pH-Sensitive Chitosan–Heparin Nanoparticles for Effective Delivery of Genetic Drugs into Epithelial Cells


 ,
,  ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Particle Preparation
2.3. Particle Structure
2.4. Oligonucleotide Loading Efficiency
2.5. Oligonucleotide Release
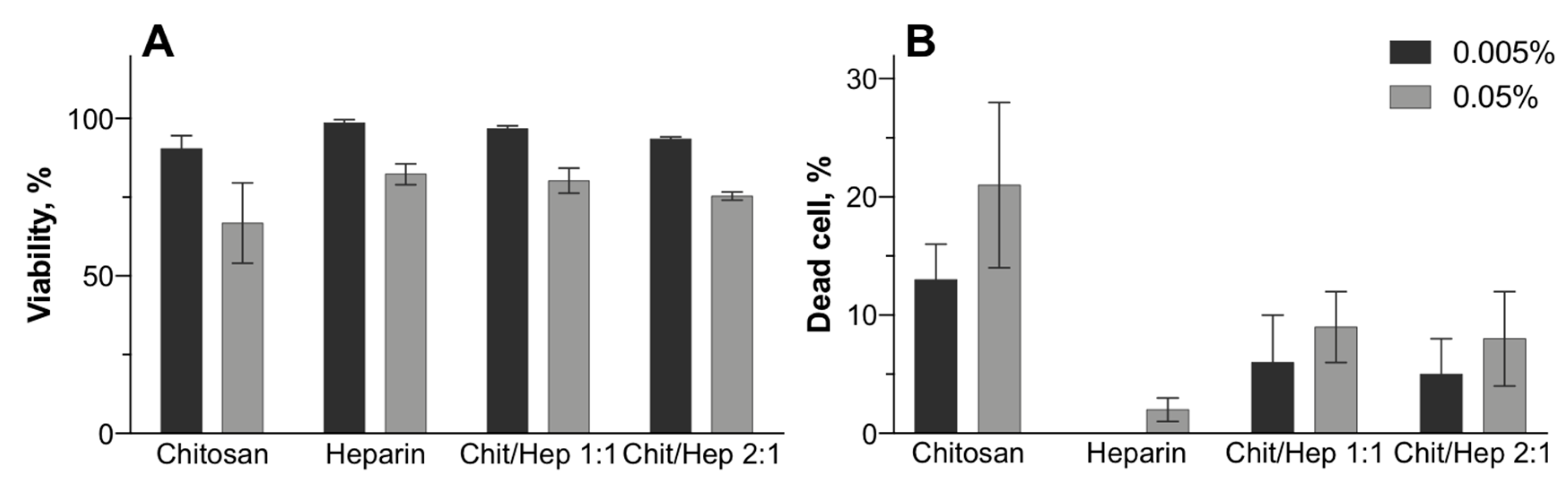
2.6. Particle Cytotoxicity
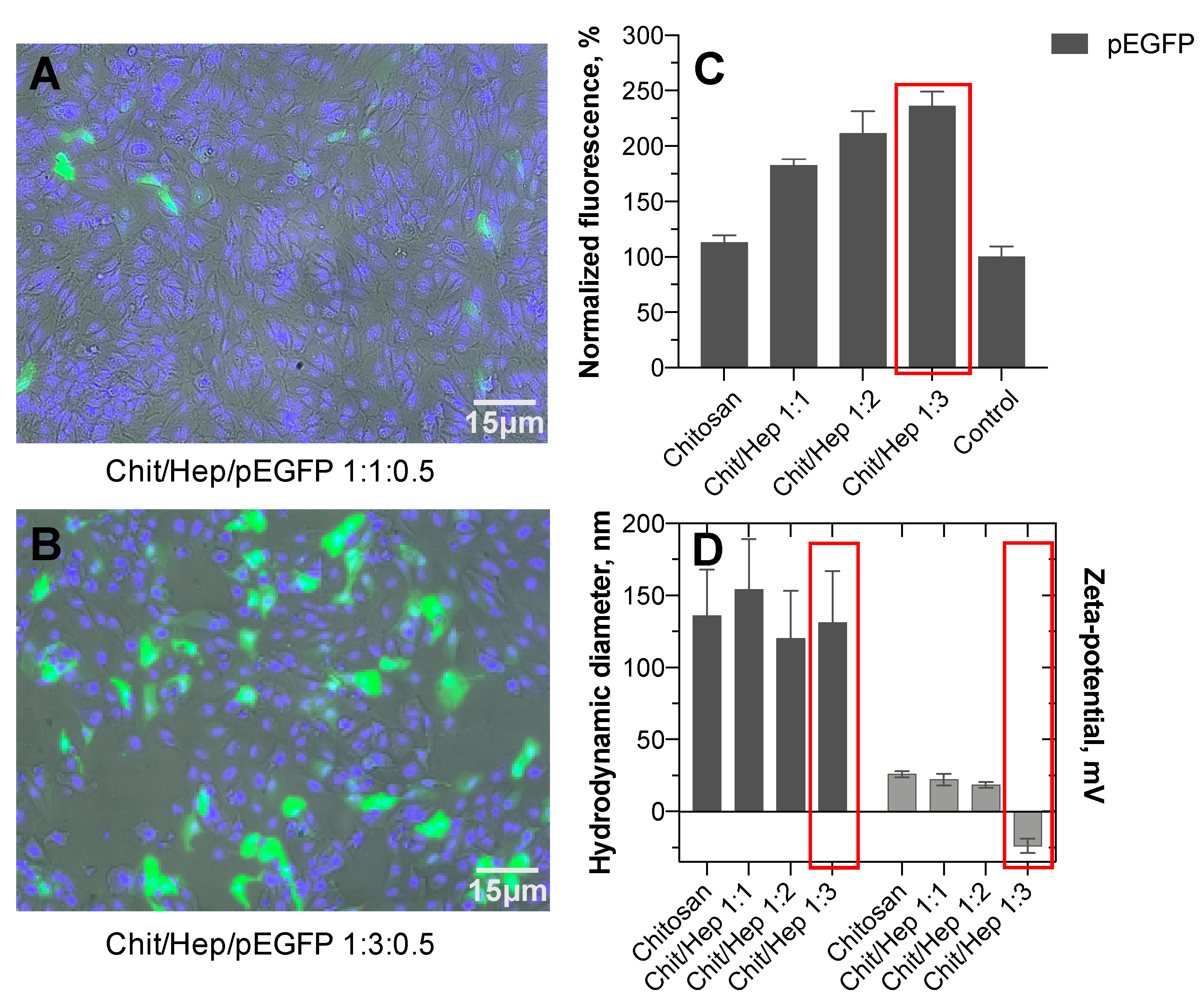
2.7. Cellular Uptake and Transfection
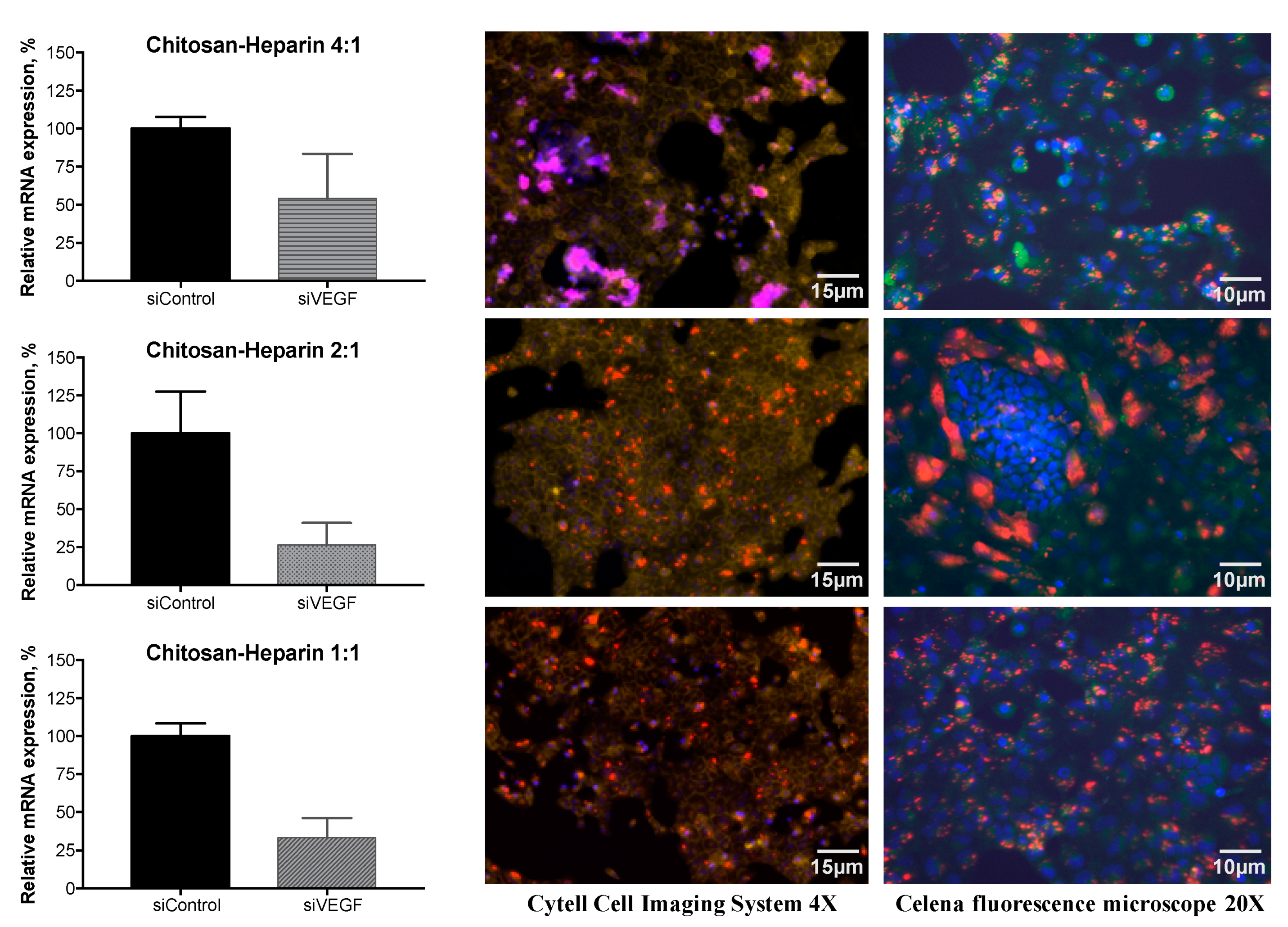
2.8. Gene Silencing of VEGF
2.9. Statistics
3. Results and Discussion
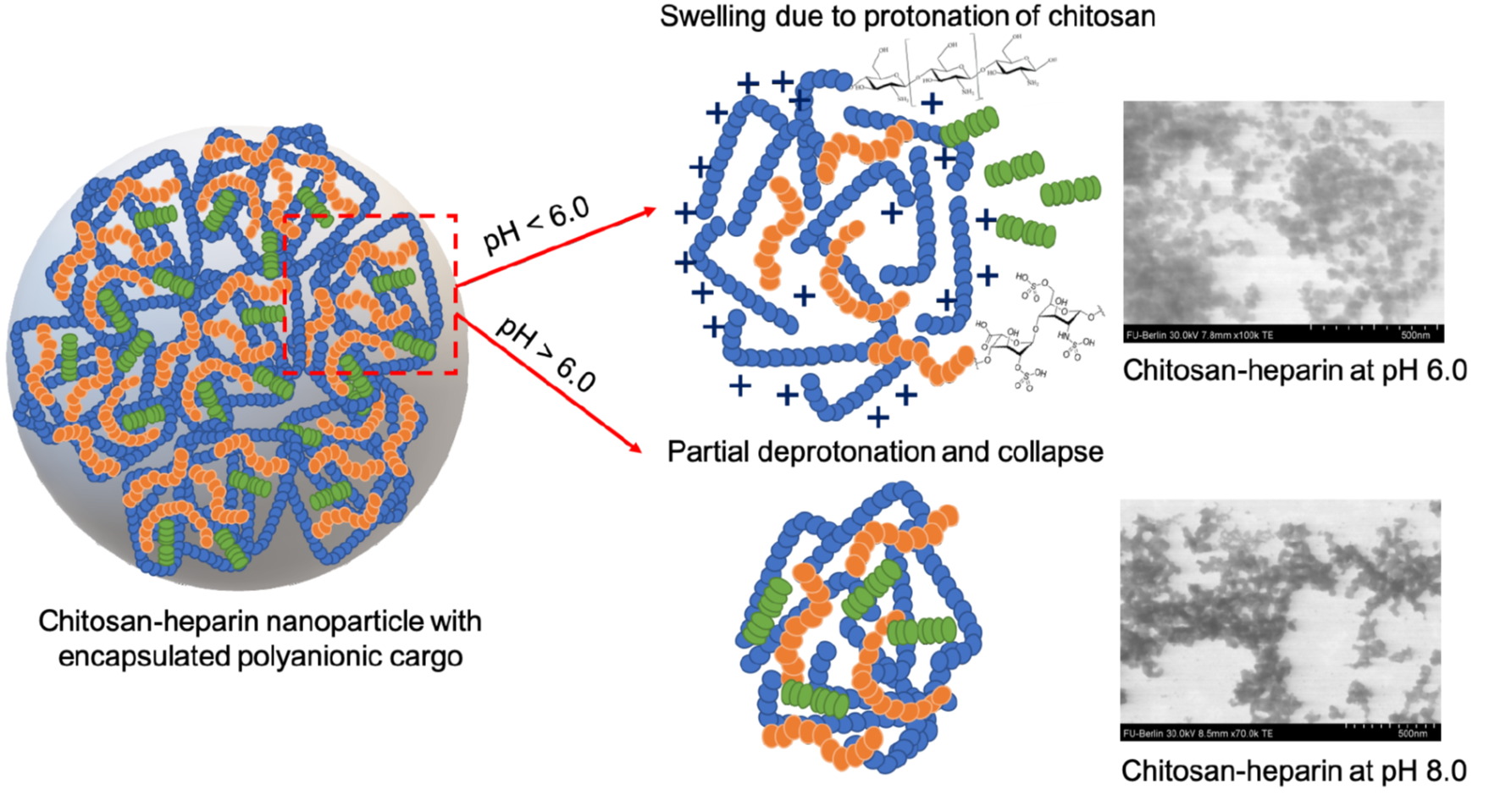
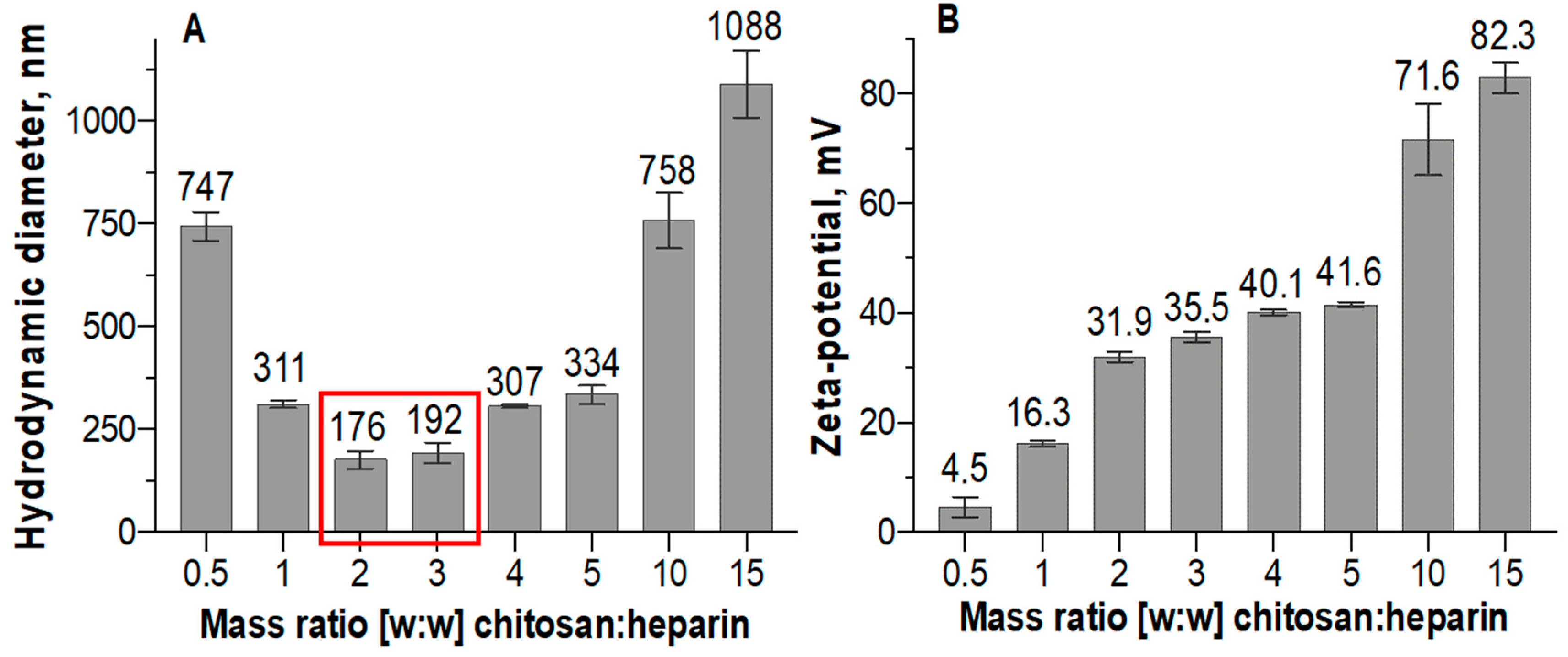
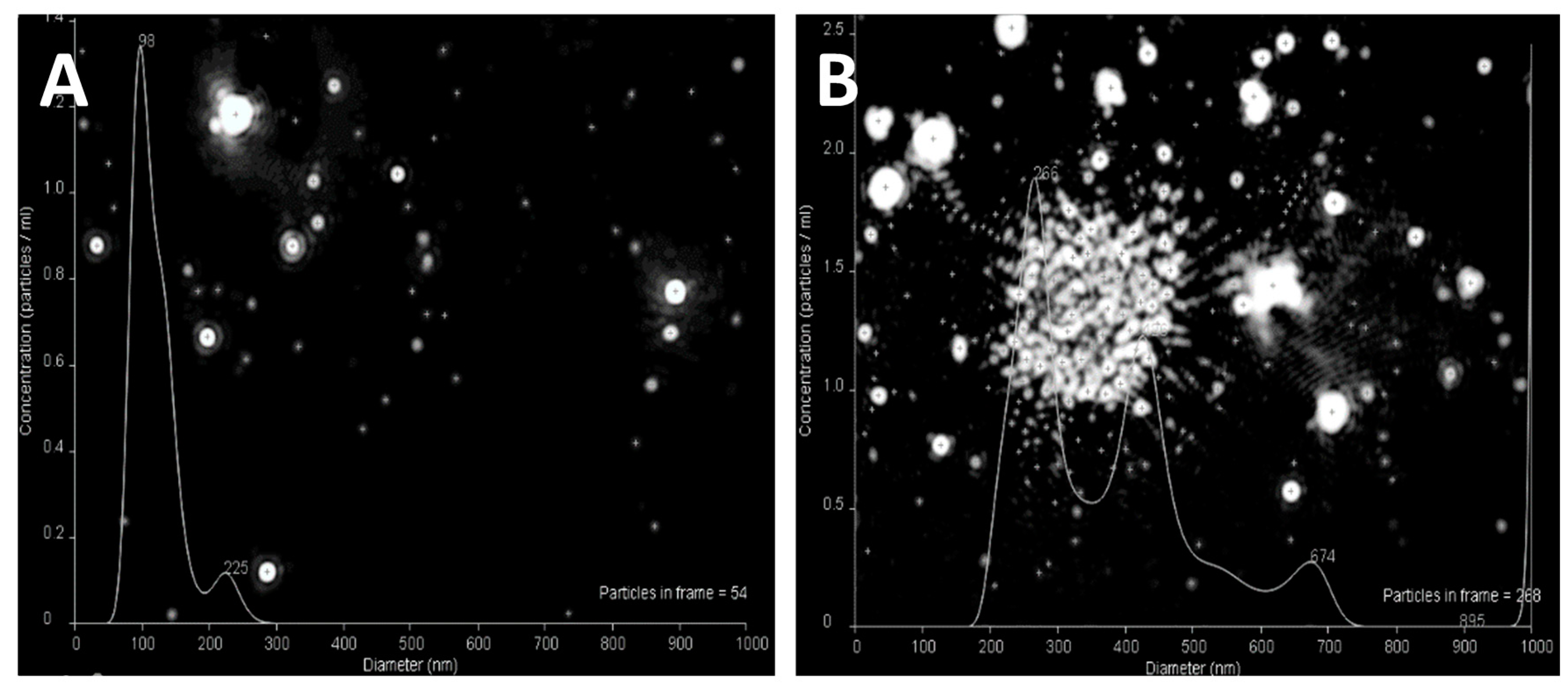
3.1. Nanoparticle Size, Shape, and Surface Charge
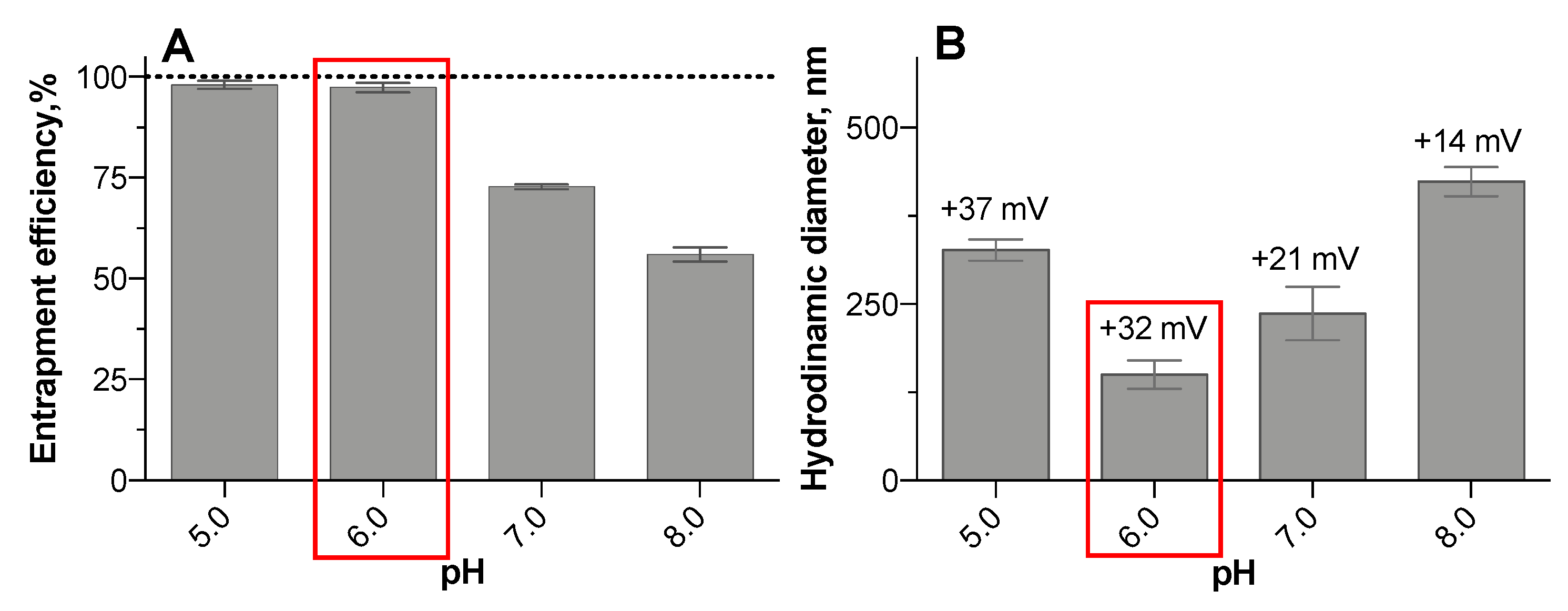
3.2. Oligonucleotide Entrapment Efficiency
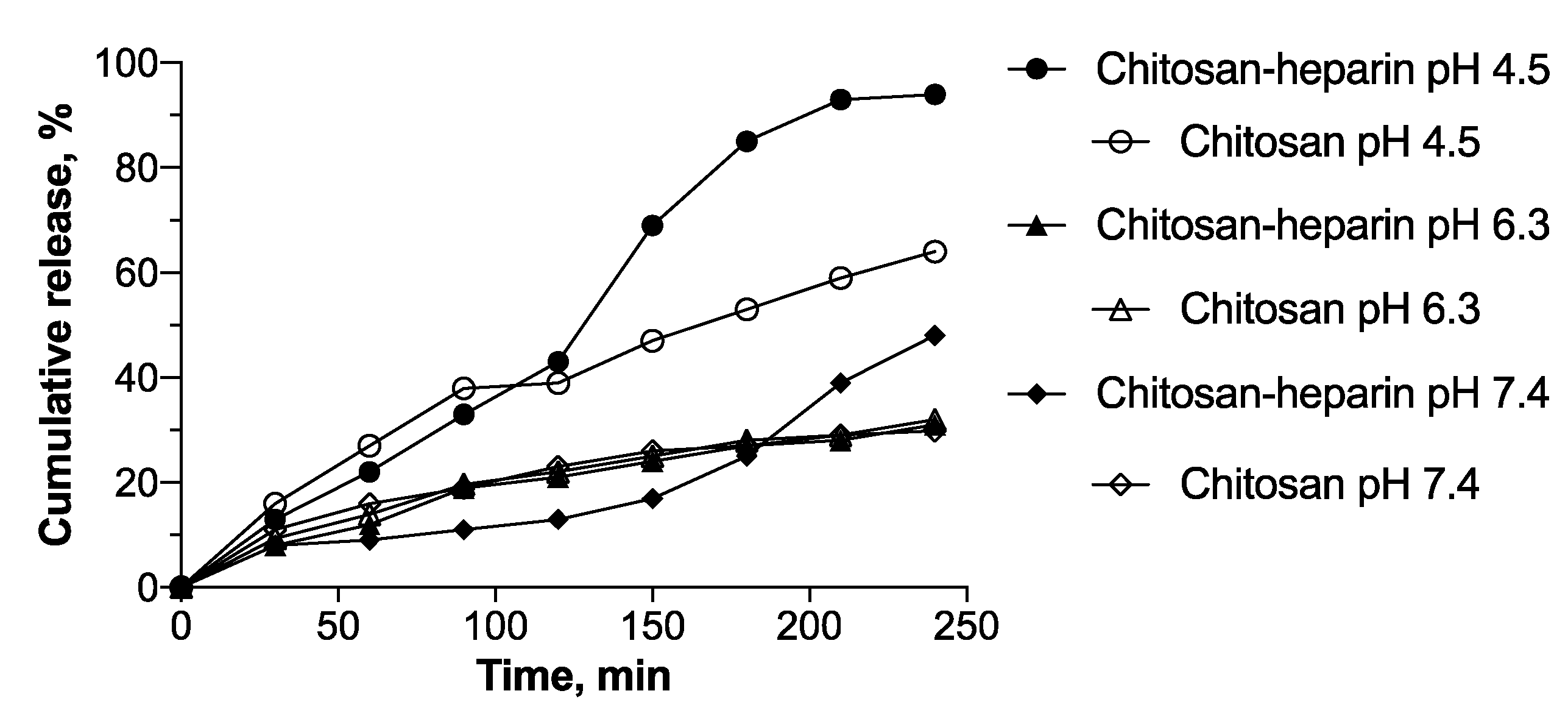
3.3. Oligonucleotide Release
3.4. Biocompatibility
3.5. Transfection Efficiency
3.6. Gene Silencing of VEGF
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lee, M.; Kim, T.; Na, J.; Jung, Y.; Jung, G.Y.; Park, N. A RNA producing DNA hydrogel as a platform for a high performance RNA interference system. Nat. Commun. 2018, 9, 4331. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Kim, S.W.; Park, T.G. Molecular design of functional polymers for gene therapy. Prog. Polym. Sci. 2007, 32, 1239–1274. [Google Scholar] [CrossRef]
- Tobío, M.; Gref, R.; Sánchez, A.; Langer, R.; Alonso, M.J. Stealth PLA-PEG nanoparticles as protein carriers for nasal administration. Pharm. Res. 1998, 15, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Germershaus, O.; Mao, S.; Sitterberg, J.; Bakowsky, U.; Kissel, T. Gene delivery using chitosan, trimethyl chitosan or polyethylenglycol-graft-trimethyl chitosan block copolymers: Establishment of structure-activity relationships in vitro. J. Control. Release 2008, 125, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ishii, T.; Okahata, Y. In vitro gene delivery mediated by chitosan. Effect of pH, serum, and molecular mass of chitosan on the transfection efficiency. Biomaterials 2001, 22, 2075–2080. [Google Scholar] [CrossRef]
- Huang, M.; Fong, C.W.; Khor, E.; Lim, L.Y. Transfection efficiency of chitosan vectors: Effect of polymer molecular weight and degree of deacetylation. J. Control. Release 2005, 106, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Lehr, C.M.; Bouwstra, J.A.; Schacht, E.H.; Junginger, H.E. In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int. J. Pharm. 1992, 78, 43–48. [Google Scholar] [CrossRef]
- Schipper, N.G.M.; Olsson, S.; Hoogstraate, J.A.; DeBoer, A.G.; Vårum, K.M.; Artursson, P. Chitosans as absorption enhancers for poorly absorbable drugs 2: Mechanism of absorption enhancement. Pharm. Res. 1997, 14, 923–929. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.A. Natural chelating polymers; alginic acid, chitin, and chitosan. Dermatol. Mon. 1973, 158, 465–473. [Google Scholar]
- Koga, D.; Mitsutomi, M.; Kono, M.; Matsumiya, M. Biochemistry of chitinases. Chitin Chitinases 1999, 87, 111–123. [Google Scholar] [CrossRef]
- Zhang, Z.; Tsai, P.C.; Ramezanli, T.; Michniak-Kohn, B.B. Polymeric nanoparticles-based topical delivery systems for the treatment of dermatological diseases. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Grenha, A. Chitosan nanoparticles: A survey of preparation methods. J. Drug Target. 2012, 20, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Hembram, K.C.; Prabha, S.; Chandra, R.; Ahmed, B.; Nimesh, S. Advances in preparation and characterization of chitosan nanoparticles for therapeutics. Artif. CellsNanomed. Biotechnol. 2016, 44, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Duceppe, N.; Tabrizian, M. Advances in using chitosan-based nanoparticles for in vitro and in vivo drug and gene delivery. Expert Opin. Drug Deliv. 2010, 7, 1191–1207. [Google Scholar] [CrossRef] [PubMed]
- Köping-Höggård, M.; Vårum, K.M.; Issa, M.; Danielsen, S.; Christensen, B.E.; Stokke, B.T.; Artursson, P. Improved chitosan-mediated gene delivery based on easily dissociated chitosan polyplexes of highly defined chitosan oligomers. Gene Ther. 2004, 11, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Douglas, K.L.; Piccirillo, C.A.; Tabrizian, M. Effects of alginate inclusion on the vector properties of chitosan-based nanoparticles. J. Control. Release 2006, 115, 354–361. [Google Scholar] [CrossRef]
- De La Fuente, M.; Seijo, B.; Alonso, M.J. Novel Hyaluronic Acid-Chitosan Nanoparticles for Ocular Gene Therapy. Investig. Opthalmol. Vis. Sci. 2008, 49, 2016–2024. [Google Scholar] [CrossRef]
- Liu, Z.; Jiao, Y.; Liu, F.; Zhang, Z. Heparin/chitosan nanoparticle carriers prepared by polyelectrolyte complexation. J. Biomed. Mater. Res. Part A 2007, 83, 806–812. [Google Scholar] [CrossRef]
- Tan, Q.; Tang, H.; Hu, J.; Hu, Y.; Zhou, X.; Tao, Y.; Wu, Z.; Hu, Y.; Zhou, X.; Tao, R. Controlled release of chitosan/heparin nanoparticle-delivered VEGF enhances regeneration of decellularized tissue-engineered scaffolds. Int. J. Nanomed. 2011, 6, 929–942. [Google Scholar] [CrossRef]
- Kubo, T.; Mihara, K.; Seyama, T.; Yanagihara, K.; Takei, Y.; Sato, Y. Lipid-Conjugated 27-Nucleotide Double-Stranded RNAs with Dicer-Substrate Potency Enhance RNAi-Mediated Gene Silencing. Mol. Pharm. 2012, 9, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Zoschke, C.; Kral, V.; Gonska, H.; Kapfer, C.; Schafer-Korting, M. Production of Organotypic NMSC Models. 2017. Available online: http://www.bb3r.de/en/forschungsplattform/Standardarbeitsvorschriften/index.html (accessed on 27 May 2019).
- Balansin Rigon, R.; Kaessmeyer, S.; Wolff, C.; Hausmann, C.; Zhang, N.; Sochorová, M.; Zoschke, C. Ultrastructural and Molecular Analysis of Ribose-Induced Glycated Reconstructed Human Skin. Int. J. Mol. Sci. 2018, 19, 3521. [Google Scholar] [CrossRef] [PubMed]
- Abcam. Ab228550 Protocol Hoechst 33258 Staining; Abcam: Cambridge, UK, 2018. [Google Scholar]
- Banerjee, A.; Qi, J.; Gogoi, R.; Wong, J.; Mitragotri, S. Role of Nanoparticle Size, Shape and Surface Chemistry in Oral Drug Delivery. J. Control. Release 2016, 238, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Almalik, A.; Donno, R.; Cadman, C.J.; Cellesi, F.; Day, P.J.; Tirelli, N. Hyaluronic acid-coated chitosan nanoparticles: Molecular weight-dependent effects on morphology and hyaluronic acid presentation. J. Control. Release 2013, 172, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Hoo, C.M.; Starostin, N.; West, P.; Mecartney, M.L. A comparison of atomic force microscopy (AFM) and dynamic light scattering (DLS) methods to characterize nanoparticle size distributions. J. Nanopart. Res. 2008, 10, 89–96. [Google Scholar] [CrossRef]
- Gross, J.; Sayle, S.; Karow, A.R.; Bakowsky, U.; Garidel, P. Nanoparticle tracking analysis of particle size and concentration detection in suspensions of polymer and protein samples: Influence of experimental and data evaluation parameters. Eur. J. Pharm. Biopharm. 2016, 104, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Rafiee, A.; Alimohammadian, M.H.; Gazori, T.; Riazi-Rad, F.; Fatemi, S.M.R.; Parizadeh, A.; Haririan, I.; Havaskary, M. Comparison of chitosan, alginate and chitosan/alginate nanoparticles with respect to their size, stability, toxicity and transfection. Asian Pac. J. Trop. Dis. 2014, 4, 372–377. [Google Scholar] [CrossRef]
- Freeman, E.C.; Weiland, L.M.; Meng, W.S. Modeling the proton sponge hypothesis: Examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J. Biomater. Sci. Polym. Ed. 2013, 24, 398–416. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef]
- Ablonczy, Z.; Crosson, C.E. VEGF modulation of retinal pigment epithelium resistance. Exp. Eye Res. 2007, 85, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Sabatini, D.M. Systematic genome-wide screens of gene function. Nat. Rev. Genet. 2004, 5, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ryther, R.C.C.; Flynt, A.S.; Phillips, J.A.; Patton, J.G. SiRNA therapeutics: Big potential from small RNAs. Gene Ther. 2005, 12, 5–11. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticle | pH | Model | |||||
|---|---|---|---|---|---|---|---|
| Zero Order | First Order | Higuchi | Hixson-Crowell | Korsmeyer–Peppas | Baker–Lonsdale | ||
| Chitosan | 4.5 | r = 0.9498 | r = 0.8150 | r = 0.9445 | r = 0.8741 | r = 0.9394 | r = 0.9430 |
| k = 13.48 | k = −0.51 | k = 28.52 | k = −0.84 | n = 1.26 | k = 8.41 | ||
| 6.3 | r = 0.9537 | r = 0.7897 | r = 0.9704 | r = 0.8662 | r = 0.9688 | r = 0.9583 | |
| k = 16.13 | k = −0.41 | k = 34.47 | k = −0.78 | n = 1.04 | k = 14.61 | ||
| 7.4 | r = 0.9811 | r = 0.9037 | r = 0.9400 | r = 0.9478 | r = 0.9489 | r = 0.8687 | |
| k = 33.80 | k = −0.47 | k = 70.43 | k = −1.09 | n = 1.12 | k = 78.14 | ||
| Chitosan–heparin | 4.5 | r = 0.9644 | r = 0.7834 | r = 0.9943 | r = 0.8651 | r = 0.9795 | r = 0.9841 |
| k = 6.52 | k = −0.33 | k = 14.01 | k = −0.57 | n = 0.86 | k = 2.42 | ||
| 6.3 | r = 0.9666 | r = 0.8193 | r = 0.9535 | r = 0.8517 | r = 0.9506 | r = 0.9639 | |
| k = 6.80 | k = −0.24 | k = 19.06 | k = −0.45 | n = 0.59 | k = 5.91 | ||
| 7.4 | r = 0.9377 | r = 0.7855 | r = 0.9839 | r = 0.8483 | r = 0.9768 | r = 0.9836 | |
| k = 14.40 | k = −0.26 | k = 31.32 | k = −0.56 | n = 0.66 | k = 16.87 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilipenko, I.; Korzhikov-Vlakh, V.; Sharoyko, V.; Zhang, N.; Schäfer-Korting, M.; Rühl, E.; Zoschke, C.; Tennikova, T. pH-Sensitive Chitosan–Heparin Nanoparticles for Effective Delivery of Genetic Drugs into Epithelial Cells. Pharmaceutics 2019, 11, 317. https://doi.org/10.3390/pharmaceutics11070317
Pilipenko I, Korzhikov-Vlakh V, Sharoyko V, Zhang N, Schäfer-Korting M, Rühl E, Zoschke C, Tennikova T. pH-Sensitive Chitosan–Heparin Nanoparticles for Effective Delivery of Genetic Drugs into Epithelial Cells. Pharmaceutics. 2019; 11(7):317. https://doi.org/10.3390/pharmaceutics11070317
Chicago/Turabian StylePilipenko, Iuliia, Viktor Korzhikov-Vlakh, Vladimir Sharoyko, Nan Zhang, Monika Schäfer-Korting, Eckart Rühl, Christian Zoschke, and Tatiana Tennikova. 2019. "pH-Sensitive Chitosan–Heparin Nanoparticles for Effective Delivery of Genetic Drugs into Epithelial Cells" Pharmaceutics 11, no. 7: 317. https://doi.org/10.3390/pharmaceutics11070317
APA StylePilipenko, I., Korzhikov-Vlakh, V., Sharoyko, V., Zhang, N., Schäfer-Korting, M., Rühl, E., Zoschke, C., & Tennikova, T. (2019). pH-Sensitive Chitosan–Heparin Nanoparticles for Effective Delivery of Genetic Drugs into Epithelial Cells. Pharmaceutics, 11(7), 317. https://doi.org/10.3390/pharmaceutics11070317