Analytical and Computational Methods for the Estimation of Drug-Polymer Solubility and Miscibility in Solid Dispersions Development



 ,
,  and
and 
Abstract
:1. Introduction
2. Analytical Techniques for the Assessment of Drug-Polymer Solubility/Miscibility
2.1. Thermal Techniques
2.2. Spectroscopic Techniques
2.3. X-ray Powder Diffraction (XRPD)
2.4. Microscopic and Imaging Techniques
2.5. Other Techniques
2.6. Techniques Used in Combination
3. Computational Methods for the Assessment of Drug-Polymer Solubility/Miscibility
3.1. Solubility Parameters
3.2. Thermodynamic Modeling
Construction of the Phase Diagram
3.3. Computational Modeling and Simulations
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AFM | Atomic Force Microscopy; |
| AFM-IR (nano IR) | Nanoscale Infrared Spectroscopy; |
| BCS | Biopharmaceutics Classification System |
| COMPASS | Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies |
| DSC | Differential Scanning Calorimetry; |
| HEC | Hydroxyethyl Cellulose; |
| HPC | Hydroxypropyl Cellulose; |
| HPMC | Hydroxypropylmethyl Cellulose; |
| HPMCAS | Hydroxypropylmethyl Cellulose Acetate Succinate; |
| HPMCP | Hydroxypropylmethyl Cellulose Phthalate; |
| HSM | Hot Stage Microscopy; |
| MD | Molecular Dynamics; |
| M-DSC | Modulated-temperature Differential Scanning Calorimetry; |
| μ-CT | Micro-computed Tomography; |
| Na CMC | Sodium Carboxymethyl Cellulose; |
| nanoTA | Nanoscale Thermal Analysis; |
| NMR | Nuclear Magnetic Resonance; |
| PAA | Polyacrylic Acid; |
| PCFF | Polymer Consistent Force Field |
| PCRM | Pure Curve Resolution Method; |
| PC-SAFT | Perturbed-Chain Statistical Associating Fluid Theory; |
| Pair Distribution Functions; | |
| PEG | Polyethylene Glycol; |
| PHPA | α,β-poly(N-5-hydroxypentyl)-l-aspartamide; |
| PLM | Polarized Light Microscopy; |
| PVA | Poly(vinyl alcohol); |
| PVP | Polyvinylpyrrolidone; |
| PVPVA | Polyvinylpyrrolidone Vinyl Acetate; |
| SEM | Scanning Electron Microscopy; |
| SSNMR | Solid State Nuclear Magnetic Resonance; |
| TASC | Thermal Analysis by Structural Characterization; |
| TEM | Transmission Electron Microscopy; |
| Tg | Glass Transition Temperature; |
| TPGS | d-α-Tocopheryl Polyethylene Glycol 1000 Succinate; |
| TSDC | Thermally stimulated depolarization current |
References
- Vo, C.L.; Park, C.; Lee, B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Kanaujia, P.; Poovizhi, P.; Ng, W.K.; Tan, R.B.H. Amorphous formulations for dissolution and bioavailability enhancement of poorly soluble APIs. Powder Technol. 2015, 285, 2–15. [Google Scholar] [CrossRef]
- Lee, T.W.; Boersen, N.A.; Hui, H.W.; Chow, S.F.; Wan, K.Y.; Chow, A.H. Delivery of poorly soluble compounds by amorphous solid dispersions. Curr. Pharm. Des. 2014, 20, 303–324. [Google Scholar] [CrossRef] [PubMed]
- Medarević, D.P.; Kachrimanis, K.; Mitrić, M.; Djuriš, J.; Djurić, Z.; Ibrić, S. Dissolution rate enhancement and physicochemical characterization of carbamazepine-poloxamer solid dispersions. Pharm. Dev. Technol. 2016, 21, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Medarević, D.P.; Kleinebudde, P.; Djuriš, J.; Djurić, Z.; Ibrić, S. Combined application of mixture experimental design and artificial neural networks in the solid dispersion development. Drug Dev. Ind. Pharm. 2016, 42, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Djuris, J.; Ioannis, N.; Ibric, S.; Djuric, Z.; Kachrimanis, K. Effect of composition in the development of carbamazepine hot-melt extruded solid dispersions by application of mixture experimental design. J. Pharm. Pharmacol. 2014, 66, 232–243. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Obi, N. Studies on absorption of eutectic mixtures. I. A comparison of the behavior of eutectic mixtures of sulphathiazole and that of ordinary sulphathiazole in man. Chem. Pharm. Bull. 1961, 9, 866–872. [Google Scholar] [CrossRef]
- Tian, B.; Wang, X.; Zhang, Y.; Zhang, K.; Zhang, Y.; Tang, X. Theoretical prediction of a phase diagram for solid dispersions. Pharm. Res. 2015, 32, 840–851. [Google Scholar] [CrossRef]
- Higashi, K.; Hayashi, H.; Yamamoto, K.; Moribe, K. The effect of drug and EUDRAGIT® S 100 miscibility in solid dispersions on the drug and polymer dissolution rate. Int. J. Pharm. 2015, 494, 9–16. [Google Scholar] [CrossRef]
- Six, K.; Leuner, C.; Dressman, J.; Verreck, G.; Peeters, J.; Blaton, N.; Augustijns, P.; Kinget, R.; Van den Mooter, G. Thermal properties of hot-stage extrudates of itraconazole and eudragit E100. Phase separation and polymorphism. J. Therm. Anal. Calor. 2002, 68, 591–601. [Google Scholar] [CrossRef]
- Marsac, P.J.; Shamblin, S.L.; Taylor, L.S. Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility. Pharm. Res. 2006, 23, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Huang, J.; Hussain, M.A. Drug-polymer solubility and miscibility: Stability consideration and practical challenges in amorphous solid dispersion development. J. Pharm. Sci. 2010, 99, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Sun, Y.; Zhang, G.G.; Yu, L. Solubility of small-molecule crystals in polymers: D-mannitol in PVP, indomethacin in PVP/VA, and nifedipine in PVP/VA. Pharm. Res. 2009, 26, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Marsac, P.J.; Li, T.; Taylor, L.S. Estimation of drug-polymer miscibility and solubility in amorphous solid dispersions using experimentally determined interaction parameters. Pharm. Res. 2009, 26, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Van Humbeeck, J.; Van den Mooter, G. Theoretical and experimental investigation on the solid solubility and miscibility of naproxen in poly(vinylpyrrolidone). Mol. Pharm. 2010, 7, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Huang, Y. A thermal analysis method to predict the complete phase diagram of drug-polymer solid dispersions. Int. J. Pharm. 2010, 399, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Inbar, P.; Chokshi, H.P.; Malick, A.W.; Choi, D.S. Prediction of the thermal phase diagram of amorphous solid dispersions by Flory-Huggins theory. J. Pharm. Sci. 2011, 100, 3196–3207. [Google Scholar] [CrossRef]
- Tian, Y.; Booth, J.; Meehan, E.; Jones, D.S.; Li, S.; Andrews, G.P. Construction of drug-polymer thermodynamic phase diagrams using Flory-Huggins interaction theory: Identifying the relevance of temperature and drug weight fraction to phase separation within solid dispersions. Mol. Pharm. 2013, 10, 236–248. [Google Scholar] [CrossRef]
- Djuris, J.; Nikolakakis, I.; Ibric, S.; Djuric, Z.; Kachrimanis, K. Preparation of carbamazepine-Soluplus solid dispersions by hot-melt extrusion, and prediction of drug-polymer miscibility by thermodynamic model fitting. Eur. J. Pharm. Biopharm. 2013, 84, 228–237. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Theoretical and experimental investigation of drug-polymer interaction and miscibility and its impact on drug supersaturation in aqueous medium. Eur. J. Pharm. Biopharm. 2016, 107, 16–31. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry Ithaca; Cornell University: New York, NY, USA, 1953. [Google Scholar]
- Cassel, B.; Packer, R. Modulated Temperature DSC and the DSC 8500: A Step Up in Performance; Technical Note; PerkinElmer, Inc.: Waltham, MA, USA, 2010. [Google Scholar]
- Karavas, E.; Ktistis, G.; Xenakis, A.; Georgarakis, E. Miscibility behavior and formation mechanism of stabilized felodipine-polyvinylpyrrolidone amorphous solid dispersions. Drug. Dev. Ind. Pharm. 2005, 31, 473–489. [Google Scholar] [CrossRef]
- Maniruzzaman, M.; Morgan, D.J.; Mendham, A.P.; Pang, J.; Snowden, M.J.; Douroumis, D. Drug-polymer intermolecular interactions in hot-melt extruded solid dispersions. Int. J. Pharm. 2013, 443, 199–208. [Google Scholar] [CrossRef]
- Zheng, X.; Yang, R.; Tang, X.; Zheng, L. Part I: Characterization of solid dispersions of nimodipine prepared by hot-melt extrusion. Drug. Dev. Ind. Pharm. 2007, 33, 791–802. [Google Scholar] [CrossRef]
- Qi, S.; Belton, P.; Nollenberger, K.; Clayden, N.; Reading, M.; Craig, D.Q. Characterisation and prediction of phase separation in hot-melt extruded solid dispersions: A thermal, microscopic and NMR relaxometry study. Pharm. Res. 2010, 27, 1869–1883. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the second-order transitions of synthetic rubbers. I. Non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Simha, R.; Boyer, R.F. On a general relation involving the glass temperature and coefficients of expansion of polymers. J. Chem. Phys. 1962, 37, 1003–1007. [Google Scholar] [CrossRef]
- Couchman, P.R.; Karasz, F.E. A Classical Thermodynamic Discussion of the Effect of Composition on Glass-Transition Temperatures. Macromolecules 1978, 11, 117–119. [Google Scholar] [CrossRef]
- Fox, T.G. Influence of Diluent and of Copolymer Composition on the Glass Temperature of a Polymer System. Bull. Am. Phys. Soc. 1956, 1, 123. [Google Scholar]
- Kalogeras, I.M. A novel approach for analyzing glass-transition temperature vs. composition patterns: Application to pharmaceutical compound + polymer systems. Eur. J. Pharm. Sci. 2011, 42, 470–483. [Google Scholar] [CrossRef]
- Baird, J.A.; Taylor, L.S. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv. Drug. Deliv. Rev. 2012, 64, 396–421. [Google Scholar] [CrossRef]
- Taylor, L.S.; Zografi, G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm. Res. 1997, 14, 1691–1698. [Google Scholar] [CrossRef]
- Prasad, D.; Chauhan, H.; Atef, E. Amorphous stabilization and dissolution enhancement of amorphous ternary solid dispersions: Combination of polymers showing drug-polymer interaction for synergistic effects. J. Pharm. Sci. 2014, 103, 3511–3523. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, X.; Suwardie, H.; Wang, P.; Gogos, C.G. Miscibility studies of indomethacin and Eudragit® E PO by thermal, rheological, and spectroscopic analysis. J. Pharm. Sci. 2012, 101, 2204–2212. [Google Scholar] [CrossRef]
- Song, Y.; Yang, X.; Chen, X.; Nie, H.; Byrn, S.; Lubach, J.W. Investigation of drug-excipient interactions in lapatinib amorphous solid dispersions using solid-state NMR spectroscopy. Mol. Pharm. 2015, 12, 857–866. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Papadimitriou, S.; Karavas, E.; Georgarakis, E.; Docoslis, A.; Bikiaris, D. Improvement in chemical and physical stability of fluvastatin drug through hydrogen bonding interactions with different polymer matrices. Curr. Drug Deliv. 2009, 6, 101–112. [Google Scholar] [CrossRef]
- Meng, F.; Trivino, A.; Prasad, D.; Chauhan, H. Investigation and correlation of drug polymer miscibility and molecular interactions by various approaches for the preparation of amorphous solid dispersions. Eur. J. Pharm. Sci. 2015, 71, 12–24. [Google Scholar] [CrossRef]
- Newman, A.; Engers, D.; Bates, S.; Ivanisevic, I.; Kelly, R.C.; Zografi, G. Characterization of amorphous API: Polymer mixtures using x-ray powder diffraction. J. Pharm. Sci. 2008, 97, 4840–4856. [Google Scholar] [CrossRef]
- Bikiaris, D.; Papageorgiou, G.Z.; Stergiou, A.; Pavlidou, E.; Karavas, E.; Kanaze, F.; Georgarakis, M. Physicochemical studies on solid dispersions of poorly water-soluble drugs: Evaluation of capabilities and limitations of thermal analysis techniques. Thermochim. Acta 2005, 439, 58–67. [Google Scholar] [CrossRef]
- Fule, R.; Amin, P. Hot melt extruded amorphous solid dispersion of posaconazole with improved bioavailability: Investigating drug-polymer miscibility with advanced characterisation. Biomed. Res. Int. 2014, 2014, 1–16. [Google Scholar] [CrossRef]
- Sun, Y.; Tao, J.; Zhang, G.G.; Yu, L. Solubilities of crystalline drugs in polymers: An improved analytical method and comparison of solubilities of indomethacin and nifedipine in PVP, PVP/VA, and PVAc. J. Pharm. Sci. 2010, 99, 4023–4031. [Google Scholar] [CrossRef]
- Mahieu, A.; Willart, J.F.; Dudognon, E.; Danède, F.; Descamps, M. A new protocol to determine the solubility of drugs into polymer matrixes. Mol. Pharm. 2013, 10, 560–566. [Google Scholar] [CrossRef]
- Tian, Y.; Jones, D.S.; Donnelly, C.; Brannigan, T.; Li, S.; Andrews, G.P. A New Method of Constructing a Drug-Polymer Temperature-Composition Phase Diagram Using Hot-Melt Extrusion. Mol. Pharm. 2018, 15, 1379–1391. [Google Scholar] [CrossRef]
- Shimizu, H.; Horiuchi, S.; Nakayama, K. Structural analysis of miscible polymer blends using thermally stimulated depolarization current method. Proc. Int. Symp. Electrets. 1999, 553–556. [Google Scholar]
- Shmeis, R.A.; Wang, Z.; Krill, S.L. A mechanistic investigation of an amorphous pharmaceutical and its solid dispersions, part I: A comparative analysis by thermally stimulated depolarization current and differential scanning calorimetry. Pharm. Res. 2004, 21, 2025–2030. [Google Scholar] [CrossRef]
- Rumondor, A.C.; Ivanisevic, I.; Bates, S.; Alonzo, D.E.; Taylor, L.S. Evaluation of drug-polymer miscibility in amorphous solid dispersion systems. Pharm. Res. 2009, 26, 2523–2534. [Google Scholar] [CrossRef]
- Aso, Y.; Yoshioka, S.; Miyazaki, T.; Kawanishi, T.; Tanaka, K.; Kitamura, S.; Takakura, A.; Hayashi, T.; Muranushi, N. Miscibility of nifedipine and hydrophilic polymers as measured by (1)H-NMR spin-lattice relaxation. Chem. Pharm. Bull. 2007, 55, 1227–1231. [Google Scholar] [CrossRef]
- Calahan, J.L.; Azali, S.C.; Munson, E.J.; Nagapudi, K. Investigation of Phase Mixing in Amorphous Solid Dispersions of AMG 517 in HPMC-AS Using DSC, Solid-State NMR, and Solution Calorimetry. Mol. Pharm. 2015, 12, 4115–4123. [Google Scholar] [CrossRef]
- Lubach, J.W.; Hau, J. Solid-State NMR Investigation of Drug-Excipient Interactions and Phase Behavior in Indomethacin-Eudragit E Amorphous Solid Dispersions. Pharm. Res. 2018, 35, 65. [Google Scholar] [CrossRef]
- Yuan, X.; Sperger, D.; Munson, E.J. Investigating miscibility and molecular mobility of nifedipine-PVP amorphous solid dispersions using solid-state NMR spectroscopy. Mol. Pharm. 2014, 11, 329–337. [Google Scholar] [CrossRef]
- Geppi, M.; Guccione, S.; Mollica, G.; Pignatello, R.; Veracini, C.A. Molecular properties of ibuprofen and its solid dispersions with Eudragit RL100 studied by solid-state nuclear magnetic resonance. Pharm. Res. 2005, 22, 1544–1555. [Google Scholar] [CrossRef]
- Li, N.; Taylor, L.S. Nanoscale Infrared, Thermal, and Mechanical Characterization of Telaprevir-Polymer Miscibility in Amorphous Solid Dispersions Prepared by Solvent Evaporation. Mol. Pharm. 2016, 13, 1123–1136. [Google Scholar] [CrossRef]
- Yoo, S.U.; Krill, S.L.; Wang, Z.; Telang, C. Miscibility/Stability Considerations in Binary Solid Dispersion Systems Composed of Functional Excipients towards the Design of Multi-Component Amorphous Systems. J. Pharm. Sci. 2009, 98, 4711–4723. [Google Scholar] [CrossRef]
- Martinez-Marcos, L.; Lamprou, D.A.; McBurney, R.T.; Halbert, G.W. A novel hot-melt extrusion formulation of albendazole for increasing dissolution properties. Int. J. Pharm. 2016, 499, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Démuth, B.; Farkas, A.; Pataki, H.; Balogh, A.; Szabó, B.; Borbás, E.; Sóti, P.L.; Vigh, T.; Kiserdei, É.; Farkas, B.; et al. Detailed stability investigation of amorphous solid dispersions prepared by single-needle and high speed electrospinning. Int. J. Pharm. 2016, 498, 234–244. [Google Scholar] [CrossRef]
- Padilla, A.M.; Ivanisevic, I.; Yang, Y.; Engers, D.; Bogner, R.H.; Pikal, M.J. The study of phase separation in amorphous freeze-dried systems. Part I: Raman mapping and computational analysis of XRPD data in model polymer systems. J. Pharm. Sci. 2011, 100, 206–222. [Google Scholar] [CrossRef]
- Qian, F.; Huang, J.; Zhu, Q.; Haddadin, R.; Gawel, J.; Garmise, R.; Hussain, M. Is a distinctive single Tg a reliable indicator for the homogeneity of amorphous solid dispersion? Int. J. Pharm. 2010, 395, 232–235. [Google Scholar] [CrossRef]
- Reading, M.; Qi, S.; Alhijjaj, M. Local Thermal Analysis by Structural Characterization (TASC). In Thermal Physics and Thermal Analysis. Hot Topics in Thermal Analysis and Calorimetry; Šesták, J., Hubík, P., Mareš, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 1–10. [Google Scholar]
- Alhijjaj, M.; Belton, P.; Fábián, L.; Wellner, N.; Reading, M.; Qi, S. Novel Thermal Imaging Method for Rapid Screening of Drug-Polymer Miscibility for Solid Dispersion Based Formulation Development. Mol. Pharm. 2018, 15, 5625–5636. [Google Scholar] [CrossRef]
- Alhijjaj, M.; Yassin, S.; Reading, M.; Zeitler, J.A.; Belton, P.; Qi, S. Characterization of Heterogeneity and Spatial Distribution of Phases in Complex Solid Dispersions by Thermal Analysis by Structural Characterization and X-ray Micro Computed Tomography. Pharm. Res. 2017, 34, 971–989. [Google Scholar] [CrossRef]
- Crowley, K.J.; Zografi, G. Water Vapor Absorption into Amorphous Hydrophobic Drug/Poly(vinylpyrrolidone) Dispersions. J. Pharm. Sci. 2002, 91, 2150–2165. [Google Scholar] [CrossRef]
- Gupta, S.S.; Parikh, T.; Meena, A.K.; Mahajan, N.; Vitez, I.; Serajuddin, A.T.M. Effect of carbamazepine on viscoelastic properties and hot melt extrudability of Soluplus®. Int. J. Pharm. 2015, 478, 232–239. [Google Scholar] [CrossRef]
- Marsac, P.J.; Rumondor, A.C.; Nivens, D.E.; Kestur, U.S.; Stanciu, L.; Taylor, L.S. Effect of temperature and moisture on the miscibility of amorphous dispersions of felodipine and poly(vinyl pyrrolidone). J. Pharm. Sci. 2010, 99, 169–185. [Google Scholar] [CrossRef]
- Janssens, S.; Denivelle, S.; Rombaut, P.; Van den Mooter, G. Influence of polyethylene glycol chain length on compatibility and release characteristics of ternary solid dispersions of itraconazole in polyethylene glycol/hydroxypropylmethylcellulose 2910 E5 blends. Eur. J. Pharm. Sci. 2008, 35, 203–210. [Google Scholar] [CrossRef]
- Gumaste, S.G.; Gupta, S.S.; Serajuddin, A.T. Investigation of Polymer-Surfactant and Polymer-Drug-Surfactant Miscibility for Solid Dispersion. AAPS J. 2016, 18, 1131–1143. [Google Scholar] [CrossRef]
- Parikh, T.; Gupta, S.S.; Meena, A.K.; Vitez, I.; Mahajan, N.; Serajuddin, A.T. Application of Film-Casting Technique to Investigate Drug-Polymer Miscibility in Solid Dispersion and Hot-Melt Extrudate. J. Pharm. Sci. 2015, 104, 2142–2152. [Google Scholar] [CrossRef]
- Alhijjaj, M.; Belton, P.; Qi, S. An investigation into the use of polymer blends to improve the printability of and regulate drug release from pharmaceutical solid dispersions prepared via fused deposition modeling (FDM) 3D printing. Eur. J. Pharm. Biopharm. 2016, 108, 111–125. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, K. Miscibility analysis of particulate solid dispersions prepared by electrospray deposition. Int. J. Pharm. 2012, 433, 71–78. [Google Scholar] [CrossRef]
- Sanchez-Rexach, E.; Meaurio, E.; Iturri, J.; Toca-Herrera, J.L.; Nir, S.; Reches, M.; Sarasua, J.R. Miscibility, interactions and antimicrobial activity of poly(ε-caprolactone)/chloramphenicol blends. Eur. Polym. J. 2018, 102, 30–37. [Google Scholar] [CrossRef]
- Al-Obaidi, H.; Lawrence, M.J.; Al-Saden, N.; Ke, P. Investigation of griseofulvin and hydroxypropylmethyl cellulose acetate succinate miscibility in ball milled solid dispersions. Int. J. Pharm. 2013, 443, 95–102. [Google Scholar] [CrossRef]
- Solanki, N.G.; Lam, K.; Tahsin, M.; Gumaste, S.G.; Shah, A.V.; Serajuddin, A.T.M. Effects of Surfactants on Itraconazole-HPMCAS Solid Dispersion Prepared by Hot-Melt Extrusion I: Miscibility and Drug Release. J. Pharm. Sci. 2019, 108, 1453–1465. [Google Scholar] [CrossRef]
- Xi, L.; Song, H.; Wang, Y.; Gao, H.; Fu, Q. Lacidipine Amorphous Solid Dispersion Based on Hot Melt Extrusion: Good Miscibility, Enhanced Dissolution, and Favorable Stability. AAPS Pharm. Sci. Tech. 2018, 19, 3076–3084. [Google Scholar] [CrossRef]
- Hu, X.Y.; Lou, H.; Hageman, M.J. Preparation of lapatinibditosylate solid dispersions using solvent rotary evaporation and hot melt extrusion for solubility and dissolution enhancement. Int. J. Pharm. 2018, 552, 154–163. [Google Scholar] [CrossRef]
- Paudel, A.; Van den Mooter, G. Influence of Solvent Composition on the Miscibility and Physical Stability of Naproxen/PVP K 25 Solid Dispersions Prepared by Cosolvent Spray-Drying. Pharm. Res. 2012, 29, 251–270. [Google Scholar] [CrossRef]
- Hildebrand, J.; Scott, R.L. The Solubility of Nonelectrolytes, 3rd ed.; Reinhold: New York, NY, USA, 1950. [Google Scholar]
- Hildebrand, J.; Scott, R.L. Regular Solutions; Prentice-Hall: Englewood Cliffs, NJ, USA, 1962. [Google Scholar]
- Scatchard, G. Equilibria in Non-electrolyte Solutions in Relation to the Vapor Pressures and Densities of the Components. Chem. Rev. 1931, 8, 321–333. [Google Scholar] [CrossRef]
- Van Krevelen, D.W.; Te Nijenhuis, K. Properties of Polymers: Their Correlation with Chemical Structure; Their Numerical Estimation and Prediction from Additive Group Contributions; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Rey-Mermet, C.; Ruelle, P.; Nam-Trân, H.; Buchmann, M.; Kesselring, U.W. Significance of partial and total cohesion parameters of pharmaceutical solids determined from dissolution calorimetric measurements. Pharm. Res. 1991, 8, 636–642. [Google Scholar] [CrossRef]
- Şen, M.; Güven, O. Determination of solubility parameter of poly(N-vinyl 2-pyrrolidon/ethylene glycol dimethacrylate) gels by swelling measurements. J. Polym. Sci. Part B 1998, 36, 213–219. [Google Scholar] [CrossRef]
- Bozdogan, A.E. A method for determination of thermodynamic and solubility parameters of polymers from temperature and molecular weight dependence of intrinsic viscosity. Polymer 2004, 45, 6415–6424. [Google Scholar] [CrossRef]
- Adamska, K.; Voelkel, A. Inverse gas chromatographic determination of solubility parameters of excipients. Int. J. Pharm. 2005, 304, 11–17. [Google Scholar] [CrossRef]
- Small, P.A. Some factors affecting the solubility of polymers. J. Appl. Chem. 1953, 3, 71–80. [Google Scholar] [CrossRef]
- Hoy, K.L. New values of the solubility parameters from vapor pressure data. J. Paint Technol. 1970, 42, 76–118. [Google Scholar]
- Van Krevelen, D.W.; Hoftyzer, P.J. Properties of Polymers: Their Estimation and Correlation with Chemical Structure, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1976; pp. 129–159. [Google Scholar]
- Fedors, R.F. A method for estimating both the solubility parameters and molar volumes of liquids. Polym. Eng. Sci. 1974, 14, 147–154. [Google Scholar] [CrossRef]
- Hansen, C.M. The universality of the solubility parameter. Ind. Eng. Chem. Prod. Res. Dev. 1969, 8, 2–11. [Google Scholar] [CrossRef]
- Hoy, K.L. The Hoy Tables of Solubility Parameters; Union Carbide Corporation, Solvents & Coatings Materials, Research & Development Department: South Charleston, WV, USA, 1985. [Google Scholar]
- Hoy, K.L. Solubility parameter as a design parameter for water-borne polymers and coatings. J. Coated Fabrics 1989, 19, 53–67. [Google Scholar] [CrossRef]
- Mavrovouniotis, M.L. Estimation of Properties from Conjugate Forms of Molecular Structures: The ABC Approach. Ind. Eng. Chem. Res. 1990, 29, 1943–1953. [Google Scholar] [CrossRef]
- Stefanis, E.; Constantinou, L.; Panayiotou, C. A Group-Contribution Method for Predicting Pure Component Properties of Biochemical and Safety Interest. Ind. Eng. Chem. Res. 2004, 43, 6253–6261. [Google Scholar] [CrossRef]
- Stefanis, E.; Panayiotou, C. Prediction of hansen solubility parameters with a new group-contribution method. Int. J. Thermophys. 2008, 29, 568–585. [Google Scholar] [CrossRef]
- Stefanis, E.; Panayiotou, C. A new expanded solubility parameter approach. Int. J. Pharm. 2012, 426, 29–43. [Google Scholar] [CrossRef]
- Just, S.; Sievert, F.; Thommes, M.; Breitkreutz, J. Improved group contribution parameter set for the application of solubility parameters to melt extrusion. Eur. J. Pharm. Biopharm. 2013, 85, 1191–1199. [Google Scholar] [CrossRef]
- Lydersen, A.L. Estimation of Critical Properties of Organic Compounds; Engineering Experiment Station Report 3; College of Engineering, University of Wisconsin: Madison, WI, USA, 1955. [Google Scholar]
- Maniruzzaman, M.; Pang, J.; Morgan, D.J.; Douroumis, D. Molecular modeling as a predictive tool for the development of solid dispersions. Mol. Pharm. 2015, 12, 1040–1049. [Google Scholar] [CrossRef]
- Piccinni, P.; Tian, Y.; McNaughton, A.; Fraser, J.; Brown, S.; Jones, D.S.; Li, S.; Andrews, G.P. Solubility parameter-based screening methods for early-stage formulation development of itraconazole amorphous solid dispersions. J. Pharm. Pharmacol. 2016, 68, 705–720. [Google Scholar] [CrossRef]
- Bagley, E.B.; Nelson, T.P.; Scigliano, J.M. Three-dimensional solubility parameters and their relationship to internal pressure measurements in polar and hydrogen bonding solvents. J. Paint Technol. 1971, 43, 35. [Google Scholar]
- Greenhalgh, D.J.; Williams, A.C.; Timmins, P.; York, P. Solubility parameters as predictors of miscibility in solid dispersions. J. Pharm. Sci. 1999, 88, 1182–1190. [Google Scholar] [CrossRef]
- Forster, A.; Hempenstall, J.; Tucker, I.; Rades, T. Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis. Int. J. Pharm. 2001, 226, 147–161. [Google Scholar] [CrossRef]
- Chan, S.Y.; Qi, S.; Craig, D.Q. An investigation into the influence of drug-polymer interactions on the miscibility, processability and structure of polyvinylpyrrolidone-based hot melt extrusion formulations. Int. J. Pharm. 2015, 496, 95–106. [Google Scholar] [CrossRef]
- Donnelly, C.; Tian, Y.; Potter, C.; Jones, D.S.; Andrews, G.P. Probing the effects of experimental conditions on the character of drug-polymer phase diagrams constructed using Flory-Huggins theory. Pharm. Res. 2015, 32, 167–179. [Google Scholar] [CrossRef]
- Bansal, K.; Baghel, U.S.; Thakral, S. Construction and Validation of Binary Phase Diagram for Amorphous Solid Dispersion Using Flory-Huggins Theory. AAPS Pharm. Sci. Tech. 2016, 17, 318–327. [Google Scholar] [CrossRef]
- Lu, J.; Cuellar, K.; Hammer, N.I.; Jo, S.; Gryczke, A.; Kolter, K.; Langley, N.; Repka, M.A. Solid-state characterization of Felodipine-Soluplus amorphous solid dispersions. Drug Dev. Ind. Pharm. 2016, 42, 485–496. [Google Scholar] [CrossRef]
- Purohit, H.S.; Taylor, L.S. Miscibility of Itraconazole-Hydroxypropyl Methylcellulose Blends: Insights with High Resolution Analytical Methodologies. Mol. Pharm. 2015, 12, 4542–4553. [Google Scholar] [CrossRef]
- He, Y.; Ho, C. Amorphous Solid Dispersions: Utilization and Challenges in Drug Discovery and Development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: New York, NY, USA, 2003. [Google Scholar]
- Yang, M.; Wang, P.; Gogos, C. Prediction of acetaminophen’s solubility in poly(ethylene oxide) at room temperature using the Flory-Huggins theory. Drug Dev. Ind. Pharm. 2013, 39, 102–108. [Google Scholar] [CrossRef]
- Prudic, A.; Ji, Y.; Sadowski, G. Thermodynamic phase behavior of API/polymer solid dispersions. Mol. Pharm. 2014, 11, 2294–2304. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Long-Term Physical Stability of PVP- and PVPVA-Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 157–171. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Impact of Polymer Type and Relative Humidity on the Long-Term Physical Stability of Amorphous Solid Dispersions. Mol. Pharm. 2017, 14, 4374–4386. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Bartels, M.; Degenhardt, M.; Sadowski, G. Physical stability of API/polymer-blend amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2018, 124, 147–157. [Google Scholar] [CrossRef]
- Luebbert, C.; Sadowski, G. Moisture-induced phase separation and recrystallization in amorphous solid dispersions. Int. J. Pharm. 2017, 532, 635–646. [Google Scholar] [CrossRef]
- Prudic, A.; Kleetz, T.; Korf, M.; Ji, Y.; Sadowski, G. Influence of copolymer composition on the phase behavior of solid dispersions. Mol. Pharm. 2014, 11, 4189–4198. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef]
- Dünweg, B. Molecular dynamics algorithms and hydrodynamic screening. J. Chem. Phys. 1993, 99, 6977–6982. [Google Scholar] [CrossRef]
- Karplus, M.; Petsko, G.A. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef]
- Cui, Y. Using molecular simulations to probe pharmaceutical materials. J. Pharm. Sci. 2011, 100, 2000–2019. [Google Scholar] [CrossRef]
- Gupta, J.; Nunes, C.; Vyas, S.; Jonnalagadda, S. Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 2014–2023. [Google Scholar] [CrossRef]
- Anwar, J.; Khan, S.; Lindfors, L. Secondary crystal nucleation: Nuclei breeding factory uncovered. Angew. Chem. Int. Ed. 2015, 54, 14681–14684. [Google Scholar] [CrossRef]
- Gupta, J.; Nunes, C.; Jonnalagadda, S. A molecular dynamics approach for predicting the glass transition temperature and plasticization effect in amorphous pharmaceuticals. Mol. Pharm. 2013, 10, 4136–4145. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Karagianni, A.; Katopodis, K.; Vardaka, E.; Kachrimanis, K. Molecular modelling and simulation of fusion-based amorphous drug dispersions in polymer/plasticizer blends. Eur. J. Pharm. Sci. 2019, 130, 260–268. [Google Scholar] [CrossRef]
- Edueng, K.; Mahlin, D.; Bergström, C.A.S. The Need for Restructuring the Disordered Science of Amorphous Drug Formulations. Pharm. Res. 2017, 34, 1754–1772. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.; Wagner, K.G.; Kornherr, A.; Zifferer, G. Molecular dynamics simulations for drug dosage form development: Thermal and solubility characteristics for hot-melt extrusion. Mol. Simul. 2008, 34, 1197–1207. [Google Scholar] [CrossRef]
- Macháčková, M.; Tokarský, J.; Čapková, P. A simple molecular modeling method for the characterization of polymeric drug carriers. Eur. J. Pharm. Sci. 2013, 48, 316–322. [Google Scholar] [Green Version]
- Barmpalexis, P.; Karagianni, A.; Kachrimanis, K. Molecular simulations for amorphous drug formulation: Polymeric matrix properties relevant to hot-melt extrusion. Eur. J. Pharm. Sci. 2018, 119, 259–267. [Google Scholar] [CrossRef]
- Ouyang, D. Investigating the molecular structures of solid dispersions by the simulated annealing method. Chem. Phys. Lett. 2012, 554, 177–184. [Google Scholar] [CrossRef]
- Xiang, T.X.; Anderson, B.D. Molecular dynamics simulation of amorphous indomethacin-poly(vinylpyrrolidone) glasses: Solubility and hydrogen bonding interactions. J. Pharm. Sci. 2013, 102, 876–891. [Google Scholar] [CrossRef]
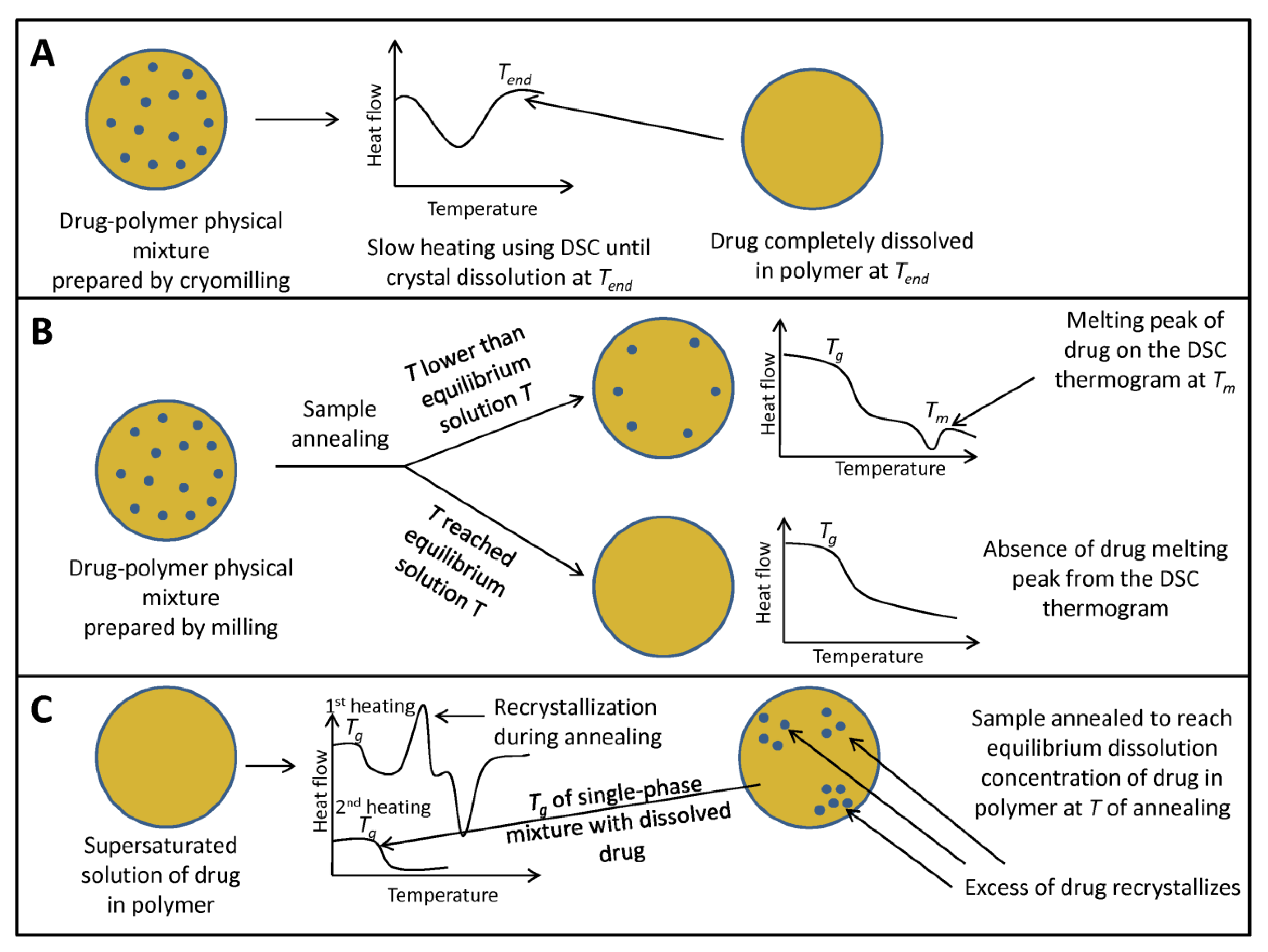
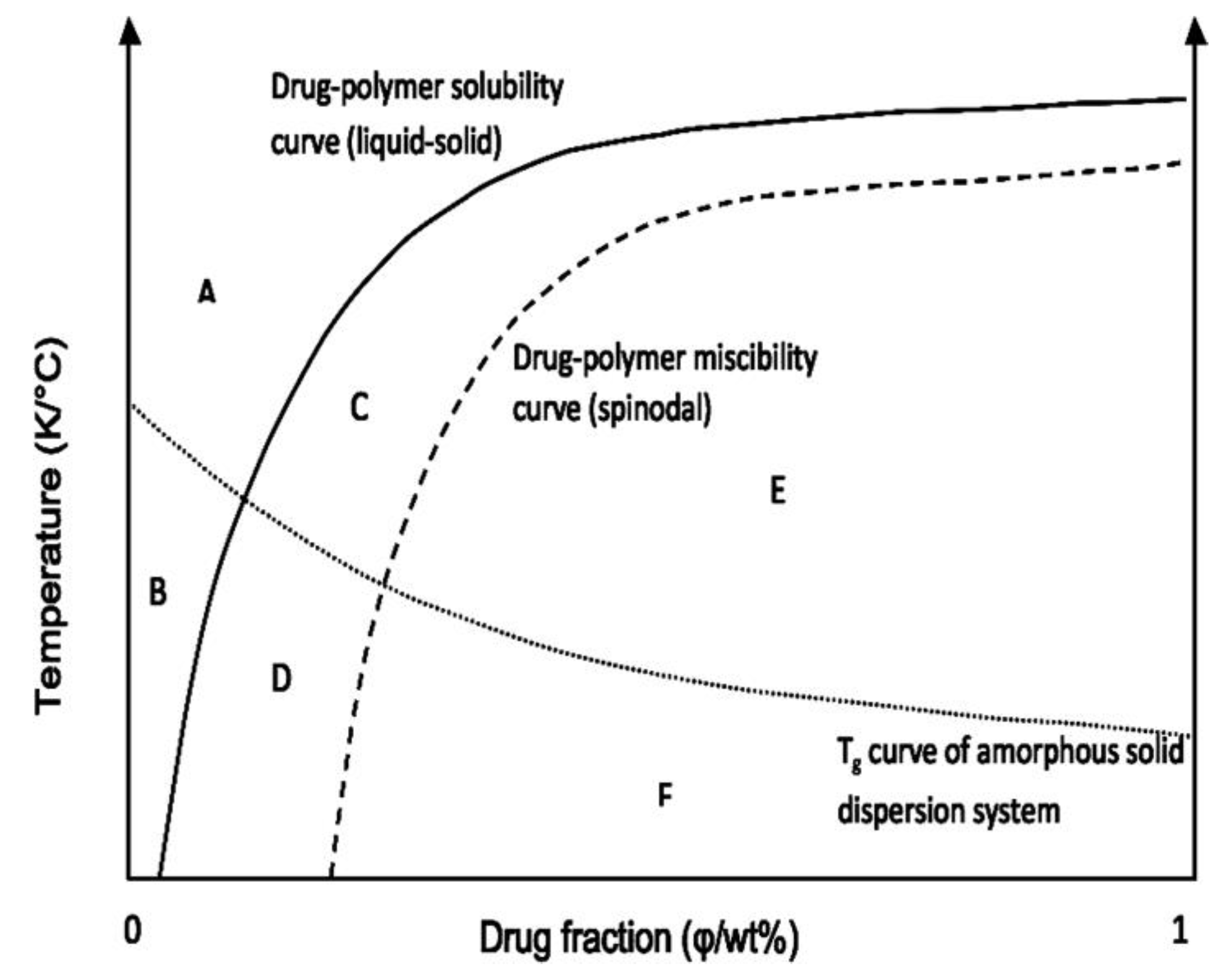

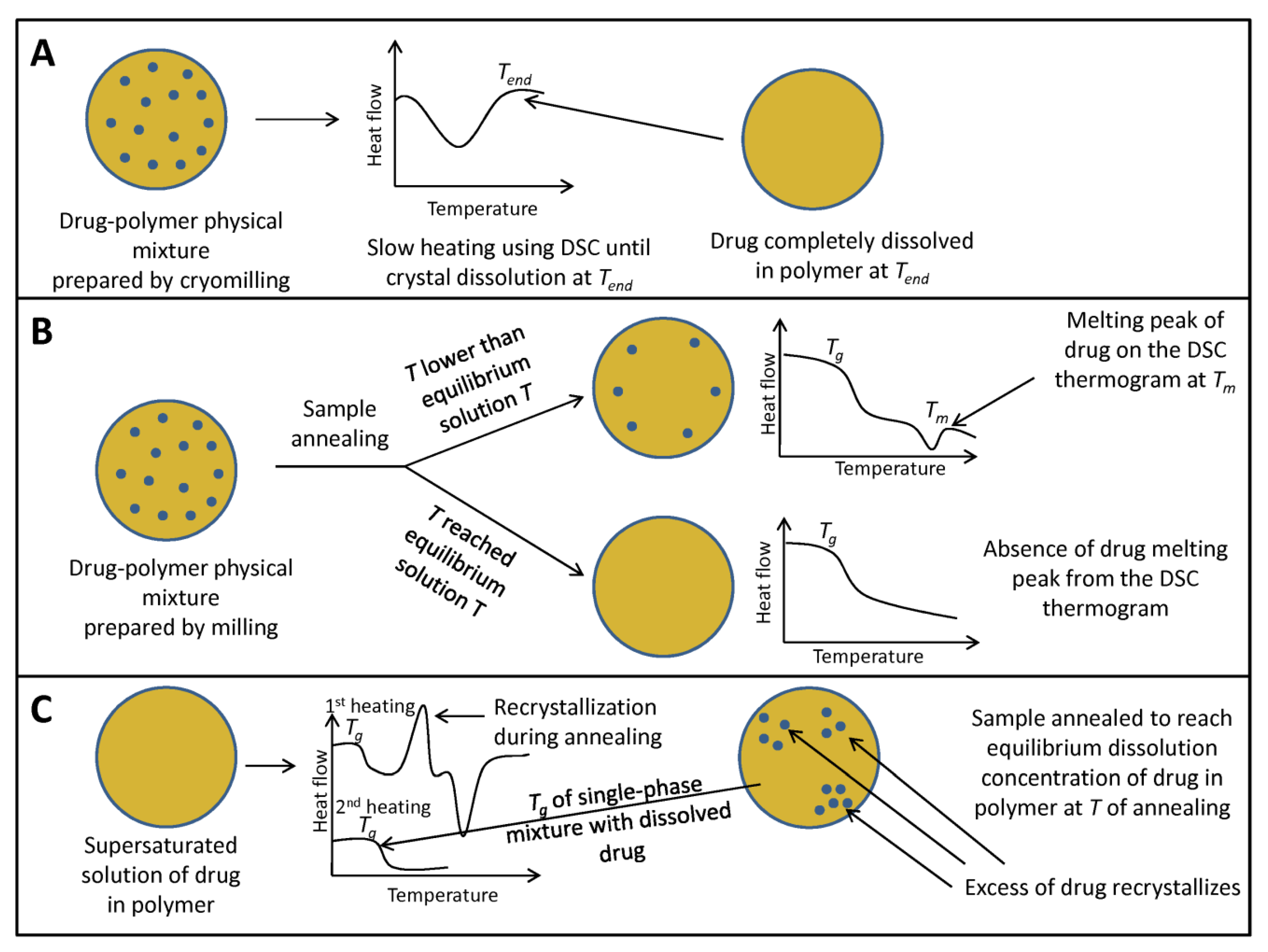{kind=link}
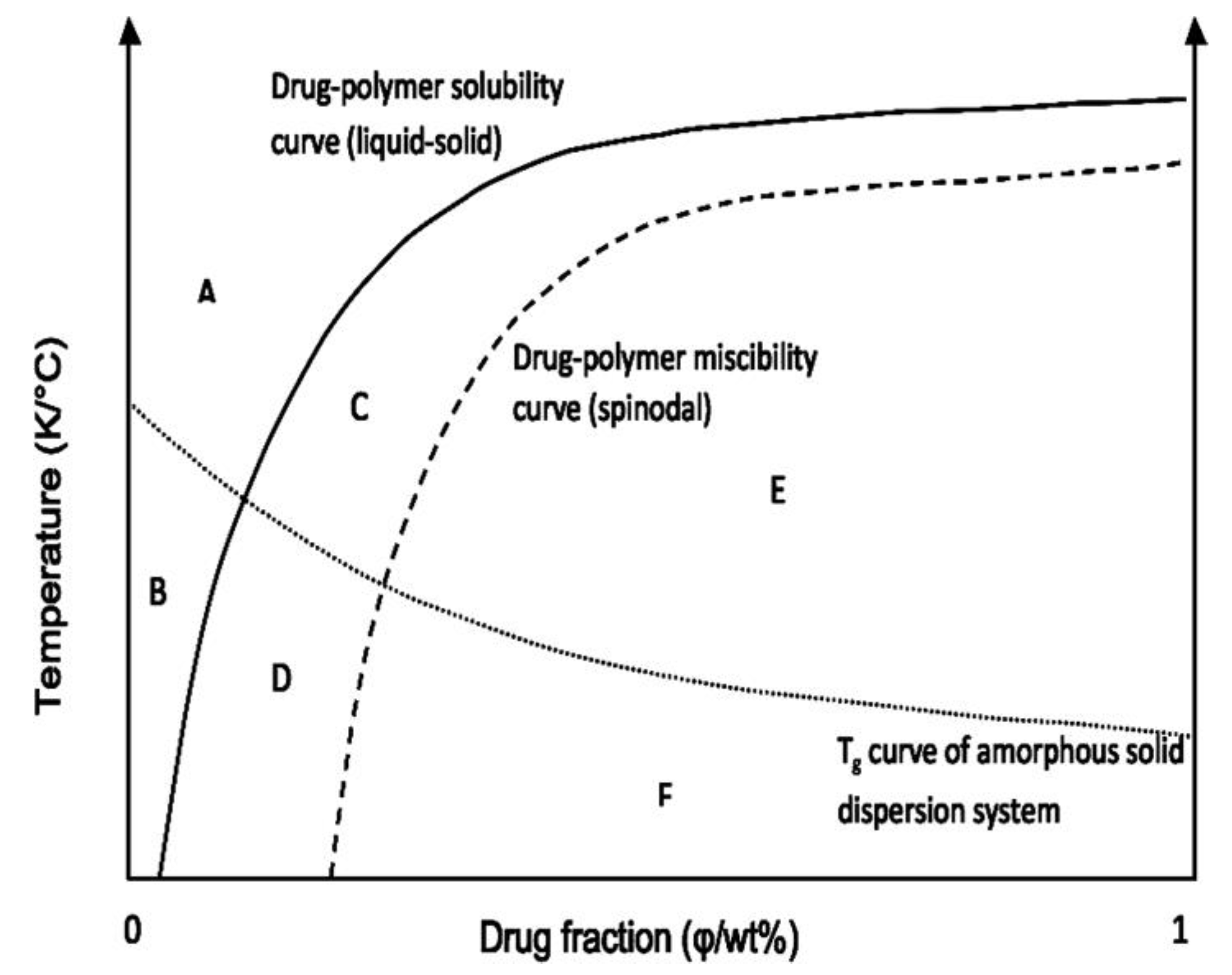{kind=link}
| Drug | Polymer(s) and Other Excipients | Method for Preparation of Solid Dispersions | Analytical Methods Used to Study Solubility/Miscibility | References |
|---|---|---|---|---|
| Albendazole | PVP | Hot-melt extrusion | DSC, XRPD, HSM, μ-CT SEM | [55] |
| Carbamazepine | Soluplus® | Hot-melt extrusion | Rheological properties, DSC, XRPD | [63] |
| Carbamazepine, Prednisolone | PVP, Eudragit® E 100 | Electrospray deposition | DSC, XRPD | [69] |
| Chloramphenicol | Poly(ε-caprolactone) | Film casting | DSC, XRPD, FT-IR, AFM | [70] |
| Diphenhydramine, Propranolol | Eudragit® L 100, Eudragit® L 100-55 | Hot-melt extrusion | XPS, DSC, XRPD, SEM | [24] |
| Felodipine | PVP | Solvent evaporation | DSC, FT-IR, XRPD | [23] |
| Eudragit® E PO | Hot-melt extrusion | SEM, DSC, M-DSC, NMR | [26] | |
| Soluplus®, HPMCAS, PVP, Eudragit® E PO, PVPVA, HPC, PAA, Na CMC, PVA, HEC | Spin coating | TASC, IR imaging | [60] | |
| Felodipine, nifedipine, ketoconazole | PVP, PAA | Solvent evaporation | DSC, FT-IR, XRPD | [47] |
| Griseofulvin | HPMCAS | Co-milling | FT-IR, XRPD, DSC | [71] |
| Ibuprofen | Eudragit® L 100 | Solvent evaporation | NMR | [52] |
| Indomethacin | PVP | Solvent evaporation | FT-IR, FT-Raman | [33] |
| Eudragit® E PO | Melting or compression methods | M-DSC, rheological properties, FT-IR | [35] | |
| Indomethacin, dextran | PVP | Solvent evaporation | XRPD, DSC | [39] |
| Indomethacin, nifedipine, d-mannitol | PVP, PVA | Co-milling | DSC, XRPD | [13] |
| Indomethacin, ursodeoxycholic acid, indapamide | PVP | Solvent evaporation | Water vapor absorption studies | [62] |
| Itraconazole | PEG and HPMC | Solvent evaporation (spray drying) | M-DSC, XRPD | [65] |
| HPMCAS and Soluplus® | Film casting | XRPD, DSC, PLM | [66] | |
| HPMCP, Soluplus®, PVPVA 64, Eudragit® E PO | [67] | |||
| HPMCAS with the addition of Poloxamer 188, Poloxamer 407, or TPGS | Film casting and hot-melt extrusion | DSC, XRPD | [72] | |
| Lacidipine | PVP K30, PVP VA64, Soluplus® | Hot-melt extrusion | XRPD, DSC, PLM, FT-IR | [73] |
| Lapatinib ditosylate | Soluplus® | Hot-melt extrusion and solvent evaporation | DSC, XRPD, SEM | [74] |
| n.a. (new chemical entity) | PVP | Solvent evaporation | TSDC, DSC | [46] |
| Naproxen | PVP | Solvent evaporation (spray drying) | M-DSC, FT-IR, XRPD | [75] |
| Nifedipine | PVP, HPMC, PHPA | Solvent evaporation (spray drying) | NMR, DSC | [48] |
| Posaconazole | Soluplus®, with the addition of PEG 4000, Poloxamer 188, Poloxamer 407 or TPGS | Hot-melt extrusion | DSC, M-DSC, SEM, AFM | [41] |
| Telaprevir | HPMC, HPMCAS, PVPVA | Solvent evaporation | AFM, AFM-IR, nanoTA, Fluorescence microscopy | [53] |
| Equations Used in the Calculation | Low Molecular Weight Substances | Amorphous Polymers |
|---|---|---|
| Additive molar functions | ||
| Auxiliary equations | ||
| Calculation of total and partial solubility parameters | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medarević, D.; Djuriš, J.; Barmpalexis, P.; Kachrimanis, K.; Ibrić, S. Analytical and Computational Methods for the Estimation of Drug-Polymer Solubility and Miscibility in Solid Dispersions Development. Pharmaceutics 2019, 11, 372. https://doi.org/10.3390/pharmaceutics11080372
Medarević D, Djuriš J, Barmpalexis P, Kachrimanis K, Ibrić S. Analytical and Computational Methods for the Estimation of Drug-Polymer Solubility and Miscibility in Solid Dispersions Development. Pharmaceutics. 2019; 11(8):372. https://doi.org/10.3390/pharmaceutics11080372
Chicago/Turabian StyleMedarević, Djordje, Jelena Djuriš, Panagiotis Barmpalexis, Kyriakos Kachrimanis, and Svetlana Ibrić. 2019. "Analytical and Computational Methods for the Estimation of Drug-Polymer Solubility and Miscibility in Solid Dispersions Development" Pharmaceutics 11, no. 8: 372. https://doi.org/10.3390/pharmaceutics11080372
APA StyleMedarević, D., Djuriš, J., Barmpalexis, P., Kachrimanis, K., & Ibrić, S. (2019). Analytical and Computational Methods for the Estimation of Drug-Polymer Solubility and Miscibility in Solid Dispersions Development. Pharmaceutics, 11(8), 372. https://doi.org/10.3390/pharmaceutics11080372