Targeting Cell Adhesion Molecules via Carbonate Apatite-Mediated Delivery of Specific siRNAs to Breast Cancer Cells In Vitro and In Vivo

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methodology
2.2.1. Cell Culture
2.2.2. siRNA Design and Sequence
2.2.3. Infrared Spectroscopy
2.2.4. Generation of CA-siRNA Complex for In Vitro Study
2.2.5. Cell Proliferation Assay by 3-(4,5-Dimethylthiazaol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT)
2.2.6. SDS-PAGE and Western Blot
2.2.7. In Vivo Tumor Regression Analysis
2.3. Data Analysis
3. Results
3.1. Elemental Analysis of CA Nanoparticles Using FT-IR Spectroscopy
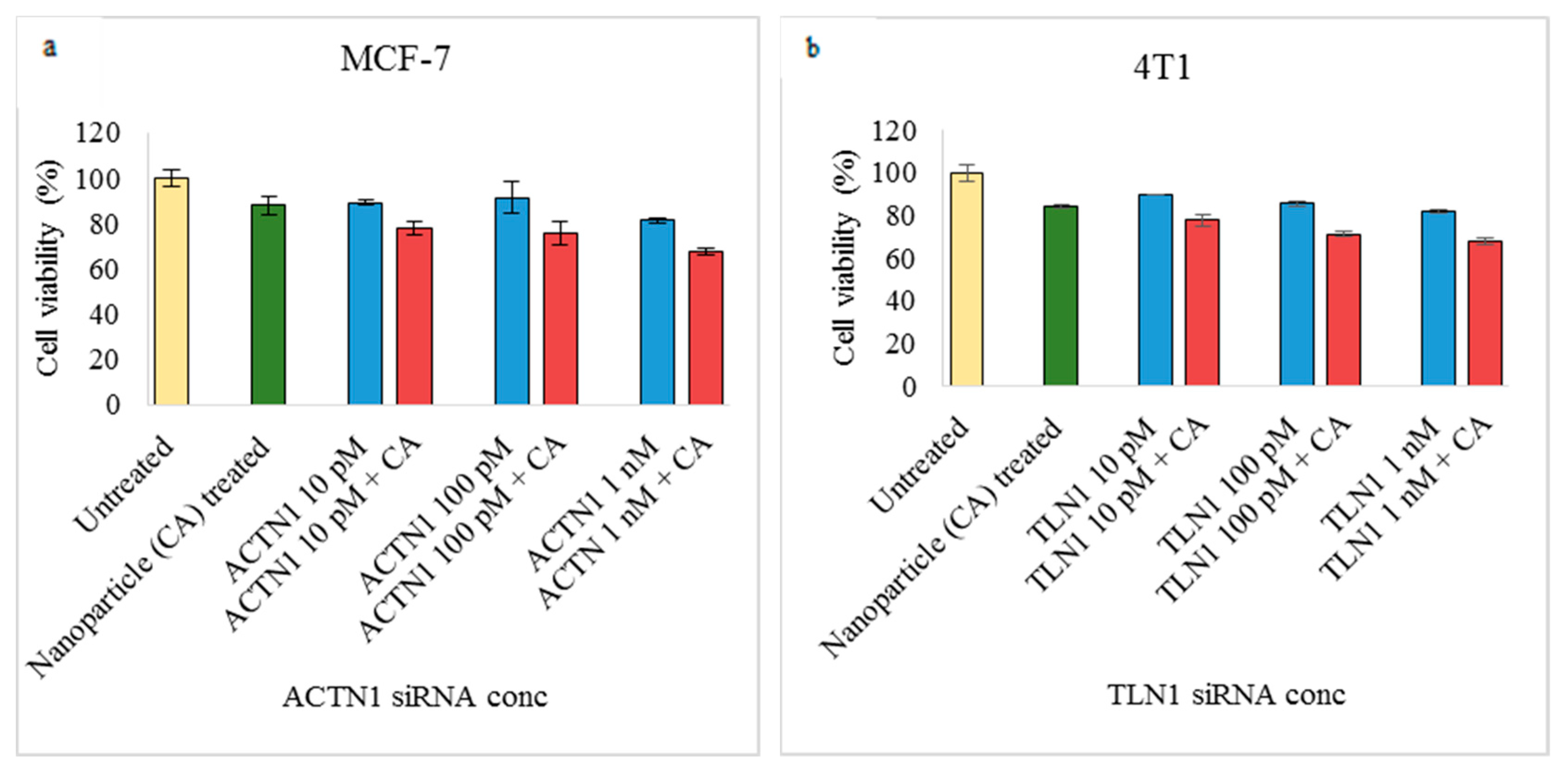
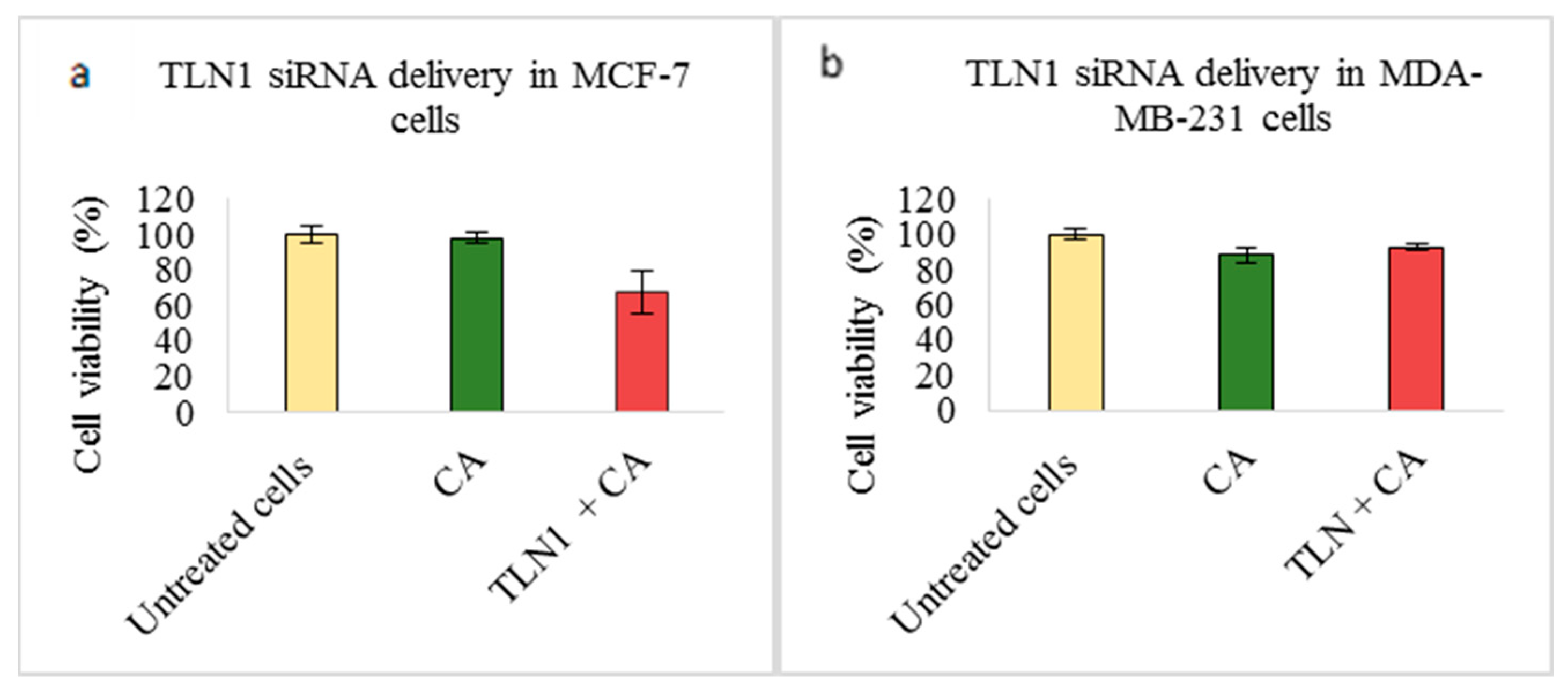
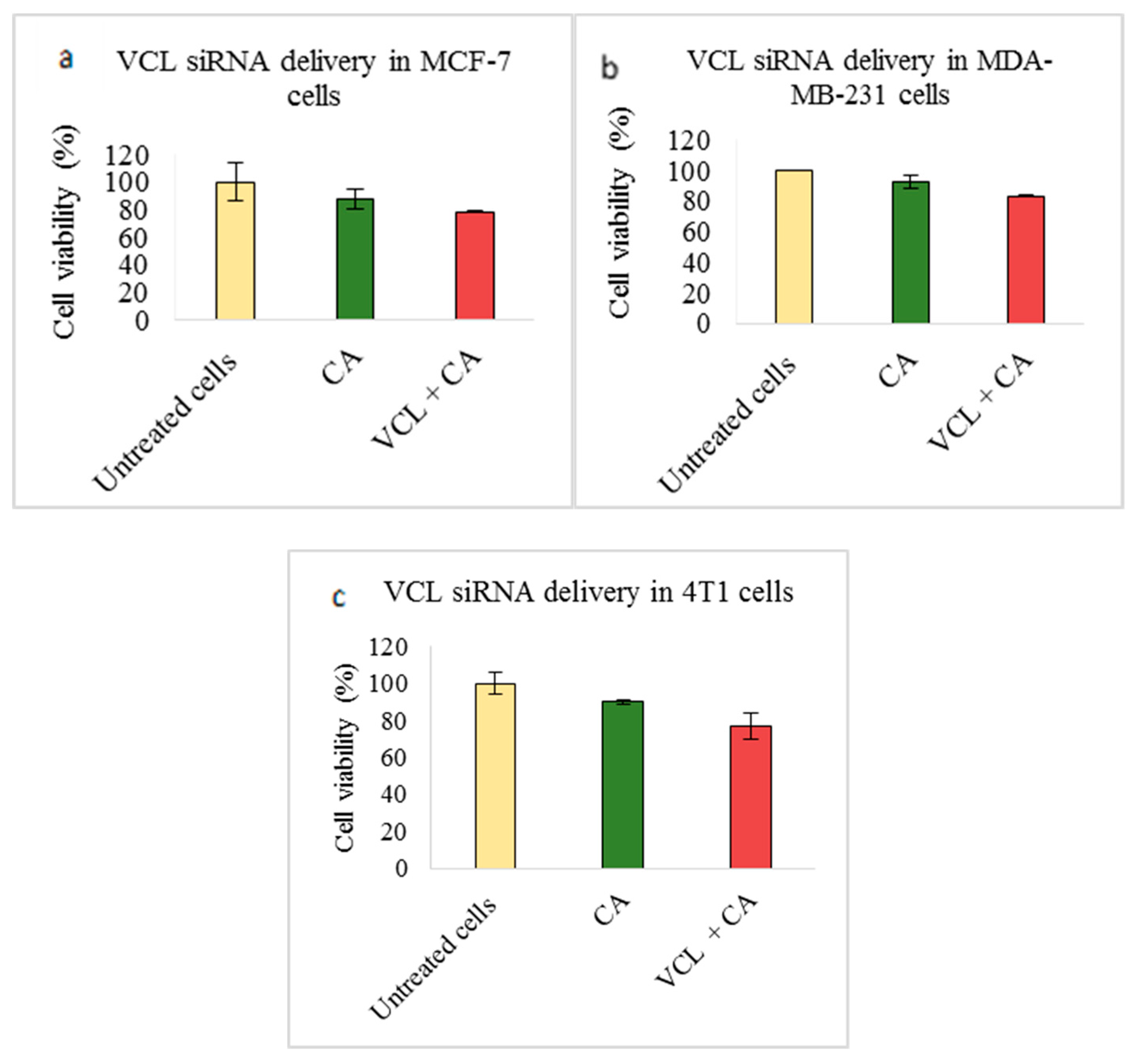
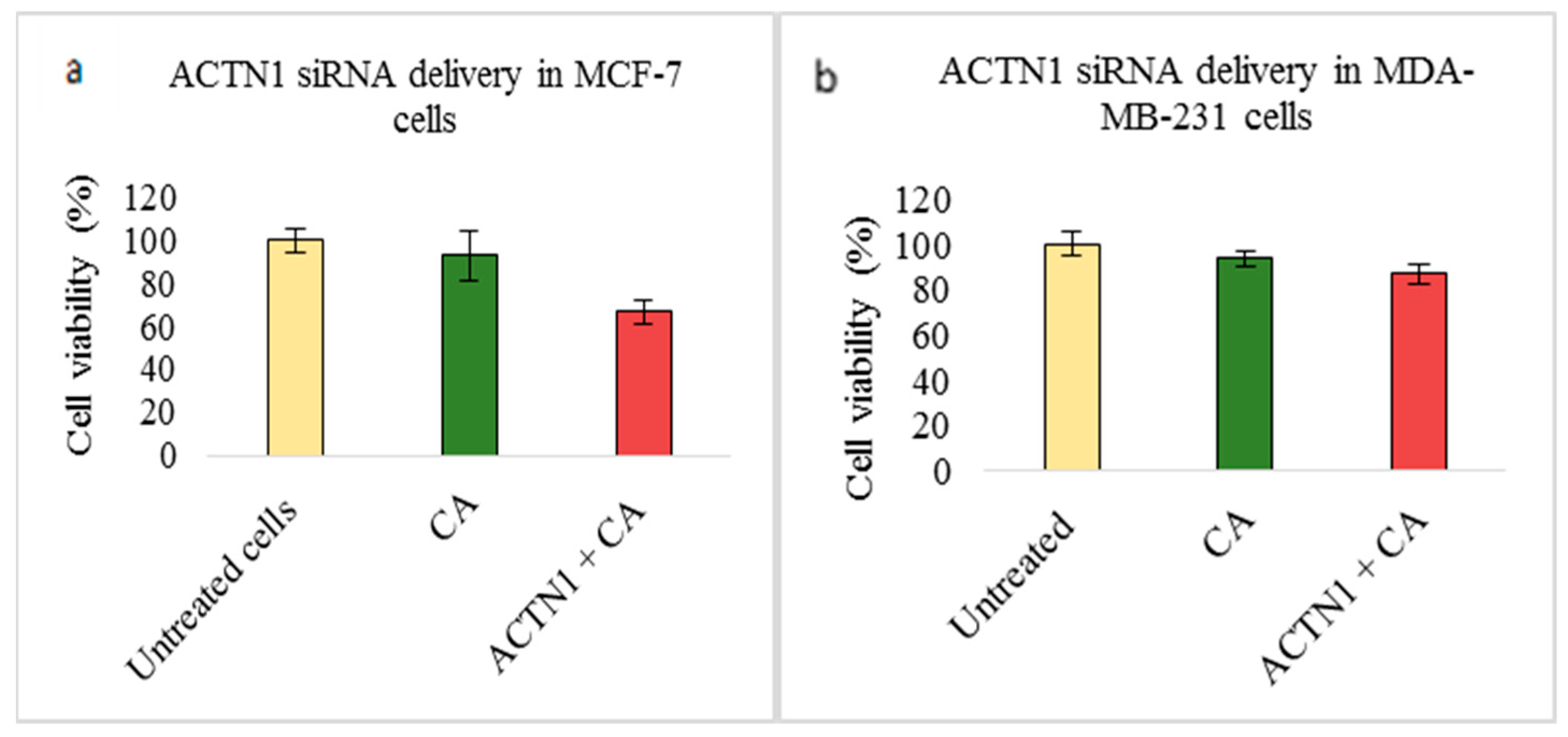
3.2. Assessment of siRNA Concentration with/without CA-Assisted Delivery in Breast Cancer Cells via the MTT Assay
3.3. Role of Additional Cell Adhesion Molecules in Proliferation and Survival of Breast Cancer Cells using the MTT Assay
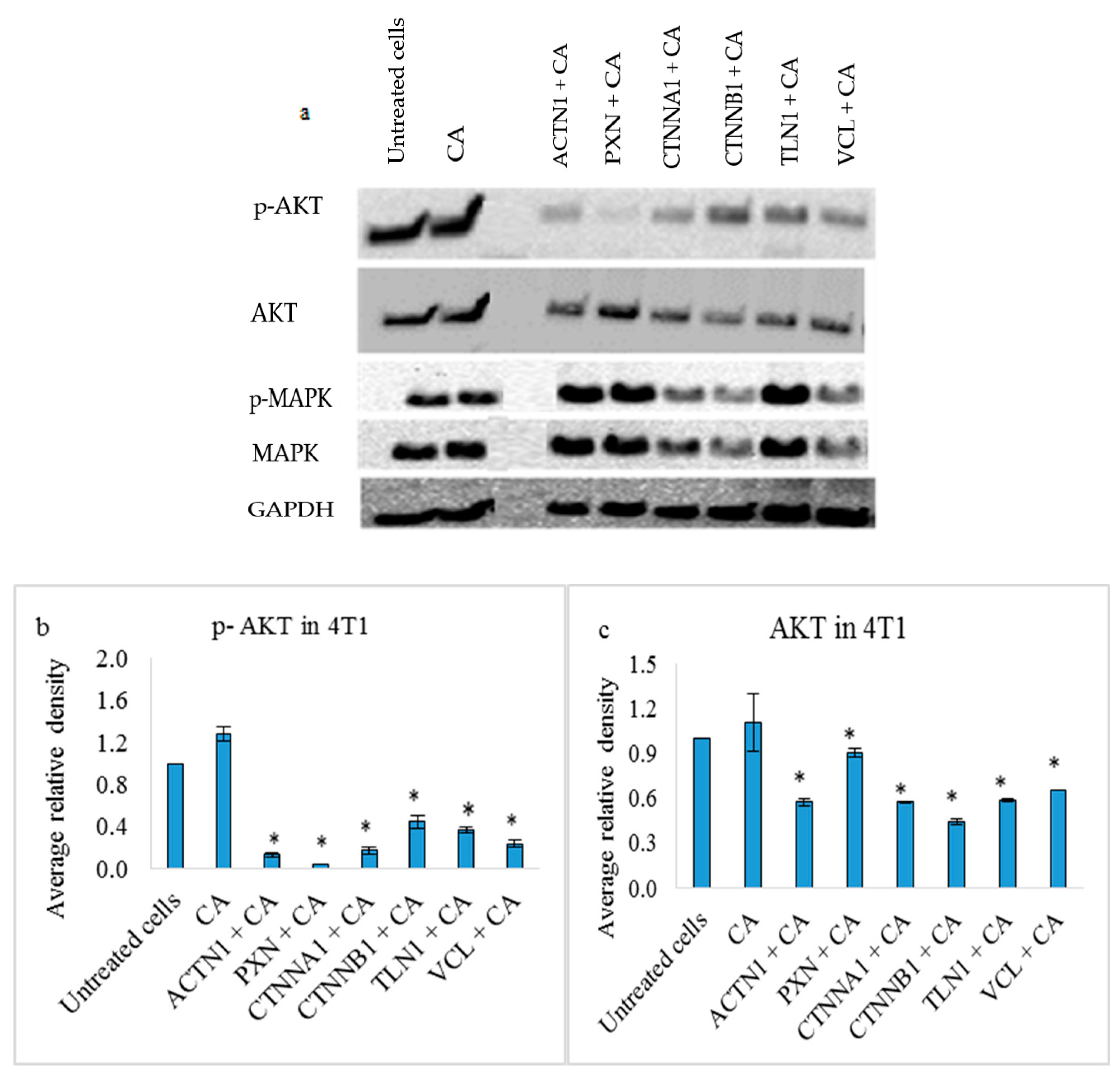
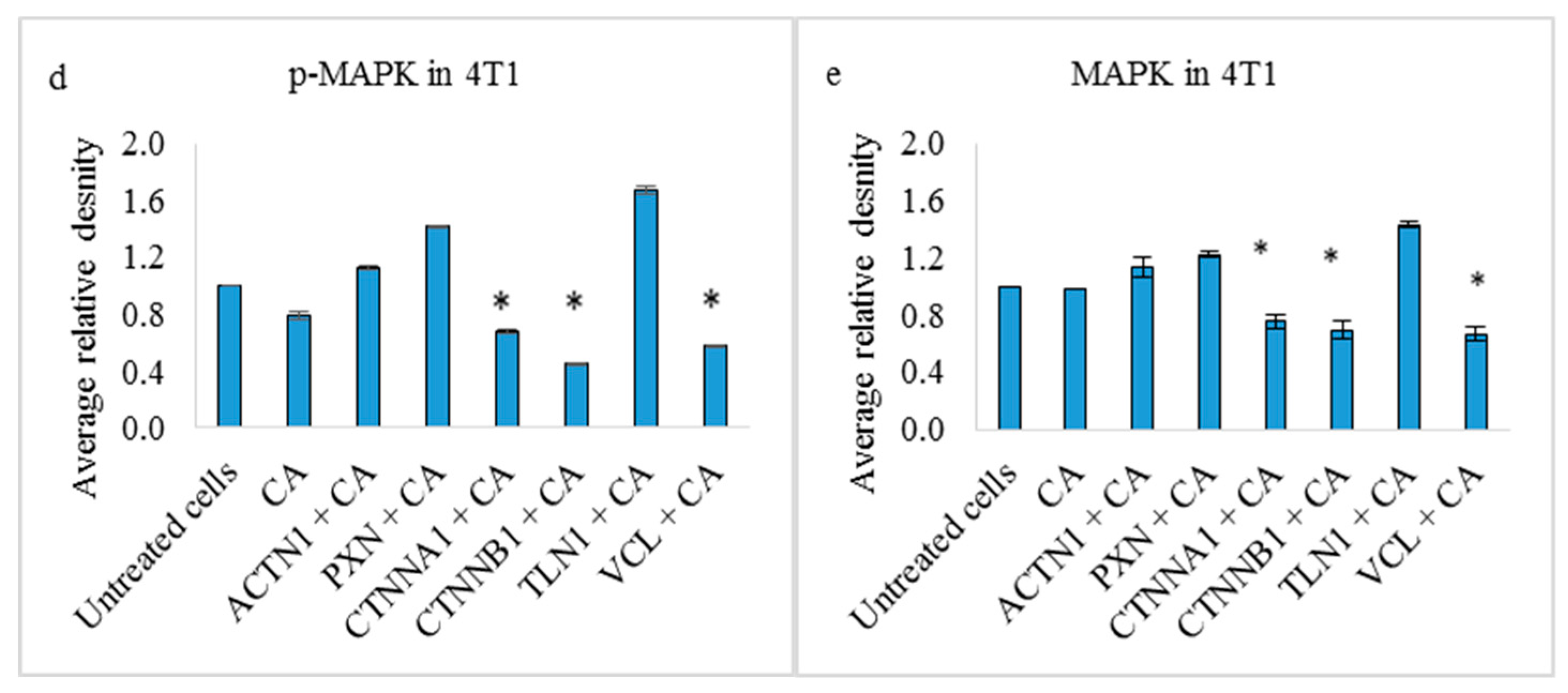
3.4. Effects on PI3-Kinase/AKT and MAPK Pathways in 4T1 Cells
3.5. Delivery of CA Loaded siRNAs against Cell Adhesion Molecules in a Murine Breast Cancer Model
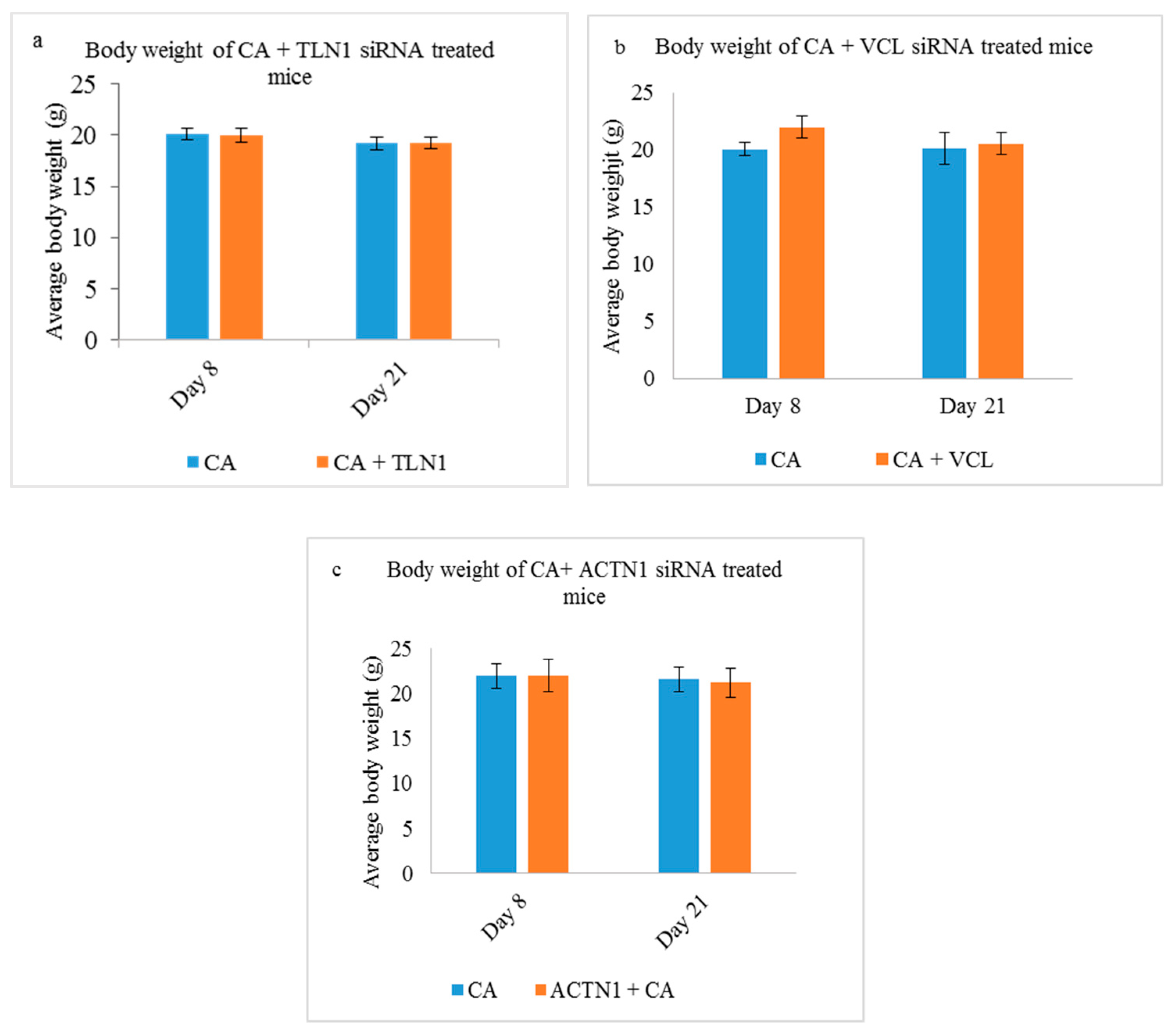
3.5.1. Body Weight
3.5.2. Tumor Regression of CA Loaded siRNAs in a Mouse Breast Cancer Model
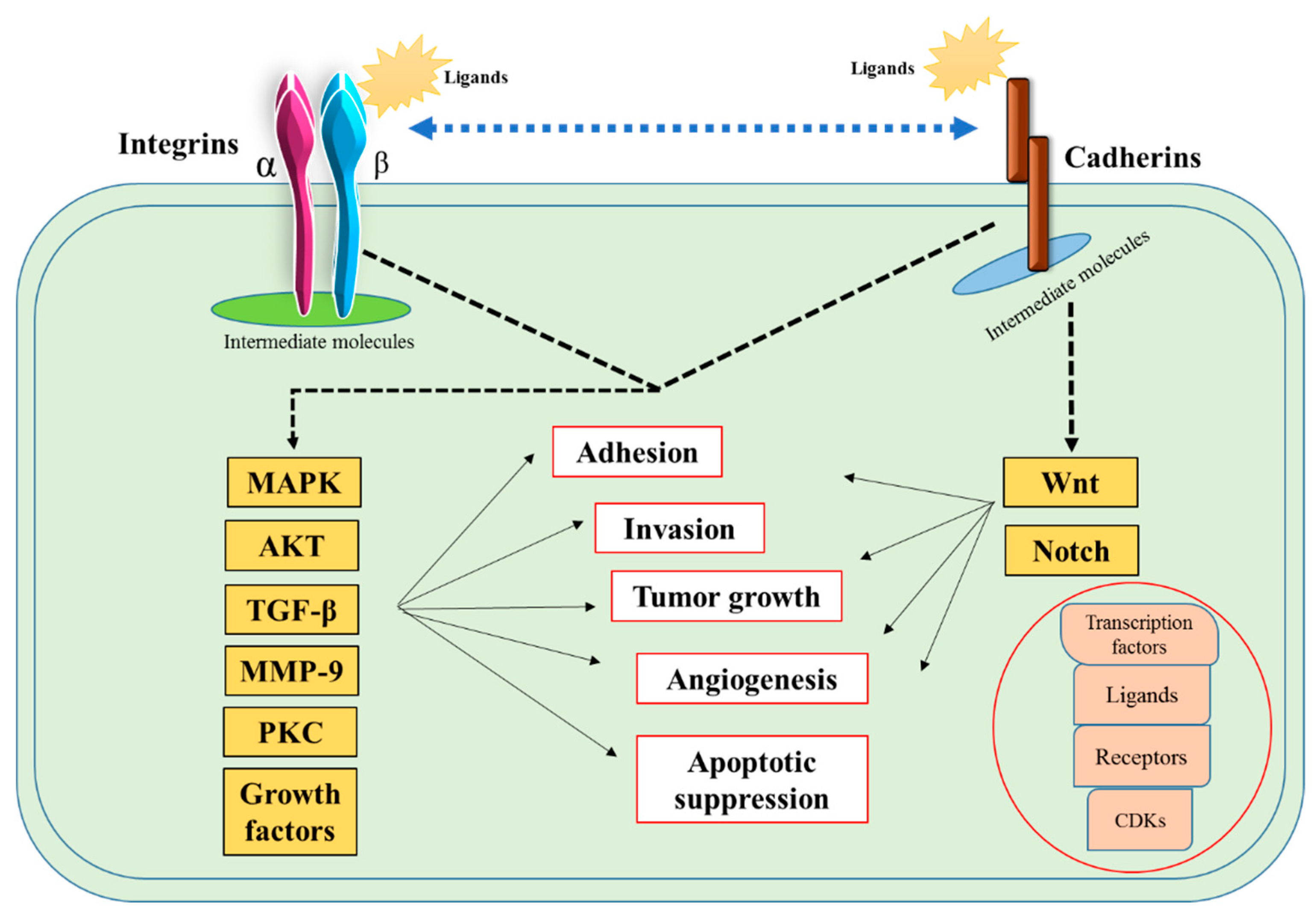
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol.Ther. 2010, 10, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Mu, P. Targeting breast cancer metastasis. Breast Cancer Basic Clin. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.; Weaver, A.M. Regulation of cancer invasiveness by the physical extracellular matrix environment. Cell adhes. Migr. 2009, 3, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; Van Roy, F. The E-cadherin/catenin complex: An important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 2001, 3, 289. [Google Scholar] [CrossRef] [PubMed]
- Cowin, P.; Rowlands, T.M.; Hatsell, S.J. Cadherins and catenins in breast cancer. Curr. Opin. Cell Biol. 2005, 17, 499–508. [Google Scholar] [CrossRef]
- Deakin, N.O.; Pignatelli, J.; Turner, C.E. Diverse roles for the paxillin family of proteins in cancer. Genes Cancer 2012, 3, 362–370. [Google Scholar] [CrossRef]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef]
- Christerson, L.B.; Vanderbilt, C.A.; Cobb, M.H. MEKK1 interacts with α-actinin and localizes to stress fibers and focal adhesions. Cell Motil. Cytoskel. 1999, 43, 186–198. [Google Scholar] [CrossRef]
- Guvakova, M.A.; Adams, J.C.; Boettiger, D. Functional role of α-actinin, PI 3-kinase and MEK1/2 in insulin-like growth factor I receptor kinase regulated motility of human breast carcinoma cells. J. Cell Sci. 2002, 115, 4149–4165. [Google Scholar] [CrossRef]
- Quick, Q.; Skalli, O. α-Actinin 1 and α-actinin 4: Contrasting roles in the survival, motility, and RhoA signaling of astrocytoma cells. Exp. Cell Res. 2010, 316, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Critchley, D.R.; Gingras, A.R. Talin at a glance. J. Cell Sci. 2008, 121, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, S.; McCann, R.O.; Dhir, R.; Kyprianou, N. Talin1 promotes tumor invasion and metastasis via focal adhesion signaling and anoikis resistance. Cancer Res. 2010, 70, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Carisey, A.; Ballestrem, C. Vinculin, an adapter protein in control of cell adhesion signalling. Eur. J. Cell Biol. 2011, 90, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.B.; Kang, L.; Roe, S.; Borgen, P.I.; Rimm, D.L. Vinculin is associated with the E-cadherin adhesion complex. J. Biol. Chem. 1997, 272, 32448–32453. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.E. Paxillin interactions. J. Cell Sci. 2000, 113, 4139–4140. [Google Scholar] [PubMed]
- Albigès-Rizo, C.; Frachet, P.; Block, M.R. Down regulation of talin alters cell adhesion and the processing of the alpha 5 beta 1 integrin. J. Cell Sci. 1995, 108, 3317–3329. [Google Scholar] [PubMed]
- Kovac, B.; Mäkelä, T.P.; Vallenius, T. Increased α-actinin-1 destabilizes E-cadherin-based adhesions and associates with poor prognosis in basal-like breast cancer. PLoS ONE 2018, 13, e0196986. [Google Scholar] [CrossRef] [PubMed]
- Bukholm, I.; Nesland, J.; Børresen-Dale, A.L. Re-expression of E-cadherin, α-catenin and β-catenin, but not of γ-catenin, in metastatic tissue from breast cancer patients. J. Pathol. 2000, 190, 15–19. [Google Scholar] [CrossRef]
- Haining, A.W.; Lieberthal, T.J.; del Río Hernández, A. Talin: A mechanosensitive molecule in health and disease. FASEB J. 2016, 30, 2073–2085. [Google Scholar] [CrossRef] [PubMed]
- Rubashkin, M.G.; Cassereau, L.; Bainer, R.; DuFort, C.C.; Yui, Y.; Ou, G.; Paszek, M.J.; Davidson, M.W.; Chen, Y.-Y.; Weaver, V.M. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Res. 2014, 74, 4597–4611. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Yoder, B.J.; Tarr, S.M.; Prescott, N.L.; Laniauskas, S.; Coleman, K.A.; Downs-Kelly, E.; Pettay, J.D.; Choueiri, T.K.; Crowe, J.P.; et al. The expression of the cytoskeletal focal adhesion protein paxillin in breast cancer correlates with HER2 overexpression and may help predict response to chemotherapy: a retrospective immunohistochemical study. Breast J. 2007, 13, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. β-catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar]
- Xu, C.-f.; Wang, J. Delivery systems for siRNA drug development in cancer therapy. Asian J. Pharm. Sci. 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Behlke, M.A. Progress towards in vivo use of siRNAs. Mol. Ther. 2006, 13, 644–670. [Google Scholar] [CrossRef] [PubMed]
- Fatemian, T.; Chowdhury, E. Cytotoxicity Enhancement in Breast Cancer Cells with Carbonate Apatite-Facilitated Intracellular Delivery of Anti-Cancer Drugs. Toxics 2018, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Pandey, H.; Rani, R.; Agarwal, V. Liposome and their applications in cancer therapy. Braz. Arch. Biol. Technol. 2016, 59, e16150477. [Google Scholar] [CrossRef]
- Li, Y.; Humphries, B.; Yang, C.; Wang, Z. Nanoparticle-Mediated Therapeutic Agent Delivery for Treating Metastatic Breast Cancer-Challenges and Opportunities. Nanomaterials 2018, 8, 361. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Kudgus, R.A.; Bhattacharya, R.; Mukherjee, P. Inorganic nanoparticles in cancer therapy. Pharm. Res. 2011, 28, 237–259. [Google Scholar] [CrossRef]
- Yalcin, S.; Gunduz, U. Nanoparticle based delivery of miRNAs to overcome drug resistance in breast cancer. J. Nanomed. Nanotechnol. 2016, 7, 414. [Google Scholar]
- Chowdhury, E. pH-sensitive nano-crystals of carbonate apatite for smart and cell-specific transgene delivery. Exp. Opin. Drug Deliv. 2007, 4, 193–196. [Google Scholar] [CrossRef]
- Chowdhury, E.; Akaike, T. High performance DNA nano-carriers of carbonate apatite: multiple factors in regulation of particle synthesis and transfection efficiency. Int. J. Nanomed. 2007, 2, 101. [Google Scholar] [CrossRef]
- Chowdhury, E.H. H-sensitive nanocrystals of carbonate apatite-a powerful and versatile tool for efficient delivery of genetic materials to mammalian cells. In Advances in Biomaterials Science and Biomedical Applications; InTechOpen, 2013. [Google Scholar]
- Hossain, S.; Stanislaus, A.; Chua, M.J.; Tada, S.; Tagawa, Y.I.; Chowdhury, E.H.; Akaike, T. Carbonate apatite-facilitated intracellularly delivered siRNA for efficient knockdown of functional genes. J. Control. Release 2010, 147, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Mozar, F.S.; Chowdhury, E.H. Surface-modification of carbonate apatite nanoparticles enhances delivery and cytotoxicity of gemcitabine and anastrozole in breast cancer cells. Pharmaceutics 2017, 9, 21. [Google Scholar] [CrossRef]
- Tiash, S.; Othman, I.; Rosli, R.; Hoque Chowdhury, E. Methotrexate-and cyclophosphamide-embedded pure and strontiumsubstituted carbonate apatite nanoparticles for augmentation of chemotherapeutic activities in breast cancer cells. Curr. Drug Deliv. 2014, 11, 214–222. [Google Scholar] [CrossRef]
- Kunnath, A.P.; Tiash, S.; Fatemian, T.; Morshed, M.; Mohamed, S.M.; Chowdhury, E.H. Intracellular delivery of ERBB2 siRNA and p53 gene synergistically inhibits the growth of established tumour in an immunocompetent mouse. J. Cancer Sci. Ther. 2014, 6, 99–104. [Google Scholar] [CrossRef]
- Tiash, S.; Kamaruzman, N.I.B.; Chowdhury, E.H. Carbonate apatite nanoparticles carry siRNA(s) targeting growth factor receptor genes egfr1 and erbb2 to regress mouse breast tumor. Drug Deliv. 2017, 24, 1721–1730. [Google Scholar] [CrossRef]
- Tiash, S.; Chowdhury, E.H. siRNAs targeting multidrug transporter genes sensitise breast tumour to doxorubicin in a syngeneic mouse model. J. Drug Target. 2019, 27, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kunnath, A.P.; Kamaruzman, N.I.; Chowdhury, E.H. Nanoparticle-facilitated intratumoral delivery of Bcl-2/IGF-1R siRNAs and p53 Gene synergistically inhibits tumor growth in immunocompetent mice. J. Nanomed. Nanotechnol. 2015, 6, 1. [Google Scholar] [CrossRef]
- Rehman, I.; Bonfield, W. Characterization of hydroxyapatite and carbonated apatite by photo acoustic FTIR spectroscopy. J. Mater. Sci. 1997, 8, 1–4. [Google Scholar] [CrossRef]
- Berzina-Cimdina, L.; Borodajenko, N. Research of calcium phosphates using Fourier transform infrared spectroscopy. In Infrared Spectroscopy-Materials Science, Engineering and Technology; IntechOpen, 2012. [Google Scholar]
- Veiderma, M.; Knubovets, R.; Tonsuaadu, K. Structural properties of apatites from Finland studied by FTIR spectroscopy. Bull. Geol. Soc. Finland 1998, 70, 69–75. [Google Scholar] [CrossRef]
- Chowdhury, E.H.; Akaike, T. pH-sensitive inorganic nano-particles and their precise cell targetibility: An efficient gene delivery and expression system. Curr. Chem. Biol. 2007, 1, 201–213. [Google Scholar]
- Stoch, A.; Jastrzebski, W.; Brożek, A.; Trybalska, B.; Cichocińska, M.; Szarawara, E. FTIR monitoring of the growth of the carbonate containing apatite layers from simulated and natural body fluids. J. Mol. Struct. 1999, 511, 287–294. [Google Scholar] [CrossRef]
- Chowdhury, E.H. pH-responsive magnesium-and carbonate-substituted apatite nano-crystals for efficient and cell-targeted delivery of transgenes. Open J. Genet. 2013, 3, 38. [Google Scholar] [CrossRef]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Modern Pathol. 2011, 24, 209. [Google Scholar] [CrossRef]
- Michaelson, J.S.; Leder, P. β-catenin is a downstream effector of Wnt-mediated tumorigenesis in the mammary gland. Oncogene 2001, 20, 5093. [Google Scholar] [CrossRef][Green Version]
- Li, L.; Li, X.; Qi, L.; Rychahou, P.; Jafari, N.; Huang, C. The role of talin2 in breast cancer tumorigenesis and metastasis. Oncotarget 2017, 8, 106876. [Google Scholar] [CrossRef][Green Version]
- Hironaka-Mitsuhashi, A.; Matsuzaki, J.; Takahashi, R.U.; Yoshida, M.; Nezu, Y.; Yamamoto, Y.; Shiino, S.; Kinoshita, T.; Ushijima, T.; Hiraoka, N.; et al. A tissue microRNA signature that predicts the prognosis of breast cancer in young women. PLoS ONE 2017, 12, e0187638. [Google Scholar] [CrossRef]
- Desiniotis, A.; Kyprianou, N. Significance of talin in cancer progression and metastasis. In International Review of Cell and Molecular Biology; Elsevier: Cambridge, MA, USA, 2011; pp. 117–147. [Google Scholar]
- Mierke, C.T.; Kollmannsberger, P.; Zitterbart, D.P.; Diez, G.; Koch, T.M.; Marg, S.; Ziegler, W.H.; Goldmann, W.H.; Fabry, B. Vinculin facilitates cell invasion into 3D collagen matrices. J. Biol. Chem. 2010. [Google Scholar] [CrossRef]
- Subauste, M.C.; Pertz, O.; Adamson, E.D.; Turner, C.E.; Junger, S.; Hahn, K.M. Vinculin modulation of paxillin–FAK interactions regulates ERK to control survival and motility. J. Cell Biol. 2004, 165, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Rajfur, Z.; Borchers, C.; Schaller, M.D.; Jacobson, K. JNK phosphorylates paxillin and regulates cell migration. Nature 2003, 424, 219. [Google Scholar] [CrossRef] [PubMed]
- Deakin, N.O.; Turner, C.E. Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol. Biol. Cell 2011, 22, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-S.; Kao, H.-Y. Alpha-actinin 4 and Tumorigenesis of Breast Cancer, in Vitamins and Hormones; Elsevier: Cambridge, MA, USA, 2013; pp. 323–351. [Google Scholar]
- Kamaruzman, N.; Tiash, S.; Ashaie, M.; Chowdhury, E. siRNAs Targeting Growth Factor Receptor and Anti-Apoptotic Genes Synergistically Kill Breast Cancer Cells through Inhibition of MAPK and PI-3 kinase pathways. Biomedicines 2018, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Tada, S.; Chowdhury, E.H.; Cho, C.S.; Akaike, T. pH-sensitive carbonate apatite as an intracellular protein transporter. Biomaterials 2010, 31, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H. Nanotherapeutics: From Laboratory to Clinic; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Park, D.; KÅresen, R.; Axcrona, U.; Noren, T.; Sauer, T. Expression pattern of adhesion molecules (E-cadherin, α-, β-, γ-catenin and claudin-7), their influence on survival in primary breast carcinoma, and their corresponding axillary lymph node metastasis. Apmis 2007, 115, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.M.; Nelson, W.J. Bench to bedside and back again: molecular mechanisms of α-catenin function and roles in tumorigenesis. In Seminars in Cancer Biology; Elsevier: Cambridge, MA, USA, 2008. [Google Scholar]
- Jevnikar, Z.; Rojnik, M.; Jamnik, P.; Doljak, B.; Fonović, U.P.; Kos, J. Cathepsin H mediates the processing of talin and regulates migration of prostate cancer cells. J. Biol. Chem. 2013, 288, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Beaty, B.T.; Wang, Y.; Bravo-Cordero, J.J.; Sharma, V.P.; Miskolci, V.; Hodgson, L.; Condeelis, J. Talin regulates moesin–NHE-1 recruitment to invadopodia and promotes mammary tumor metastasis. J. Cell Biol. 2014, 205, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.P.; Dai, W.; Ren, Y.H.; Xu, Y.C.; Zhang, S.M.; Qian, Y.B. Both Talin-1 and Talin-2 correlate with malignancy potential of the human hepatocellular carcinoma MHCC-97 L cell. BMC Cancer 2016, 16, 45. [Google Scholar] [CrossRef]
- Lifschitz-Mercer, B.; Czernobilsky, B.; Feldberg, E.; Geiger, B. Expression of the adherens junction protein vinculin in human basal and squamous cell tumors: Relationship to invasiveness and metastatic potential. Human Pathol. 1997, 28, 1230–1236. [Google Scholar] [CrossRef]
- Shao, H.; Wu, C.; Wells, A. Phosphorylation of alpha-actinin-4 upon epidermal growth factor (EGF) exposure regulates its interaction with actin. J. Biol. Chem. 2010, 285, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Honda, K.; Tsuda, H.; Hiraoka, N.; Imoto, I.; Kosuge, T.; Umaki, T.; Onozato, T.; Shitashige, M.; Yamaguchi, U.; et al. Expression and gene amplification of actinin-4 in invasive ductal carcinoma of the pancreas. Clin. Cancer Res. 2008, 14, 5348–5356. [Google Scholar] [CrossRef] [PubMed]
- Welsch, T.; Keleg, S.; Bergmann, F.; Bauer, S.; Hinz, U.; Schmidt, J. Actinin-4 expression in primary and metastasized pancreatic ductal adenocarcinoma. Pancreas 2009, 38, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.H.; Downey, C.; Basson, M.D. siRNA-mediated reduction of α-actinin-1 inhibits pressure-induced murine tumor cell wound implantation and enhances tumor-free survival. Neoplasia 2008, 10, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.H.; Haimovich, B.; Basson, M.D. α-Actinin-1 phosphorylation modulates pressure-induced colon cancer cell adhesion through regulation of focal adhesion kinase-Src interaction. Am. J. Physiol.-Cell Physiol. 2007, 293, C1862–C1874. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product Name | Gene Name | Code | Target Sequence |
|---|---|---|---|
| Hs_CTNNA1_6 | Catenin alpha 1 | CTNNA1 | AAGTGGATAAGCTGAACATTA |
| Hs_CTNNB1_5 | Catenin beta 1 | CTNNB1 | CTCGGGATGTTCACAACCGAA |
| Hs_PXN_6 | Paxillin | PXN | CCGACTGAAACTGGAACCCTT |
| Hs_ACTN1_5 | Actinin 1 | ACTN1 | AACAAATCTGAATACGGCTTT |
| Hs_TLN1_5 | Talin 1 | TLN1 | AACAGAGACCCCTGAAGATCC |
| Hs_VCL_5 | Vinculin | VCL | AAAGATGATTGACGAGAGACA |
| CA + siRNAs | MCF-7 | MDA-MB-231 | 4T1 | |||
|---|---|---|---|---|---|---|
| Cell Viability (%) ± S.D. | Actual Cytotoxicity (%) ± S.D. | Cell Viability (%) ± S.D. | Actual Cytotoxicity (%) ± S.D. | Cell Viability (%) ± S.D. | Actual Cytotoxicity (%) ± S.D. | |
| CTNNA1 | 92.65 ± 5.0 | 2.03 ± 6.2 | 81.65 ± 6.1 | −2.28 ± 4.9 | 80.76 ± 5.4 | 13.42 ± 8.9 |
| CTNNB1 | 76.46 ± 10.8 | 14.54 ± 9.3 | 86.93 ± 3.1 | 8.15 ± 7.6 | 77.06 ± 3.0 | 16.03 ± 5.3 |
| TLN1 | 67.65 ± 11.8 | 30.2 ± 12.1 | 92.5 ± 1.5 | −4.39 ± 2.9 | 66.46 ± 11.6 | 23.53 ± 10.4 |
| VCL | 78.34 ± 0.8 | 8.74 ± 7.40 | 83.3 ± 0.6 | 9.37 ± 3.9 | 70.67 ± 16.4 | 19.38 ± 16.1 |
| PXN | 57.60 ± 7.3 | 29.76 ± 0.8 | 82.92 ± 1.2 | 13.82 ± 3.1 | 67.65 ± 0.5 | 27.07 ± 8.9 |
| ACTN1 | 66.67 ± 5.2 | 25.97 ± 8.9 | 87.15 ± 4.2 | 6.59 ± 0.4 | 71.81 ± 3.4 | 18.54 ± 6.3 |
| Day 9 | Day 11 | Day 13 | Day 15 | Day 17 | Day 19 | Day 21 |
|---|---|---|---|---|---|---|
| 3.74 | 3.41 | 2.65 | 2.60 | 3.40 | 1.44 | 0.88 |
| Day 10 | Day 11 | Day 13 | Day 15 | Day 17 | Day 19 | Day 21 |
|---|---|---|---|---|---|---|
| 4.8 | 3.8 | 3.7 | 5.1 | 2.7 | 4.2 | 4.8 |
| Day 10 | Day 12 | Day 14 | Day 16 | Day 19 | Day 21 |
|---|---|---|---|---|---|
| 1.58 | 2.58 | 2.11 | 2.29 | 2.04 | 1.91 |
| Day 10 | Day 12 | Day 14 | Day 16 | Day 19 | Day 21 |
|---|---|---|---|---|---|
| 2.18 | 3.61 | 1.85 | 2.02 | 2.16 | 1.64 |
| Day 10 | Day 12 | Day 14 | Day 16 | Day 19 | Day 21 |
|---|---|---|---|---|---|
| 4.80 | 4.02 | 5.02 | 5.61 | 6.19 | 5.92 |
| Day 10 | Day 11 | Day 13 | Day 15 | Day 17 | Day 19 | Day 21 |
|---|---|---|---|---|---|---|
| 3.02 | 4.42 | 4.64 | 4.49 | 4.50 | 3.42 | 3.19 |
| Proteins | Critical Signaling Pathways | Expression in | References |
|---|---|---|---|
| Catenins | WNT | Adenocarcinomas, TNBC/Basal-like breast cancers, Metastasis | [48,49] |
| Talin | P13K-AKT, RAS, FAK-MAPK | TNBC/Basal-like breast cancers, HER2-overexpressing, Metastasis | [13,50,51,52] |
| Vinculin | FAK-MAPK, PI3K | TNBC | [14,21,53,54] |
| Paxillin | Src/FAK/PI3K, ERK, Rho/ROCK, JNK, p38 MAPK | Adenocarcinomas, TNBC/Basal-like breast cancers, HER2 overexpressing, Metastasis | [7,8,22,55,56] |
| Actinin | P13K-AKT, MAPK | Adenocarcinomas, TNBC/Basal-like breast cancers | [9,10,18,57] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashaie, M.A.; Islam, R.A.; Kamaruzman, N.I.; Ibnat, N.; Tha, K.K.; Chowdhury, E.H. Targeting Cell Adhesion Molecules via Carbonate Apatite-Mediated Delivery of Specific siRNAs to Breast Cancer Cells In Vitro and In Vivo. Pharmaceutics 2019, 11, 309. https://doi.org/10.3390/pharmaceutics11070309
Ashaie MA, Islam RA, Kamaruzman NI, Ibnat N, Tha KK, Chowdhury EH. Targeting Cell Adhesion Molecules via Carbonate Apatite-Mediated Delivery of Specific siRNAs to Breast Cancer Cells In Vitro and In Vivo. Pharmaceutics. 2019; 11(7):309. https://doi.org/10.3390/pharmaceutics11070309
Chicago/Turabian StyleAshaie, Maeirah Afzal, Rowshan Ara Islam, Nur Izyani Kamaruzman, Nabilah Ibnat, Kyi Kyi Tha, and Ezharul Hoque Chowdhury. 2019. "Targeting Cell Adhesion Molecules via Carbonate Apatite-Mediated Delivery of Specific siRNAs to Breast Cancer Cells In Vitro and In Vivo" Pharmaceutics 11, no. 7: 309. https://doi.org/10.3390/pharmaceutics11070309
APA StyleAshaie, M. A., Islam, R. A., Kamaruzman, N. I., Ibnat, N., Tha, K. K., & Chowdhury, E. H. (2019). Targeting Cell Adhesion Molecules via Carbonate Apatite-Mediated Delivery of Specific siRNAs to Breast Cancer Cells In Vitro and In Vivo. Pharmaceutics, 11(7), 309. https://doi.org/10.3390/pharmaceutics11070309