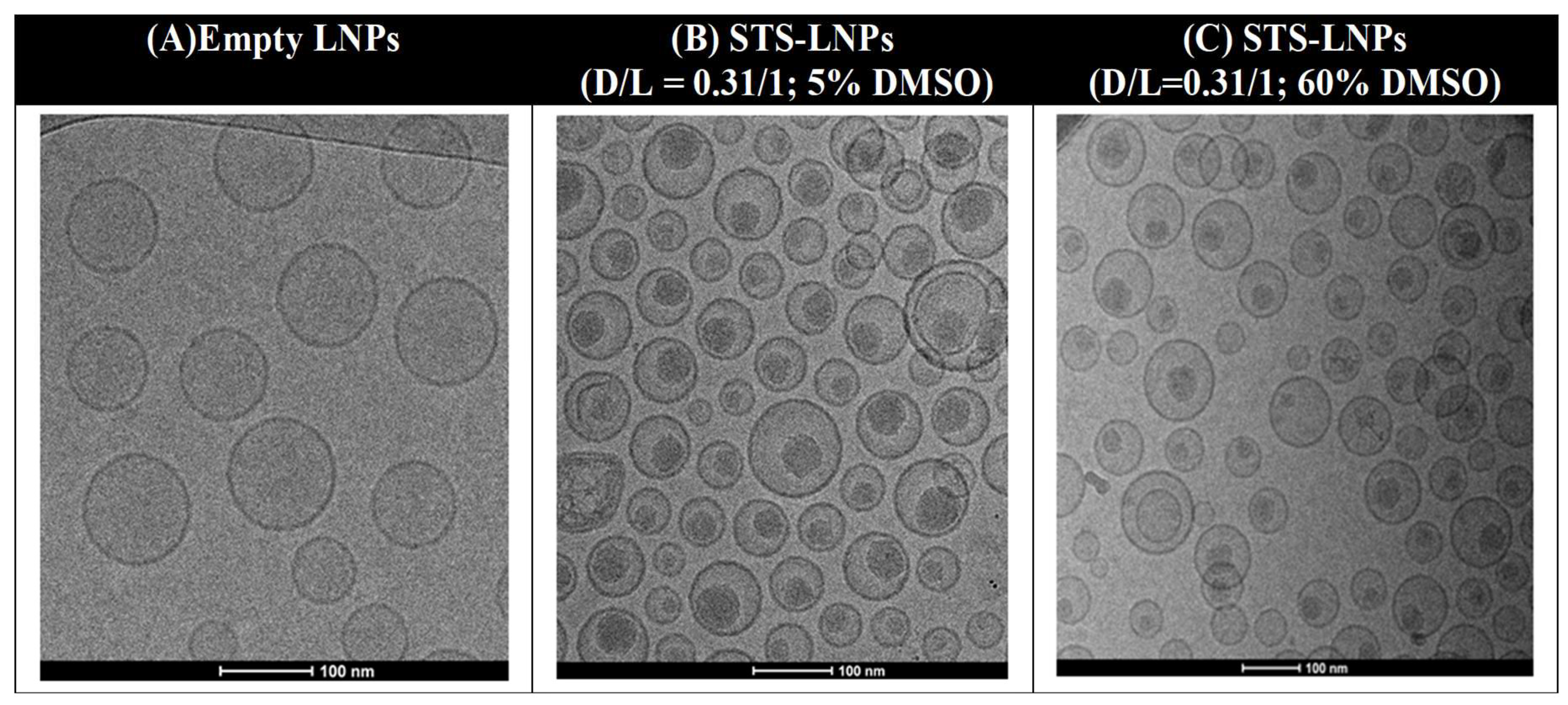
2.3. Proof-of-Principle with a Weak Base Drug
Staurosporine and Liposomal Loading
2.4. Proof-of-Principle with a Weak Acid Drug
2.4.1. Introduction
2.4.2. Gambogic Acid (GA)
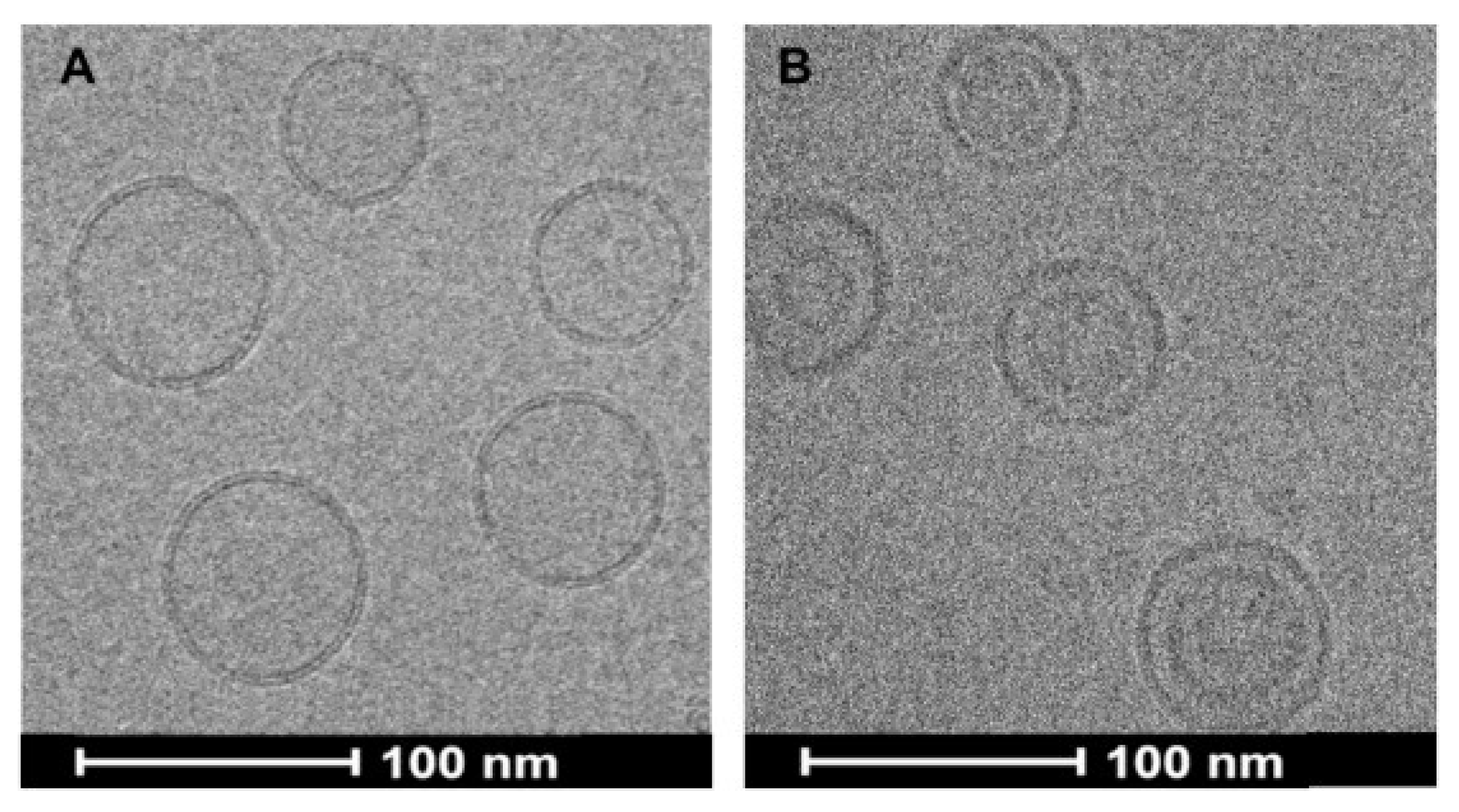
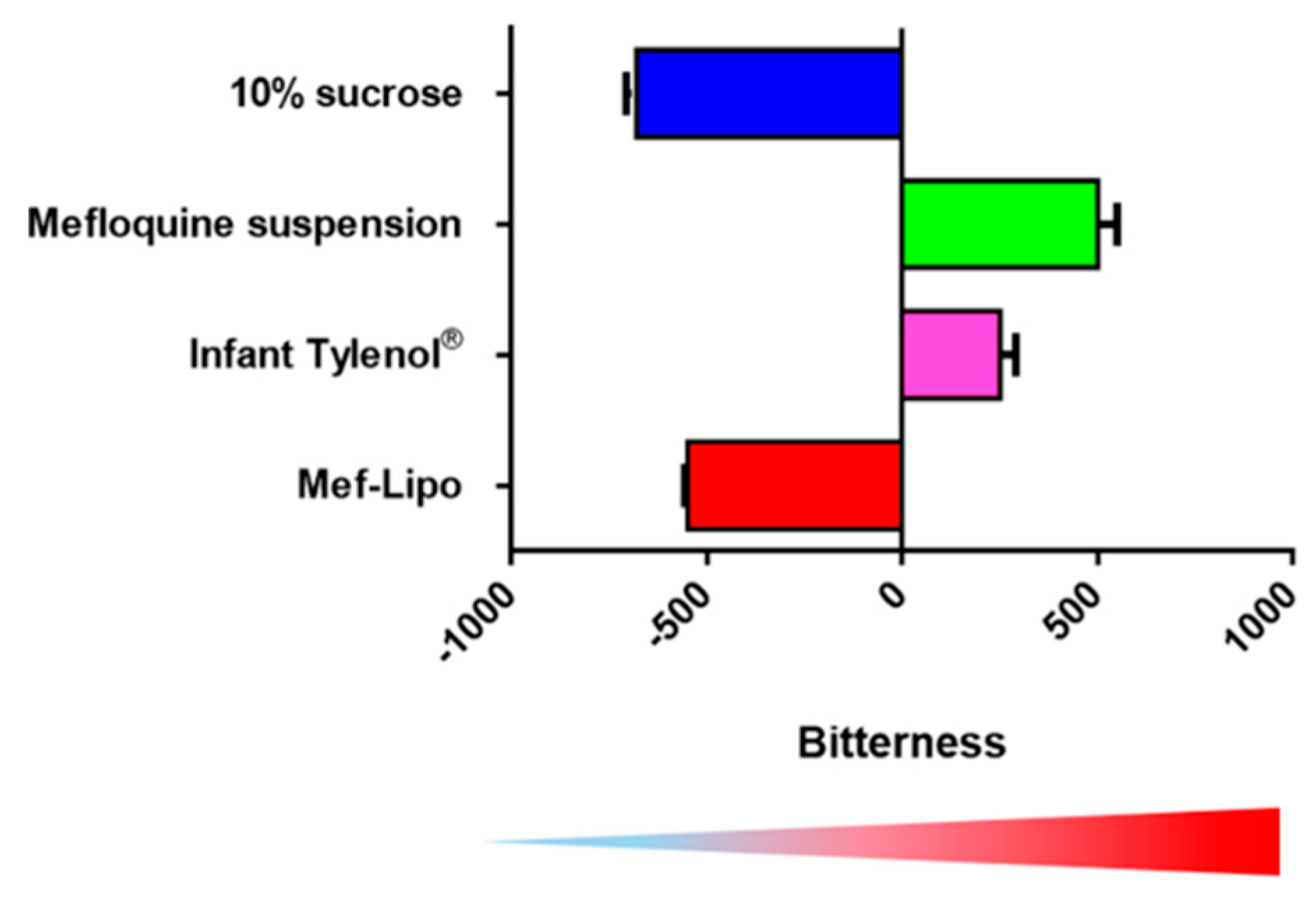
3. Pediatric Formulation
4. Perspectives and Future Directions
Funding
Conflicts of Interest
References
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Dressman, J.B. The developability classification system: Application of biopharmaceutics concepts to formulation development. J. Pharm. Sci. 2010, 99, 4940–4954. [Google Scholar] [CrossRef] [PubMed]
- McNamara, K.; Tofail, S.A.M. Nanoparticles in biomedical applications. Adv. Phys. X 2017, 2, 54–88. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation. Methods Mol. Biol. 2017, 1522, 17–22. [Google Scholar] [CrossRef]
- Cortesi, R.; Esposito, E.; Gambarin, S.; Telloli, P.; Menegatti, E.; Nastruzzi, C. Preparation of liposomes by reverse-phase evaporation using alternative organic solvents. J. Microencapsul. 1999, 16, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Lee, R.J.; Lee, L.J. Microfluidic methods for production of liposomes. Methods Enzymol. 2009, 465, 129–141. [Google Scholar] [CrossRef]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Maeki, M.; Kimura, N.; Sato, Y.; Harashima, H.; Tokeshi, M. Advances in microfluidics for lipid nanoparticles and extracellular vesicles and applications in drug delivery systems. Adv. Drug Deliv. Rev. 2018, 128, 84–100. [Google Scholar] [CrossRef]
- Dimov, N.; Kastner, E.; Hussain, M.; Perrie, Y.; Szita, N. Formation and purification of tailored liposomes for drug delivery using a module-based micro continuous-flow system. Sci. Rep. UK 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Omri, A. The effect of different lipid components on the in vitro stability and release kinetics of liposome formulations. Drug Deliv. 2004, 11, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.L.; Tang, W.H.; Szeitz, A.; Kulkarni, J.; Cullis, P.; Li, S.D. Systemic study of solvent-assisted active loading of gambogic acid into liposomes and its formulation optimization for improved delivery. Biomaterials 2018, 166, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.C.; May, J.P.; Chen, I.W.; Undzys, E.; Li, S.D. A Study of Liposomal Formulations to Improve the Delivery of Aquated Cisplatin to a Multidrug Resistant Tumor. Pharm. Res. 2015, 32, 3261–3268. [Google Scholar] [CrossRef]
- Bally, M.B.; Mayer, L.D.; Loughrey, H.; Redelmeier, T.; Madden, T.D.; Wong, K.; Harrigan, P.R.; Hope, M.J.; Cullis, P.R. Dopamine accumulation in large unilamellar vesicle systems induced by transmembrane ion gradients. Chem. Phys. Lipids 1988, 47, 97–107. [Google Scholar] [CrossRef]
- Mayer, L.D.; Bally, M.B.; Cullis, P.R. Uptake of Adriamycin into Large Unilamellar Vesicles in Response to a Ph Gradient. Biochim. Biophys. Acta 1986, 857, 123–126. [Google Scholar] [CrossRef]
- Deamer, D.W.; Prince, R.C.; Crofts, A.R. The response of fluorescent amines to pH gradients across liposome membranes. Biochim. Biophys. Acta 1972, 274, 323–335. [Google Scholar] [CrossRef]
- Haran, G.; Cohen, R.; Bar, L.K.; Barenholz, Y. Transmembrane Ammonium-Sulfate Gradients in Liposomes Produce Efficient and Stable Entrapment of Amphipathic Weak Bases. Biochim. Biophys. Acta 1993, 1151, 201–215. [Google Scholar] [CrossRef]
- Clerc, S.; Barenholz, Y. Loading of amphipathic weak acids into liposomes in response to transmembrane calcium acetate gradients. Biochim. Biophys. Acta 1995, 1240, 257–265. [Google Scholar] [CrossRef]
- Bhatt, P.; Lalani, R.; Vhora, I.; Patil, S.; Amrutiya, J.; Misra, A.; Mashru, R. Liposomes encapsulating native and cyclodextrin enclosed paclitaxel: Enhanced loading efficiency and its pharmacokinetic evaluation. Int. J. Pharm. 2018, 536, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Cipolla, D.; Rades, T.; Boyd, B.J. Drug nanocrystallisation within liposomes. J. Control. Release 2018, 288, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.L.; Tang, W.H.; Chen, W.C.; Diako, C.; Ross, C.F.; Li, S.D. Development of a Rapidly Dissolvable Oral Pediatric Formulation for Mefloquine Using Liposomes. Mol. Pharm. 2017, 14, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.L.; Chen, W.C.; Roy, A.; Undzys, E.; Li, S.D. A Simple and Improved Active Loading Method to Efficiently Encapsulate Staurosporine into Lipid-Based Nanoparticles for Enhanced Therapy of Multidrug Resistant Cancer. Pharm. Res. 2016, 33, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; Redwood, S.M.; Ohnuma, T.; Holland, J.F.; Droller, M.J.; Liu, B.C.S. Inhibition of Invasion of Invasive Human Bladder-Carcinoma Cells by Protein-Kinase-C Inhibitor Staurosporine. J. Natl. Cancer Inst. 1990, 82, 1753–1756. [Google Scholar] [CrossRef]
- Akinaga, S.; Gomi, K.; Morimoto, M.; Tamaoki, T.; Okabe, M. Antitumor-Activity of Ucn-01, a Selective Inhibitor of Protein-Kinase-C, in Murine and Human Tumor-Models. Cancer Res. 1991, 51, 4888–4892. [Google Scholar] [PubMed]
- Mukthavaram, R.; Jiang, P.F.; Saklecha, R.; Simberg, D.; Bharati, I.S.; Nomura, N.; Chao, Y.; Pastorino, S.; Pingle, S.C.; Fogal, V.; et al. High-efficiency liposomal encapsulation of a tyrosine kinase inhibitor leads to improved in vivo toxicity and tumor response profile. Int. J. Nanomed. 2013, 8, 3991–4006. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Guo, Q.L.; You, Q.D.; Zhao, L.; Gu, H.Y. Gambogic acid inhibits proliferation of human lung carcinoma SPC-A1 cells in vivo and in vitro and represses telomerase activity and telomerase reverse transcriptase mRNA expression in the cells. Biol. Pharm. Bull. 2004, 27, 1769–1774. [Google Scholar] [CrossRef]
- Li, X.F.; Liu, S.T.; Huang, H.B.; Liu, N.N.; Zhao, C.; Liao, S.Y.; Yang, C.S.; Liu, Y.R.; Zhao, C.G.; Li, S.J.; et al. Gambogic Acid Is a Tissue-Specific Proteasome Inhibitor In Vitro and In Vivo. Cell Rep. 2013, 3, 211–222. [Google Scholar] [CrossRef]
- Ishaq, M.; Khan, M.A.; Sharma, K.; Sharma, G.; Dutta, R.K.; Majumdar, S. Gambogic acid induced oxidative stress dependent caspase activation regulates both apoptosis and autophagy by targeting various key molecules (NF-kappa B, Beclin-1, p62 and NBR1) in human bladder cancer cells. Biochim. Biophys. Acta 2014, 1840, 3374–3384. [Google Scholar] [CrossRef]
- Cai, L.L.; Qiu, N.; Xiang, M.L.; Tong, R.S.; Yan, J.F.; He, L.; Shi, J.Y.; Chen, T.; Wen, J.L.; Wang, W.W.; et al. Improving aqueous solubility and antitumor effects by nanosized gambogic acid-mPEG(2000) micelles. Int. J. Nanomed. 2014, 9, 243–255. [Google Scholar] [CrossRef]
- Doddapaneni, R.; Patel, K.; Owaid, I.H.; Singh, M. Tumor neovasculature-targeted cationic PEGylated liposomes of gambogic acid for the treatment of triple-negative breast cancer. Drug Deliv. 2016, 23, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qian, H.Q.; Yang, M.; Li, R.T.; Hu, J.; Li, L.; Yu, L.X.; Liu, B.R.; Qian, X.P. Gambogic acid-loaded biomimetic nanoparticles in colorectal cancer treatment. Int. J. Nanomed. 2017, 12, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.K.; Yang, Y.; Cai, H.X.; Wang, F.; Peng, D.Y.; He, L.Q. Gambogic Acid-Loaded Electrosprayed Particles for Site-Specific Treatment of Hepatocellular Carcinoma. Mol. Pharm. 2014, 11, 4107–4117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.H.; Zou, Z.Y.; Ren, W.; Qian, H.Q.; Cheng, Q.F.; Ji, L.L.; Liu, B.R.; Liu, Q. Gambogic acid-loaded PEG-PCL nanoparticles act as an effective antitumor agent against gastric cancer. Pharm. Dev. Technol. 2018, 23, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Charrois, G.J.R.; Allen, T.M. Drug release rate influences the pharmacokinetics, biodistribution, therapeutic activity, and toxicity of pegylated liposomal doxorubicin formulations in murine breast cancer. Biochim. Biophys. Acta 2004, 1663, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.I.; Yeh, M.K. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar] [CrossRef]
- Kheirolomoom, A.; Mahakian, L.M.; Lai, C.Y.; Lindfors, H.A.; Seo, J.W.; Paoli, E.E.; Watson, K.D.; Haynam, E.M.; Ingham, E.S.; Xing, L.; et al. Copper-Doxorubicin as a Nanoparticle Cargo Retains Efficacy with Minimal Toxicity. Mol. Pharm. 2010, 7, 1948–1958. [Google Scholar] [CrossRef]
- Dicko, A.; Kwak, S.; Frazier, A.A.; Mayer, L.D.; Liboiron, B.D. Biophysical characterization of a liposomal formulation of cytarabine and daunorubeticin. Int. J. Pharm. 2010, 391, 248–259. [Google Scholar] [CrossRef]
- Lupescu, A.; Jilani, K.; Zelenak, C.; Zbidah, M.; Shaik, N.; Lang, F. Induction of Programmed Erythrocyte Death by Gambogic Acid. Cell. Physiol. Biochem. 2012, 30, 428–438. [Google Scholar] [CrossRef]
- Breman, J.G.; Alilio, M.S.; Mills, A. Conquering the intolerable burden of malaria: What’s new, what’s needed: A summary. Am. J. Trop. Med. Hyg. 2004, 71, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Caminade, C.; Kovats, S.; Rocklov, J.; Tompkins, A.M.; Morse, A.P.; Colon-Gonzalez, F.J.; Stenlund, H.; Martens, P.; Lloyd, S.J. Impact of climate change on global malaria distribution. Proc. Natl. Acad. Sci. USA 2014, 111, 3286–3291. [Google Scholar] [CrossRef] [PubMed]
- Schlagenhauf, P.; Adamcova, M.; Regep, L.; Schaerer, M.T.; Bansod, S.; Rhein, H.G. Use of mefloquine in children—A review of dosage, pharmacokinetics and tolerability data. Malar. J. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- White, N.J. Antimalarial drug resistance. J. Clin. Investig. 2004, 113, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- du Plessis, L.H.; Helena, C.; van Huysteen, E.; Wiesner, L.; Kotze, A.F. Formulation and evaluation of Pheroid vesicles containing mefloquine for the treatment of malaria. J. Pharm. Pharmcol. 2014, 66, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Mbela, T.K.M.; Deharo, E.; Haemers, A.; Ludwig, A. Submicron oil-in-water emulsion formulations for mefloquine and halofantrine: Effect of electric-charge inducers on antimalarial activity in mice. J. Pharm. Pharmacol. 1998, 50, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Schwartzberg, L.S.; Navari, R.M. Safety of Polysorbate 80 in the Oncology Setting. Adv. Ther. 2018, 35, 754–767. [Google Scholar] [CrossRef] [PubMed]
- Daeihamed, M.; Dadashzadeh, S.; Haeri, A.; Akhlaghi, M.F. Potential of Liposomes for Enhancement of Oral Drug Absorption. Curr. Drug Deliv. 2017, 14, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Kannan, V.; Balabathula, P.; Thoma, L.A.; Wood, G.C. Effect of sucrose as a lyoprotectant on the integrity of paclitaxel-loaded liposomes during lyophilization. J. Liposome Res. 2015, 25, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.L.; Tang, W.H.; Li, S.D. Cancer theranostic applications of lipid-based nanoparticles. Drug Discov. Today 2018, 23, 1159–1166. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pauli, G.; Tang, W.-L.; Li, S.-D. Development and Characterization of the Solvent-Assisted Active Loading Technology (SALT) for Liposomal Loading of Poorly Water-Soluble Compounds. Pharmaceutics 2019, 11, 465. https://doi.org/10.3390/pharmaceutics11090465
Pauli G, Tang W-L, Li S-D. Development and Characterization of the Solvent-Assisted Active Loading Technology (SALT) for Liposomal Loading of Poorly Water-Soluble Compounds. Pharmaceutics. 2019; 11(9):465. https://doi.org/10.3390/pharmaceutics11090465
Chicago/Turabian StylePauli, Griffin, Wei-Lun Tang, and Shyh-Dar Li. 2019. "Development and Characterization of the Solvent-Assisted Active Loading Technology (SALT) for Liposomal Loading of Poorly Water-Soluble Compounds" Pharmaceutics 11, no. 9: 465. https://doi.org/10.3390/pharmaceutics11090465
APA StylePauli, G., Tang, W.-L., & Li, S.-D. (2019). Development and Characterization of the Solvent-Assisted Active Loading Technology (SALT) for Liposomal Loading of Poorly Water-Soluble Compounds. Pharmaceutics, 11(9), 465. https://doi.org/10.3390/pharmaceutics11090465