Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods



Abstract
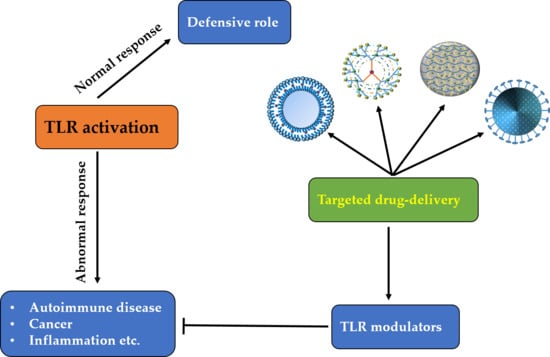
1. Introduction
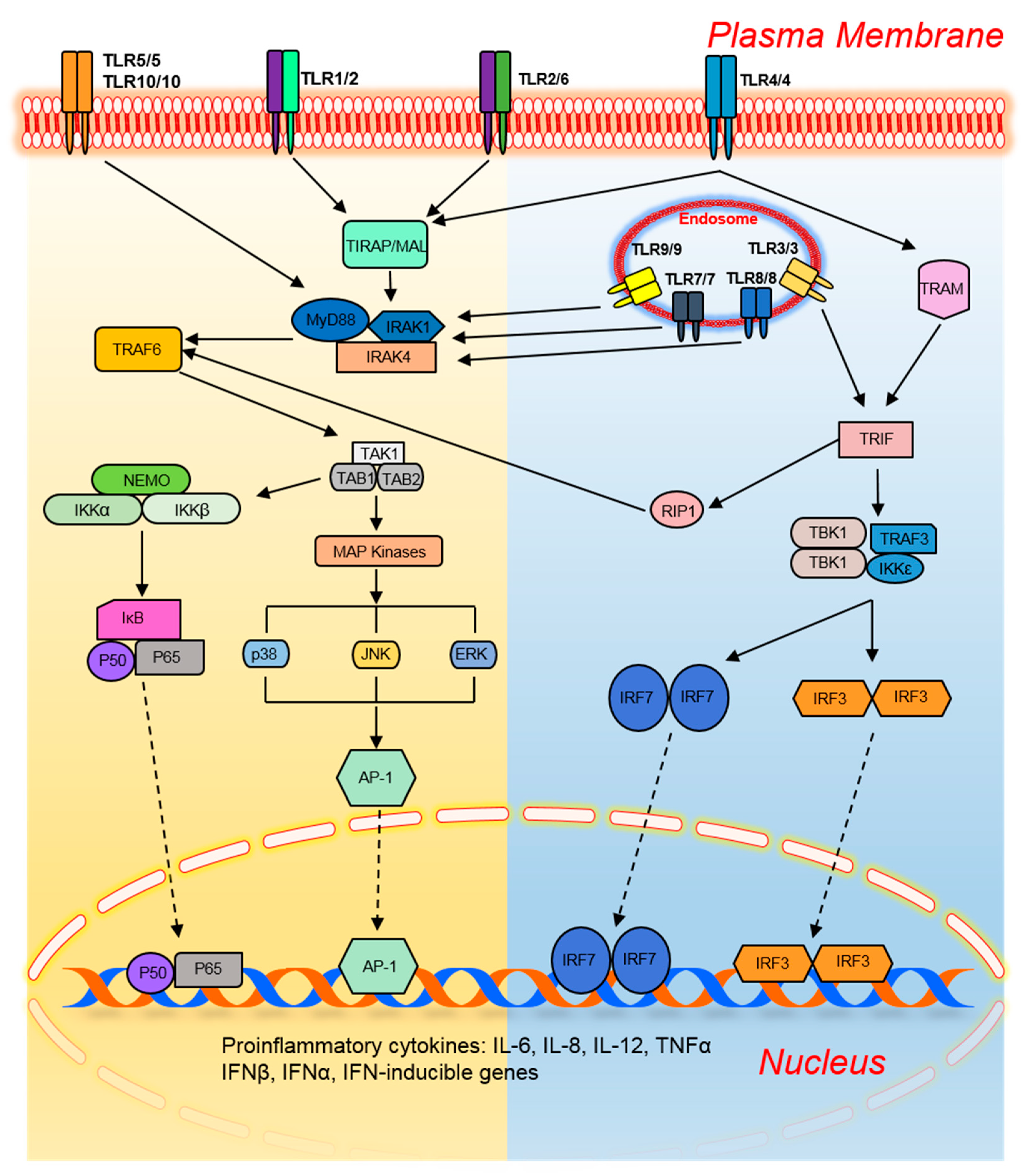
2. Toll-Like Receptors and Their Signaling Pathways
2.1. The MyD88-Dependent Pathway
2.2. The TRIF-Dependent Pathway
3. Representative Diseases Associated with TLRs
4. TLR-Targeting Therapeutics
4.1. TLR1/2 and TLR2/6
4.2. TLR3
4.3. TLR4
4.4. TLR5
4.5. TLR7 and TLR8
4.6. TLR9
4.7. TLR10–TLR13
5. Controlled Drug Delivery Systems
5.1. The Diffusion-Controlled System
5.2. The Solvent-Activated System
5.3. The Chemically Controlled System
5.4. The Magnetically Controlled System
5.5. A Targeted Drug Delivery System
5.5.1. Active Targeted Drug Delivery
5.5.2. Passive Targeted Drug Delivery
5.6. Examples of Targeted Drug Delivery
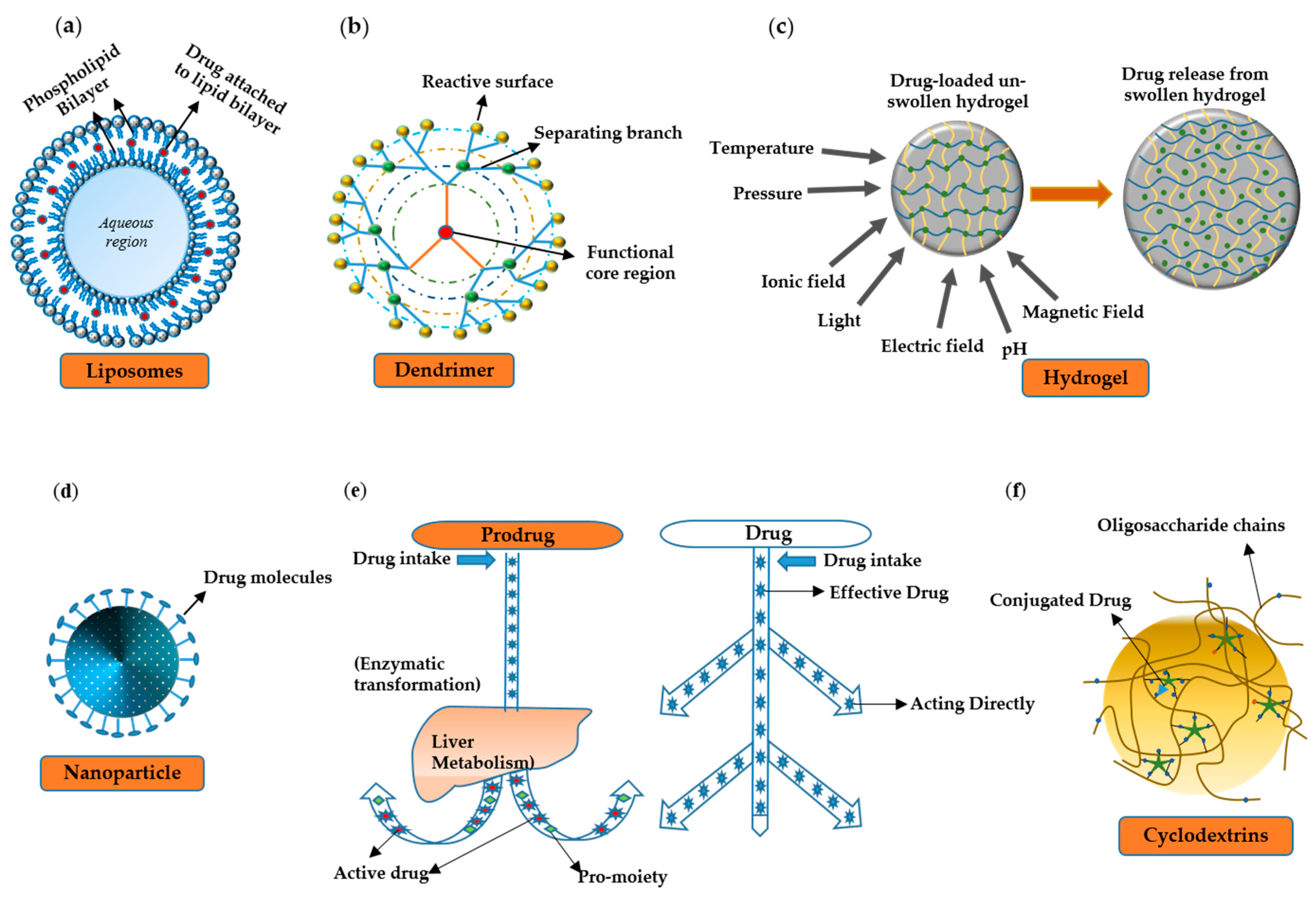
6. Drug Delivery Vehicles
6.1. Liposomes
6.2. Hydrogels
6.3. Prodrugs
6.4. The Nanoparticle System
6.5. Dendrimers
6.6. Cyclodextrins
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Lill, J.R. Introduction to Biotherapeutics. In Analytical Characterization of Biotherapeutics, 1st ed.; Wiley: Hoboken, NJ, USA, 2017; pp. 1–14. [Google Scholar]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Palfi, S.; Gurruchaga, J.M.; Ralph, G.S.; Lepetit, H.; Lavisse, S.; Buttery, P.C.; Watts, C.; Miskin, J.; Kelleher, M.; Deeley, S.; et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 2014, 383, 1138–1146. [Google Scholar] [CrossRef]
- Sahel, J.A.; Roska, B. Gene therapy for blindness. Annu. Rev. Neurosci. 2013, 36, 467–488. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318. [Google Scholar] [CrossRef]
- Aiuti, A.; Bacchetta, R.; Seger, R.; Villa, A.; Cavazzana-Calvo, M. Gene therapy for primary immunodeficiencies: Part 2. Curr. Opin. Immunol. 2012, 24, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Chiriaco, M.; Farinelli, G.; Capo, V.; Zonari, E.; Scaramuzza, S.; Di Matteo, G.; Sergi, L.S.; Migliavacca, M.; Hernandez, R.J.; Bombelli, F.; et al. Dual-regulated lentiviral vector for gene therapy of X-linked chronic granulomatosis. Mol. Ther. 2014, 22, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed]
- Sleep, D. Albumin and its application in drug delivery. Expert Opin. Drug Deliv. 2015, 12, 793–812. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhang, J.; Xu, Q.; Huang, Z.; Wang, Y.; Shen, Q. Hyaluronic acid-coated cationic nanostructured lipid carriers for oral vincristine sulfate delivery. Drug Dev. Ind. Pharm. 2017, 43, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shi, S.; Zhang, Z.; Gong, T.; Sun, X. Coating solid lipid nanoparticles with hyaluronic acid enhances antitumor activity against melanoma stem-like cells. Theranostics 2015, 5, 755. [Google Scholar] [CrossRef] [PubMed]
- Almalik, A.; Benabdelkamel, H.; Masood, A.; Alanazi, I.O.; Alradwan, I.; Majrashi, M.A.; Alfadda, A.A.; Alghamdi, W.M.; Alrabiah, H.; Tirelli, N.; et al. Hyaluronic acid coated chitosan nanoparticles reduced the immunogenicity of the formed protein corona. Sci. Rep. 2017, 7, 10542. [Google Scholar] [CrossRef]
- Martens, T.F.; Remaut, K.; Deschout, H.; Engbersen, J.F.; Hennink, W.E.; Van Steenbergen, M.J.; Demeester, J.; De Smedt, S.C.; Braeckmans, K. Coating nanocarriers with hyaluronic acid facilitates intravitreal drug delivery for retinal gene therapy. J. Control. Release 2015, 202, 83–92. [Google Scholar] [CrossRef]
- Wang, T.; Hou, J.; Su, C.; Zhao, L.; Shi, Y. Hyaluronic acid-coated chitosan nanoparticles induce ROS-mediated tumor cell apoptosis and enhance antitumor efficiency by targeted drug delivery via CD44. J. Nanobiotechnol. 2017, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Neumann, P.; Corbo, M.; Loh, E. Recent advances in biotherapeutics drug discovery and development. In Drug Discovery and Development-Present and Future; IntechOpen: London, UK, 2011. [Google Scholar]
- Reichert, J.M. Antibody-based therapeutics to watch in 2011. In MAbs; Taylor & Francis: Didcot, UK, 2011; pp. 76–99. [Google Scholar]
- Oliveira, P.H.; Mairhofer, J.; Alves, P.M.; Lara, A.R.; Kontoravdi, C. Advances in the Development of Biotherapeutics. Biomed Res. Int. 2015, 2015, 793876. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G. Biopharmaceutical benchmarks 2014. Nat. Biotechnol. 2014, 32, 992. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Metrics for antibody therapeutics development. In MAbs; Taylor & Francis: Didcot, UK, 2010; pp. 695–700. [Google Scholar]
- Hummel, G.; Reineke, U.; Reimer, U. Translating peptides into small molecules. Mol. Biosyst. 2006, 2, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131. [Google Scholar] [CrossRef]
- Sharma, S.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hernandez, L.D.; Galán, J.E.; Janeway, C.A., Jr.; Medzhitov, R.; Flavell, R.A. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 2002, 110, 191–202. [Google Scholar] [CrossRef]
- Suzuki, N.; Suzuki, S.; Duncan, G.S.; Millar, D.G.; Wada, T.; Mirtsos, C.; Takada, H.; Wakeham, A.; Itie, A.; Li, S.; et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 2002, 416, 750. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitin signalling in the NF-κB pathway. Nat. Cell Biol. 2005, 7, 758. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Sanjo, H.; Uematsu, S.; Kaisho, T.; Hoshino, K.; Takeuchi, O.; Kobayashi, M.; Fujita, T.; et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature 2002, 420, 324. [Google Scholar] [CrossRef] [PubMed]
- Schnare, M.; Holt, A.C.; Takeda, K.; Akira, S.; Medzhitov, R. Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr. Biol. 2000, 10, 1139–1142. [Google Scholar] [CrossRef]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Eugene, C.Y.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Kaisho, T.; Takeuchi, O.; Sato, S.; Sanjo, H.; Hoshino, K.; Horiuchi, T.; Tomizawa, H.; Takeda, K.; Akira, S. Small anti-viral compounds activate immune cells via the TLR7 MyD88–dependent signaling pathway. Nat. Immunol. 2002, 3, 196. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takeuchi, O.; Fujita, T.; Inoue, J.I.; Mühlradt, P.F.; Sato, S.; Hoshino, K.; Akira, S. Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharide-inducible genes. J. Immunol. 2001, 167, 5887–5894. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3–mediated interferon-β induction. Nat. Immunol. 2003, 4, 161. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Uematsu, S.; Hoshino, K.; Kaisho, T.; Takeuchi, O.; Takeda, K.; Akira, S. TRAM is specifically involved in the Toll-like receptor 4–mediated MyD88-independent signaling pathway. Nat. Immunol. 2003, 4, 1144. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Burns, K.; Hofmann, K.; Blancheteau, V.; Martinon, F.; Kelliher, M.; Tschopp, J. RIP1 is an essential mediator of Toll-like receptor 3–induced NF-κB activation. Nat. Immunol. 2004, 5, 503. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sugiyama, M.; Yamamoto, M.; Watanabe, Y.; Kawai, T.; Takeda, K.; Akira, S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 2003, 171, 4304–4310. [Google Scholar] [PubMed]
- Pradere, J.P.; Dapito, D.H.; Schwabe, R.F. The Yin and Yang of Toll-like receptors in cancer. Oncogene 2014, 33, 3485. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: A journey from molecular to nano therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H. TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 2011, 11, 807. [Google Scholar] [CrossRef]
- Joosten, L.A.; Abdollahi-Roodsaz, S.; Dinarello, C.A.; O’neill, L.; Netea, M.G. Toll-like receptors and chronic inflammation in rheumatic diseases: New developments. Nat. Rev. Rheumatol. 2016, 12, 344. [Google Scholar] [CrossRef]
- Duffy, L.; O’Reilly, S.C. Toll-like receptors in the pathogenesis of autoimmune diseases: Recent and emerging translational developments. Immunotargets Ther. 2016, 5, 69. [Google Scholar]
- Lin, Y.T.; Verma, A.; P Hodgkinson, C. Toll-like receptors and human disease: Lessons from single nucleotide polymorphisms. Curr. Genom. 2012, 13, 633–645. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef] [PubMed]
- Hansbro, P.M.; Haw, T.J.; Starkey, M.R.; Miyake, K. Toll-Like Receptors in COPD. Eur. Resp. J. 2017, 49, 1700739. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S. Sepsis, severe sepsis and septic shock: Changes in incidence, pathogens and outcomes. Expert Rev. Anti Infect. Ther. 2012, 10, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.J.; Williams, S.N.; DeFrances, C.J.; Golosinskiy, A. Inpatient Care for Septicemia or Sepsis: A Challenge for Patients and Hospitals; NCHS Data Brief, No 62; National Center for Health Statistics: Hyattsville, MD, USA, 2010. [Google Scholar]
- Friedman, G.; Silva, E.; Vincent, J.L. Has the mortality of septic shock changed with time? Crit. Care Med. 1998, 26, 2078–2086. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L. Germs. N Engl J Med. 1972, 287, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Savva, A.; Roger, T. Targeting toll-like receptors: Promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front. Immunol. 2013, 4, 387. [Google Scholar] [CrossRef]
- Kesimer, M.; Ford, A.A.; Ceppe, A.; Radicioni, G.; Cao, R.; Davis, C.W.; Doerschuk, C.M.; Alexis, N.E.; Anderson, W.H.; Henderson, A.G.; et al. Airway mucin concentration as a marker of chronic bronchitis. N. Engl. J. Med. 2017, 377, 911–922. [Google Scholar] [CrossRef]
- Pomerenke, A.; Lea, S.R.; Herrick, S.; Lindsay, M.A.; Singh, D. Characterization of TLR-induced inflammatory responses in COPD and control lung tissue explants. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 2409. [Google Scholar] [CrossRef]
- Schrijver, I.A.; Melief, M.J.; Tak, P.P.; Hazenberg, M.P.; Laman, J.D. Antigen-presenting cells containing bacterial peptidoglycan in synovial tissues of rheumatoid arthritis patients coexpress costimulatory molecules and cytokines. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2000, 43, 2160–2168. [Google Scholar] [CrossRef]
- Van Der Heijden, I.M.; Wilbrink, B.; Tchetverikov, I.; Schrijver, I.A.; Schouls, L.M.; Hazenberg, M.P.; Breedveld, F.C.; Tak, P.P. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2000, 43, 593–598. [Google Scholar] [CrossRef]
- Barrat, F.J.; Meeker, T.; Gregorio, J.; Chan, J.H.; Uematsu, S.; Akira, S.; Chang, B.; Duramad, O.; Coffman, R.L. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med. 2005, 202, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Raber, M.L.; Dunand-Sauthier, I.; Wu, T.; Li, Q.Z.; Uematsu, S.; Akira, S.; Reith, W.; Mohan, C.; Kotzin, B.L.; Izui, S. Critical role of TLR7 in the acceleration of systemic lupus erythematosus in TLR9-deficient mice. J. Autoimmun. 2010, 34, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, K.M.; Christensen, S.R.; Shupe, J.; Kashgarian, M.; Kim, D.; Elkon, K.; Shlomchik, M.J. TLR9 regulates TLR7-and MyD88-dependent autoantibody production and disease in a murine model of lupus. J. Immunol. 2010, 184, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Spachidou, M.; Bourazopoulou, E.; Maratheftis, C.; Kapsogeorgou, E.; Moutsopoulos, H.; Tzioufas, A.; Manoussakis, M. Expression of functional Toll-like receptors by salivary gland epithelial cells: Increased mRNA expression in cells derived from patients with primary Sjögren’s syndrome. Clin. Exp. Immunol. 2007, 147, 497–503. [Google Scholar] [CrossRef]
- Karlsen, M.; Jonsson, R.; Brun, J.; Appel, S.; Hansen, T. TLR-7 and-9 stimulation of peripheral blood B cells indicate altered TLR signalling in primary Sjögren’s syndrome patients by increased secretion of cytokines. Scand. J. Immunol. 2015, 82, 523–531. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860. [Google Scholar] [CrossRef]
- Fukata, M.; Hernandez, Y.; Conduah, D.; Cohen, J.; Chen, A.; Breglio, K.; Goo, T.; Hsu, D.; Xu, R.; Abreu, M.T. Innate immune signaling by Toll-like receptor-4 (TLR4) shapes the inflammatory microenvironment in colitis-associated tumors. Inflamm. Bowel Dis. 2009, 15, 997–1006. [Google Scholar] [CrossRef]
- Fukata, M.; Shang, L.; Santaolalla, R.; Sotolongo, J.; Pastorini, C.; España, C.; Ungaro, R.; Harpaz, N.; Cooper, H.S.; Elson, G.; et al. Constitutive activation of epithelial TLR4 augments inflammatory responses to mucosal injury and drives colitis-associated tumorigenesis. Inflamm. Bowel Dis. 2010, 17, 1464–1473. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Yusuf, N.; Nasti, T.H.; Long, J.A.; Naseemuddin, M.; Lucas, A.P.; Xu, H.; Elmets, C.A. Protective role of Toll-like receptor 4 during the initiation stage of cutaneous chemical carcinogenesis. Cancer Res. 2008, 68, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Cataisson, C.; Salcedo, R.; Hakim, S.; Moffitt, B.A.; Wright, L.; Yi, M.; Stephens, R.; Dai, R.M.; Lyakh, L.; Schenten, D.; et al. IL-1R–MyD88 signaling in keratinocyte transformation and carcinogenesis. J. Exp. Med. 2012, 209, 1689–1702. [Google Scholar] [CrossRef] [PubMed]
- Chochi, K.; Ichikura, T.; Kinoshita, M.; Majima, T.; Shinomiya, N.; Tsujimoto, H.; Kawabata, T.; Sugasawa, H.; Ono, S.; Seki, S.; et al. Helicobacter pylori augments growth of gastric cancers via the lipopolysaccharide-toll-like receptor 4 pathway whereas its lipopolysaccharide attenuates antitumor activities of human mononuclear cells. Clin. Cancer Res. 2008, 14, 2909–2917. [Google Scholar] [CrossRef] [PubMed]
- Harmey, J.H.; Bucana, C.D.; Lu, W.; Byrne, A.M.; McDonnell, S.; Lynch, C.; Bouchier-Hayes, D.; Dong, Z. Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int. J. Cancer 2002, 101, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, H.; Feng, P.; Zhou, X.; Wen, H.; Xie, X.; Shen, H.; Zhu, X. Reduced expression of Toll-like receptor 4 inhibits human breast cancer cells proliferation and inflammatory cytokines secretion. J. Exp. Clin. Cancer Res. 2010, 29, 92. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Yusuf, N. Expression of toll-like receptors on breast tumors: Taking a toll on tumor microenvironment. Int. J. Breast Cancer 2012, 2012, 716564. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhao, J.; Li, H.; He, K.L.; Chen, Y.; Mayer, L.; Unkeless, J.C.; Xiong, H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005, 65, 5009–5014. [Google Scholar] [CrossRef]
- Huang, Y.; Cai, B.; Xu, M.; Qiu, Z.; Tao, Y.; Zhang, Y.; Wang, J.; Xu, Y.; Zhou, Y.; Yang, J.; et al. Gene silencing of Toll-like receptor 2 inhibits proliferation of human liver cancer cells and secretion of inflammatory cytokines. PLoS ONE 2012, 7, e38890. [Google Scholar] [CrossRef]
- Huang, B.; Zhao, J.; Shen, S.; Li, H.; He, K.L.; Shen, G.X.; Mayer, L.; Unkeless, J.; Li, D.; Yuan, Y.; et al. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. 2007, 67, 4346–4352. [Google Scholar] [CrossRef]
- Song, E.J.; Kang, M.J.; Kim, Y.S.; Kim, S.M.; Lee, S.E.; Kim, C.H.; Kim, D.J.; Park, J.H. Flagellin promotes the proliferation of gastric cancer cells via the Toll-like receptor 5. Int. J. Mol. Med. 2011, 28, 115–119. [Google Scholar]
- Cherfils-Vicini, J.; Platonova, S.; Gillard, M.; Laurans, L.; Validire, P.; Caliandro, R.; Magdeleinat, P.; Mami-Chouaib, F.; Dieu-Nosjean, M.C.; Fridman, W.H.; et al. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J. Clin. Investig. 2010, 120, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Wu, M.S.; Lin, J.T.; Chen, C.C. Helicobacter pylori-induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase-2 induction through TLR2/TLR9 and promoter regulation. J. Immunol. 2005, 175, 8242–8252. [Google Scholar] [CrossRef] [PubMed]
- Bhatelia, K.; Singh, K.; Singh, R. TLRs: Linking inflammation and breast cancer. Cell. Signal. 2014, 26, 2350–2357. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, H.; Peng, G. TLR-mediated metabolic reprogramming in the tumor microenvironment: Potential novel strategies for cancer immunotherapy. Cell. Mol. Immunol. 2018, 15, 428. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Tan, R.S.; Ho, B.; Leung, B.P.; Ding, J.L. TLR cross-talk confers specificity to innate immunity. Int. Rev. Immunol. 2014, 33, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, M.; Hasturk, H.; Liang, Y.; Shin, H.; Hetzel, J.T.; Kantarci, A.; Rubin, D.; McDonnell, M.E.; Van Dyke, T.E.; Ganley-Leal, L.M.; et al. TLR cross-talk specifically regulates cytokine production by B cells from chronic inflammatory disease patients. J. Immunol. 2009, 183, 7461–7470. [Google Scholar] [CrossRef] [PubMed]
- Ratanji, K.D.; Derrick, J.P.; Dearman, R.J.; Kimber, I. Immunogenicity of therapeutic proteins: Influence of aggregation. J. Immunotoxicol. 2014, 11, 99–109. [Google Scholar] [CrossRef]
- Jostock, T.; Knopf, H.P. Mammalian stable expression of biotherapeutics. In Therapeutic Proteins; Springer, Humana Press: Totowa, NJ, USA, 2012; pp. 227–238. [Google Scholar]
- Mócsai, A.; Kovács, L.; Gergely, P. What is the future of targeted therapy in rheumatology: Biologics or small molecules? BMC Med. 2014, 12, 43. [Google Scholar] [CrossRef]
- Jayasena, S.D. Aptamers: An emerging class of molecules that rival antibodies in diagnostics. Clin. Chem. 1999, 45, 1628–1650. [Google Scholar]
- Zähringer, U.; Lindner, B.; Inamura, S.; Heine, H.; Alexander, C. TLR2–promiscuous or specific? A critical re-evaluation of a receptor expressing apparent broad specificity. Immunobiology 2008, 213, 205–224. [Google Scholar] [CrossRef] [PubMed]
- Flo, T.H.; Halaas, Ø.; Torp, S.; Ryan, L.; Lien, E.; Dybdahl, B.; Sundan, A.; Espevik, T. Differential expression of Toll-like receptor 2 in human cells. J. Leukoc. Biol. 2001, 69, 474–481. [Google Scholar] [PubMed]
- Reilly, M.; Miller, R.; Thomson, M.; Patris, V.; Ryle, P.; McLoughlin, L.; Mutch, P.; Gilboy, P.; Miller, C.; Broekema, M.; et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin. Pharmacol. Ther. 2013, 94, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Dar, P.; Kalaivanan, R.; Sied, N.; Mamo, B.; Kishore, S.; Suryanarayana, V.; Kondabattula, G. Montanide ISA™ 201 adjuvanted FMD vaccine induces improved immune responses and protection in cattle. Vaccine 2013, 31, 3327–3332. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Durai, P.; Govindaraj, R.G.; Choi, S. Structure and dynamic behavior of Toll-like receptor 2 subfamily triggered by malarial glycosylphosphatidylinositols of Plasmodium falciparum. FEBS J. 2013, 280, 6196–6212. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Nan, X.; Jin, M.S.; Youn, S.J.; Ryu, Y.H.; Mah, S.; Han, S.H.; Lee, H.; Paik, S.G.; Lee, J.O. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 2009, 31, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Guo, M.; Wang, X.; Li, J.; Wang, Y.; Ye, L.; Dai, M.; Zhou, L.; Persidsky, Y.; Ho, W. TLR3 activation efficiency by high or low molecular mass poly I: C. Innate Immun. 2013, 19, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Seya, T. TLR3: Interferon induction by double-stranded RNA including poly (I: C). Adv. Drug Deliv. Rev. 2008, 60, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dewan, V.; Yin, H. Discovery of small molecules as multi-toll-like receptor agonists with proinflammatory and anticancer activities. J. Med. Chem. 2017, 60, 5029–5044. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Wang, X.; Yin, H. Small-molecule inhibitors of the TLR3/dsRNA complex. J. Am. Chem. Soc. 2011, 133, 3764–3767. [Google Scholar] [CrossRef] [PubMed]
- Bunting, R.A.; Duffy, K.E.; Lamb, R.J.; San Mateo, L.R.; Smalley, K.; Raymond, H.; Liu, X.; Petley, T.; Fisher, J.; Beck, H.; et al. Novel antagonist antibody to TLR3 blocks poly (I: C)-induced inflammation in vivo and in vitro. Cell. Immunol. 2011, 267, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Silkoff, P.E.; Flavin, S.; Gordon, R.; Loza, M.J.; Sterk, P.J.; Lutter, R.; Diamant, Z.; Turner, R.B.; Lipworth, B.J.; Proud, D.; et al. Toll-like receptor 3 blockade in rhinovirus-induced experimental asthma exacerbations: A randomized controlled study. J. Allergy Clin. Immunol. 2018, 141, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Botos, I.; Wang, Y.; Leonard, J.N.; Shiloach, J.; Segal, D.M.; Davies, D.R. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science 2008, 320, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Choe, J.; Kelker, M.S.; Wilson, I.A. Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science 2005, 309, 581–585. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature 2009, 458, 1191. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shin, H.J.; Shah, M.; Cho, H.Y.; Anwar, M.A.; Achek, A.; Kwon, H.K.; Lee, B.; Yoo, T.H.; Choi, S. TLR4/MD2 specific peptides stalled in vivo LPS-induced immune exacerbation. Biomaterials 2017, 126, 49–60. [Google Scholar] [CrossRef]
- Wang, Y.; Su, L.; Morin, M.D.; Jones, B.T.; Whitby, L.R.; Surakattula, M.M.; Huang, H.; Shi, H.; Choi, J.H.; Wang, K.W.; et al. TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc. Natl. Acad. Sci. USA 2016, 113, E884–E893. [Google Scholar] [CrossRef]
- Ohto, U.; Fukase, K.; Miyake, K.; Satow, Y. Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 2007, 316, 1632–1634. [Google Scholar] [CrossRef]
- Arias, M.A.; Van Roey, G.A.; Tregoning, J.S.; Moutaftsi, M.; Coler, R.N.; Windish, H.P.; Reed, S.G.; Carter, D.; Shattock, R.J. Glucopyranosyl lipid adjuvant (GLA), a synthetic TLR4 agonist, promotes potent systemic and mucosal responses to intranasal immunization with HIVgp140. PLoS ONE 2012, 7, e41144. [Google Scholar] [CrossRef]
- Romero, C.D.; Varma, T.K.; Hobbs, J.B.; Reyes, A.; Driver, B.; Sherwood, E.R. The Toll-like receptor 4 agonist monophosphoryl lipid a augments innate host resistance to systemic bacterial infection. Infect. Immun. 2011, 79, 3576–3587. [Google Scholar] [CrossRef] [PubMed]
- Monnet, E.; Lapeyre, G.; van Poelgeest, E.; Jacqmin, P.; de Graaf, K.; Reijers, J.; Moerland, M.; Burggraaf, J.; de Min, C. Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin. Pharmacol. Ther. 2017, 101, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Feuillet, V.; Medjane, S.; Mondor, I.; Demaria, O.; Pagni, P.P.; Galán, J.E.; Flavell, R.A.; Alexopoulou, L. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 12487–12492. [Google Scholar] [CrossRef] [PubMed]
- Parkunan, S.M.; Astley, R.; Callegan, M.C. Role of TLR5 and flagella in Bacillus intraocular infection. PLoS ONE 2014, 9, e100543. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T.S. How flagellin and toll-like receptor 5 contribute to enteric infection. Infect. Immun. 2007, 75, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Mett, V.; Komarova, E.; Greene, K.; Bespalov, I.; Brackett, C.; Gillard, B.; Gleiberman, A.; Toshkov, I.; Aygün-Sunar, S.; Johnson, C.; et al. Mobilan: A recombinant adenovirus carrying Toll-like receptor 5 self-activating cassette for cancer immunotherapy. Oncogene 2018, 37, 439. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.N.; Treanor, J.J.; Strout, C.; Johnson, C.; Fitzgerald, T.; Kavita, U.; Ozer, K.; Tussey, L.; Shaw, A. Induction of a potent immune response in the elderly using the TLR-5 agonist, flagellin, with a recombinant hemagglutinin influenza–flagellin fusion vaccine (VAX125, STF2. HA1 SI). Vaccine 2011, 29, 4897–4902. [Google Scholar] [CrossRef]
- Yang, H.; Brackett, C.M.; Morales-Tirado, V.M.; Li, Z.; Zhang, Q.; Wilson, M.W.; Benjamin, C.; Harris, W.; Waller, E.K.; Gudkov, A.V.; et al. The Toll-like receptor 5 agonist entolimod suppresses hepatic metastases in a murine model of ocular melanoma via an NK cell-dependent mechanism. Oncotarget 2016, 7, 2936. [Google Scholar] [CrossRef]
- Toshkov, I.A.; Gleiberman, A.S.; Mett, V.L.; Hutson, A.D.; Singh, A.K.; Gudkov, A.V.; Burdelya, L.G. Mitigation of radiation-induced epithelial damage by the TLR5 agonist entolimod in a mouse model of fractionated head and neck irradiation. Radiat. Res. 2017, 187, 570–580. [Google Scholar] [CrossRef]
- Mizel, S.B.; Bates, J.T. Flagellin as an adjuvant: Cellular mechanisms and potential. J. Immunol. 2010, 185, 5677–5682. [Google Scholar] [CrossRef]
- Blohmke, C.J.; Victor, R.E.; Hirschfeld, A.F.; Elias, I.M.; Hancock, D.G.; Lane, C.R.; Davidson, A.G.F.; Wilcox, P.G.; Smith, K.D.; Overhage, J.; et al. Innate immunity mediated by TLR5 as a novel antiinflammatory target for cystic fibrosis lung disease. J. Immunol. 2008, 180, 7764–7773. [Google Scholar] [CrossRef] [PubMed]
- Song, W.S.; Jeon, Y.J.; Namgung, B.; Hong, M.; Yoon, S.I. A conserved TLR5 binding and activation hot spot on flagellin. Sci. Rep. 2017, 7, 40878. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Abel, K.; Lantz, K.; Krieg, A.M.; McChesney, M.B.; Miller, C.J. The Toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J. Virol. 2005, 79, 14355–14370. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.; Tsitoura, D.; Quint, D.; Powley, W.; Lee, L. Safety and pharmacodynamics of intranasal GSK 2245035, a TLR 7 agonist for allergic rhinitis: A randomized trial. Clin. Exp. Allergy 2017, 47, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.L.; Ponsaerts, P.; Berneman, Z.N.; Van Tendeloo, V.F. The use of TLR7 and TLR8 ligands for the enhancement of cancer immunotherapy. Oncologist 2008, 13, 859–875. [Google Scholar] [CrossRef]
- Gorden, K.B.; Gorski, K.S.; Gibson, S.J.; Kedl, R.M.; Kieper, W.C.; Qiu, X.; Tomai, M.A.; Alkan, S.S.; Vasilakos, J.P. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J. Immunol. 2005, 174, 1259–1268. [Google Scholar] [CrossRef]
- Janssen, H.L.; Brunetto, M.R.; Kim, Y.J.; Ferrari, C.; Massetto, B.; Nguyen, A.H.; Joshi, A.; Woo, J.; Lau, A.H.; Gaggar, A.; et al. Safety, efficacy and pharmacodynamics of vesatolimod (GS-9620) in virally suppressed patients with chronic hepatitis B. J. Hepatol. 2018, 68, 431–440. [Google Scholar] [CrossRef]
- Lu, H.; Dietsch, G.N.; Matthews, M.A.H.; Yang, Y.; Ghanekar, S.; Inokuma, M.; Suni, M.; Maino, V.C.; Henderson, K.E.; Howbert, J.J.; et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin. Cancer Res. 2012, 18, 499–509. [Google Scholar] [CrossRef]
- Tanji, H.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Isobe, T.; Miyake, K.; Shimizu, T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 2015, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural analysis reveals that Toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Akira, S. TLR7 structure: Cut in Z-loop. Immunity 2016, 45, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Tanji, H.; Ohto, U.; Motoi, Y.; Shibata, T.; Miyake, K.; Shimizu, T. Autoinhibition and relief mechanism by the proteolytic processing of Toll-like receptor 8. Proc. Natl. Acad. Sci. USA 2016, 113, 3012–3017. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Ortiz, Z.G.; Specht, C.A.; Wang, J.P.; Lee, C.K.; Bartholomeu, D.C.; Gazzinelli, R.T.; Levitz, S.M. Toll-like receptor 9-dependent immune activation by unmethylated CpG motifs in Aspergillus fumigatus DNA. Infect. Immun. 2008, 76, 2123–2129. [Google Scholar] [CrossRef] [PubMed]
- Ashkar, A.A.; Rosenthal, K.L. Toll-like receptor 9, CpG DNA and innate immunity. Curr. Mol. Med. 2002, 2, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Candia, A.F.; Delaney, S.; Floettmann, S.; Wong, C.; Campbell, J.D.; Kell, S.; Lum, J.; Hessel, E.M.; Traquina, P. First-in-human study with the inhaled TLR9 oligonucleotide agonist AZD1419 results in interferon responses in the lung, and is safe and well-tolerated. Clin. Pharmacol. Ther. 2018, 104, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Casale, T.; Cole, J.; Beck, E.; Vogelmeier, C.; Willers, J.; Lassen, C.; Hammann-Haenni, A.; Trokan, L.; Saudan, P.; Wechsler, M. CYT 003, a TLR 9 agonist, in persistent allergic asthma–a randomized placebo-controlled Phase 2b study. Allergy 2015, 70, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Ruzsa, A.; Sen, M.; Evans, M.; Lee, L.W.; Hideghety, K.; Rottey, S.; Klimak, P.; Holeckova, P.; Fayette, J.; Csoszi, T.; et al. Phase 2, open-label, 1: 1 randomized controlled trial exploring the efficacy of EMD 1201081 in combination with cetuximab in second-line cetuximab-naive patients with recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN). Investig. New Drugs 2014, 32, 1278–1284. [Google Scholar] [CrossRef]
- Muthusamy, N.; Breidenbach, H.; Andritsos, L.; Flynn, J.; Jones, J.; Ramanunni, A.; Mo, X.; Jarjoura, D.; Byrd, J.C.; Heerema, N.A. Enhanced detection of chromosomal abnormalities in chronic lymphocytic leukemia by conventional cytogenetics using CpG oligonucleotide in combination with pokeweed mitogen and phorbol myristate acetate. Cancer Genet. 2011, 204, 77–83. [Google Scholar] [CrossRef]
- Ohto, U.; Shibata, T.; Tanji, H.; Ishida, H.; Krayukhina, E.; Uchiyama, S.; Miyake, K.; Shimizu, T. Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature 2015, 520, 702. [Google Scholar] [CrossRef] [PubMed]
- Yarovinsky, F. Innate immunity to Toxoplasma gondii infection. Nat. Rev. Immunol. 2014, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.-H.; Ulevitch, R.J. Identification of hTLR10: A novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta BBA Gene Struct. Expr. 2001, 1518, 157–161. [Google Scholar] [CrossRef]
- Oosting, M.; Cheng, S.C.; Bolscher, J.M.; Vestering-Stenger, R.; Plantinga, T.S.; Verschueren, I.C.; Arts, P.; Garritsen, A.; van Eenennaam, H.; Sturm, P.; et al. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc. Natl. Acad. Sci. USA 2014, 111, E4478–E4484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, G.; Hayden, M.S.; Greenblatt, M.B.; Bussey, C.; Flavell, R.A.; Ghosh, S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science 2004, 303, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.B.; Zhao, H.; Xia, J.; Wu, D.; Nervi, S.; Stinson, S.F.; Majerova, E.; Bramhall, C.; Zaharevitz, D.W. Poly (ethylene glycol) prodrugs of the CDK inhibitor, alsterpaullone (NSC 705701): Synthesis and pharmacokinetic studies. Bioconjug. Chem. 2004, 15, 1076–1083. [Google Scholar] [CrossRef]
- Duncan, R.; Gac-Breton, S.; Keane, R.; Musila, R.; Sat, Y.; Satchi, R.; Searle, F. Polymer–drug conjugates, PDEPT and PELT: Basic principles for design and transfer from the laboratory to clinic. J. Control. Release 2001, 74, 135–146. [Google Scholar] [CrossRef]
- Kopeček, J.; Kopečková, P.; Minko, T.; Lu, Z.R.; Peterson, C. Water soluble polymers in tumor targeted delivery. J. Control. Release 2001, 74, 147–158. [Google Scholar] [CrossRef]
- Avgoustakis, K.; Beletsi, A.; Panagi, Z.; Klepetsanis, P.; Karydas, A.; Ithakissios, D. PLGA–mPEG nanoparticles of cisplatin: In vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control. Release 2002, 79, 123–135. [Google Scholar] [CrossRef]
- Huang, G.; Gao, J.; Hu, Z.; John, J.V.S.; Ponder, B.C.; Moro, D. Controlled drug release from hydrogel nanoparticle networks. J. Control. Release 2004, 94, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Gombotz, W.R.; Pettit, D.K. Biodegradable polymers for protein and peptide drug delivery. Bioconjug. Chem. 1995, 6, 332–351. [Google Scholar] [CrossRef] [PubMed]
- Brannon-Peppas, L. Polymers in controlled drug delivery. Med. Plast. Biomater. Mag. 1997, 4, 34–44. [Google Scholar]
- Sharma, S.; Singh, S.; Bhardwaj, S.; Gaurave, K.; Gupta, G. Osmotic controlled drug delivery system. Latest Rev. 2008, 6, 3–7. [Google Scholar]
- Chasin, M. Biodegradable Polymers as Drug Delivery Systems, 1st ed.; Informa HealthCare; Marcel Dekker Inc.: New York, NY, USA, 1990; Volume 45. [Google Scholar]
- Vadlapudi, A.; CholKAr, K.; Dasari, S.; Mitra, A. Ocular drug delivery. Drug Deliv. Jones Bartlett Learn. Burlingt. Ma USA 2015, 1, 219–263. [Google Scholar]
- Urbina, M.C.; Zinoveva, S.; Miller, T.; Sabliov, C.M.; Monroe, W.T.; Kumar, C.S. Investigation of magnetic nanoparticle—Polymer composites for multiple-controlled drug delivery. J. Phys. Chem. 2008, 112, 11102–11108. [Google Scholar] [CrossRef]
- Galvin, P.; Thompson, D.; Ryan, K.B.; McCarthy, A.; Moore, A.C.; Burke, C.S.; Dyson, M.; MacCraith, B.D.; Gun’ko, Y.K.; Byrne, M.T.; et al. Nanoparticle-based drug delivery: Case studies for cancer and cardiovascular applications. Cell. Mol. Life Sci. 2012, 69, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Noyhouzer, T.; L’Homme, C.; Beaulieu, I.; Mazurkiewicz, S.; Kuss, S.; Kraatz, H.B.; Canesi, S.; Mauzeroll, J. Ferrocene-modified phospholipid: An innovative precursor for redox-triggered drug delivery vesicles selective to cancer cells. Langmuir 2016, 32, 4169–4178. [Google Scholar] [CrossRef]
- van Vlerken, L.E.; Vyas, T.K.; Amiji, M.M. Poly (ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm. Res. 2007, 24, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297. [Google Scholar]
- Ahsan, F.; Rivas, I.P.; Khan, M.A.; Suárez, A.I.T. Targeting to macrophages: Role of physicochemical properties of particulate carriers—liposomes and microspheres—on the phagocytosis by macrophages. J. Control. Release 2002, 79, 29–40. [Google Scholar] [CrossRef]
- Chono, S.; Tanino, T.; Seki, T.; Morimoto, K. Influence of particle size on drug delivery to rat alveolar macrophages following pulmonary administration of ciprofloxacin incorporated into liposomes. J. Drug Target. 2006, 14, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Chono, S.; Tauchi, Y.; Morimoto, K. Influence of particle size on the distributions of liposomes to atherosclerotic lesions in mice. Drug Dev. Ind. Pharm. 2006, 32, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Aramaki, Y.; Tsuchiya, S. Physicochemical properties of liposomes affecting apoptosis induced by cationic liposomes in macrophages. Pharm. Res. 2003, 20, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Iwaoka, S.; Nakamura, T.; Takano, S.; Tsuchiya, S.; Aramaki, Y. Cationic liposomes induce apoptosis through p38 MAP kinase–caspase-8–Bid pathway in macrophage-like RAW264. 7 cells. J. Leukoc. Biol. 2006, 79, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, Y.; Takano, S.; Tsuchiya, S. Cationic liposomes induce macrophage apoptosis through mitochondrial pathway. Arch. Biochem. Biophys. 2001, 392, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Arisaka, M.; Nakamura, T.; Yamada, A.; Negishi, Y.; Aramaki, Y. Involvement of protein kinase Cδ in induction of apoptosis by cationic liposomes in macrophage-like RAW264. 7 cells. FEBS Lett. 2010, 584, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.; Raz, A.; Fogler, W.; Kirsh, R.; Bugelski, P.; Poste, G. Design of liposomes to improve delivery of macrophage-augmenting agents to alveolar macrophages. Cancer Res. 1980, 40, 4460–4466. [Google Scholar] [PubMed]
- Sun, P.; Zhong, M.; Shi, X.; Li, Z. Anionic LPD complexes for gene delivery to macrophage: Preparation, characterization and transfection in vitro. J. Drug Target. 2008, 16, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Andreakos, E.; Rauchhaus, U.; Stavropoulos, A.; Endert, G.; Wendisch, V.; Benahmed, A.S.; Giaglis, S.; Karras, J.; Lee, S.; Gaus, H.; et al. Amphoteric liposomes enable systemic antigen-presenting cell–directed delivery of CD40 antisense and are therapeutically effective in experimental arthritis. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2009, 60, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Alam, R.; Dixit, V.; Kang, H.M. Cell-targeting and cell-penetrating peptides for delivery of therapeutic and imaging agents. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Karathanasis, E.; Geigerman, C.M.; Parkos, C.A.; Chan, L.; Bellamkonda, R.V.; Jaye, D.L. Selective targeting of nanocarriers to neutrophils and monocytes. Ann. Biomed. Eng. 2009, 37, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, L.; Burritt, J.B.; Jesaitis, A.J.; Nusrat, A.; Liang, T.W.; Gewirtz, A.T.; Schnell, F.J.; Parkos, C.A. Cell-specific peptide binding by human neutrophils. Blood 1999, 93, 1738–1748. [Google Scholar] [PubMed]
- Qin, J.; Chen, D.; Hu, H.; Qiao, M.; Zhao, X.; Chen, B. Body distributioin of RGD-mediated liposome in brain-targeting drug delivery. Yakugaku Zasshi 2007, 127, 1497–1501. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qin, J.; Chen, D.; Hu, H.; Cui, Q.; Qiao, M.; Chen, B. Surface modification of RGD-liposomes for selective drug delivery to monocytes/neutrophils in brain. Chem. Pharm. Bull. 2007, 55, 1192–1197. [Google Scholar] [CrossRef]
- Yan, X.; Scherphof, G.L.; Kamps, J.A. Liposome opsonization. J. Liposome Res. 2005, 15, 109–139. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C. Recognition by macrophages and liver cells of opsonized phospholipid vesicles and phospholipid headgroups. Pharm. Res. 2001, 18, 1–8. [Google Scholar] [CrossRef]
- Wijagkanalan, W.; Higuchi, Y.; Kawakami, S.; Teshima, M.; Sasaki, H.; Hashida, M. Enhanced anti-inflammation of inhaled dexamethasone palmitate using mannosylated liposomes in an endotoxin-induced lung inflammation model. Mol. Pharmacol. 2008, 74, 1183–1192. [Google Scholar] [CrossRef]
- Kuramoto, Y.; Kawakami, S.; Zhou, S.; Fukuda, K.; Yamashita, F.; Hashida, M. Use of mannosylated cationic liposomes/immunostimulatory CpG DNA complex for effective inhibition of peritoneal dissemination in mice. J. Gene Med. A Cross Discip. J. Res. Sci. Gene Transf. Its Clin. Appl. 2008, 10, 392–399. [Google Scholar] [CrossRef]
- Steinhagen, F.; Kinjo, T.; Bode, C.; Klinman, D.M. TLR-based immune adjuvants. Vaccine 2011, 29, 3341–3355. [Google Scholar] [CrossRef]
- Kaczanowska, S.; Joseph, A.M.; Davila, E. TLR agonists: Our best frenemy in cancer immunotherapy. J. Leukoc. Biol. 2013, 93, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Mandraju, R.; Murray, S.; Forman, J.; Pasare, C. Differential ability of surface and endosomal TLRs to induce CD8 T cell responses in vivo. J. Immunol. 2014, 192, 4303–4315. [Google Scholar] [CrossRef]
- Oh, J.Z.; Kurche, J.S.; Burchill, M.A.; Kedl, R.M. TLR7 enables cross-presentation by multiple dendritic cell subsets through a type I IFN-dependent pathway. Blood 2011, 118, 3028–3038. [Google Scholar] [CrossRef] [PubMed]
- De Titta, A.; Ballester, M.; Julier, Z.; Nembrini, C.; Jeanbart, L.; Van Der Vlies, A.J.; Swartz, M.A.; Hubbell, J.A. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proc. Natl. Acad. Sci. USA 2013, 110, 19902–19907. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.T.; Keller, S.; Manganiello, M.J.; Cheng, C.; Lee, C.C.; Opara, C.; Convertine, A.; Stayton, P.S. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano 2013, 7, 3912–3925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chiu, Y.C.; Tostanoski, L.H.; Jewell, C.M. Polyelectrolyte multilayers assembled entirely from immune signals on gold nanoparticle templates promote antigen-specific T cell response. ACS Nano 2015, 9, 6465–6477. [Google Scholar] [CrossRef] [PubMed]
- Napolitani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1–polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769. [Google Scholar] [CrossRef]
- Pradhan, P.; Qin, H.; Leleux, J.A.; Gwak, D.; Sakamaki, I.; Kwak, L.W.; Roy, K. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials 2014, 35, 5491–5504. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-delivered transforming growth factor-β siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef]
- Peppas, N.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Gupta, P.; Vermani, K.; Garg, S. Hydrogels: From controlled release to pH-responsive drug delivery. Drug Discov. Today 2002, 7, 569–579. [Google Scholar] [CrossRef]
- Nayak, S.; Lyon, L.A. Soft nanotechnology with soft nanoparticles. Angew. Chem. Int. Ed. 2005, 44, 7686–7708. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.K.; Drumright, R.; Siegwart, D.J.; Matyjaszewski, K. The development of microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2008, 33, 448–477. [Google Scholar] [CrossRef]
- Kageyama, S.; Kitano, S.; Hirayama, M.; Nagata, Y.; Imai, H.; Shiraishi, T.; Akiyoshi, K.; Scott, A.M.; Murphy, R.; Hoffman, E.W.; et al. Humoral immune responses in patients vaccinated with 1–146 HER2 protein complexed with cholesteryl pullulan nanogel. Cancer Sci. 2008, 99, 601–607. [Google Scholar] [CrossRef]
- Kitano, S.; Kageyama, S.; Nagata, Y.; Miyahara, Y.; Hiasa, A.; Naota, H.; Okumura, S.; Imai, H.; Shiraishi, T.; Masuya, M.; et al. HER2-specific T-cell immune responses in patients vaccinated with truncated HER2 protein complexed with nanogels of cholesteryl pullulan. Clin. Cancer Res. 2006, 12, 7397–7405. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Khondee, S.; Linz, T.H.; Berkland, C. Poly (N-vinylformamide) nanogels capable of pH-sensitive protein release. Macromolecules 2008, 41, 6546–6554. [Google Scholar] [CrossRef]
- Goh, S.L.; Murthy, N.; Xu, M.; Fréchet, J.M. Cross-linked microparticles as carriers for the delivery of plasmid DNA for vaccine development. Bioconjug. Chem. 2004, 15, 467–474. [Google Scholar] [CrossRef]
- Standley, S.M.; Mende, I.; Goh, S.L.; Kwon, Y.J.; Beaudette, T.T.; Engleman, E.G.; Fréchet, J.M. Incorporation of CpG oligonucleotide ligand into protein-loaded particle vaccines promotes antigen-specific CD8 T-cell immunity. Bioconjug. Chem. 2007, 18, 77–83. [Google Scholar] [CrossRef]
- Scott, R.C.; Crabbe, D.; Krynska, B.; Ansari, R.; Kiani, M.F. Aiming for the heart: Targeted delivery of drugs to diseased cardiac tissue. Expert Opin. Drug Deliv. 2008, 5, 459–470. [Google Scholar] [CrossRef]
- Trafton, A. Tumors targeted using tiny gold particles. Mit Tech Talk 2009, 53, 4. [Google Scholar]
- Gujral, S.; Khatri, S. A review on basic concept of drug targeting and drug carrier system. Int. J. Adv. Pharm. Biol. Chem. 2013, 2, 130–136. [Google Scholar]
- Agnihotri, J.; Saraf, S.; Khale, A. Targeting: New potential carriers for targeted drug delivery system. Int. J. Pharm. Sci. Rev. Res. 2011, 8, 117–123. [Google Scholar]
- Bawarski, W.E.; Chidlowsky, E.; Bharali, D.J.; Mousa, S.A. Emerging nanopharmaceuticals. Nanomed. Nanotechnol. Biol. Med. 2008, 4, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Jing, N.; Jin, Y. Preparation and in vitro evaluation of liposomal chloroquine diphosphate loaded by a transmembrane pH-gradient method. Int. J. Pharm. 2008, 361, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.M.; He, K.; Dagher, R.; Sridhara, R.; Farrell, A.; Justice, R.; Pazdur, R. Liposomal Doxorubicin in Combination with Bortezomib for Relapsed or Refractory Multiple Myeloma: Page 3 of 3. Oncology 2007, 21. [Google Scholar]
- Touitou, E.; Levi-Schaffer, F.; Dayan, N.; Alhaique, F.; Riccieri, F. Modulation of caffeine skin delivery by carrier design: Liposomes versus permeation enhancers. Int. J. Pharm. 1994, 103, 131–136. [Google Scholar] [CrossRef]
- Yarosh, D.B. Liposomes in investigative dermatology. Photodermatol. Photoimmunol. Photomed. Rev. Artic. 2001, 17, 203–212. [Google Scholar] [CrossRef]
- Klein, R. The detection of oxidation in liposome preparations. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1970, 210, 486–489. [Google Scholar] [CrossRef]
- Wong, M.; Thompson, T.E. Aggregation of dipalmitoylphosphatidylcholine vesicles. Biochemistry 1982, 21, 4133–4139. [Google Scholar] [CrossRef]
- Payne, N.I.; Ambrose, C.V.; Timmins, P.; Ward, M.D.; Ridgway, F. Proliposomes: A novel solution to an old problem. J. Pharm. Sci. 1986, 75, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Deo, M.; Sant, V.; Parekh, S.; Khopade, A.; Banakar, U. Proliposome-based transdermal delivery of levonorgestrel. J. Biomater. Appl. 1997, 12, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Katare, O.; Vyas, S.; Dixit, V. Proliposomes of indomethacin for oral administration. J. Microencapsul. 1991, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.N.; Kim, S.K.; Shim, C.K. Proliposomes as an intranasal dosage form for the sustained delivery of propranolol. J. Control. Release 1995, 34, 203–210. [Google Scholar] [CrossRef]
- Junping, W.; Maitani, Y.; Takayama, K.; Nagai, T. In vivo evaluation of doxorubicin carried with long circulating and remote loading proliposome. Int. J. Pharm. 2000, 203, 61–69. [Google Scholar] [CrossRef]
- Wei, N.; Lu, B. Preparation, morphology and in vitro release of chitosan coated liposomes of fluorouracil for colon targeting. Acta Pharm. Sin. 2003, 38, 53–56. [Google Scholar]
- Ye, Z.; Hu, Q.; Liang, W. Preparation of interferon-alpha-containing liposomes by the powder bed grinding method. Zhejiang Da Xue Xue Bao. Yi Xue Ban J. Zhejiang Univ. Med. Sci. 2002, 31, 433–436. [Google Scholar]
- Yang, Z.; Hino, T.; Kawashima, Y. Studies on the size of rehydrated new liposome from scutellaria proliposome. J. China Pharm. Univ. 1993, 24, 161. [Google Scholar]
- Chen, Q.; Huang, Y.; Gu, X.; Chen, C.; Huang, D. A study on the preparation of proliposomes by spray drying method. J. Shenyang Pharm. Univ. 1997, 14, 166–169. [Google Scholar]
- Zhang, J.; Zhu, J. A novel method to prepare liposomes containing amikacin. J. Microencapsul. 1999, 16, 511–516. [Google Scholar] [PubMed]
- Pardakhty, A.; Moazeni, E. Nano-niosomes in drug, vaccine and gene delivery: A rapid overview. Nanomed. J. 2013, 1, 1–12. [Google Scholar]
- Moghassemi, S.; Hadjizadeh, A. Nano-niosomes as nanoscale drug delivery systems: An illustrated review. J. Control. Release 2014, 185, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.P.; Rajeshwarrao, P. Nonionic surfactant vesicular systems for effective drug delivery—an overview. Acta Pharm. Sin. B 2011, 1, 208–219. [Google Scholar] [CrossRef]
- Uchegbu, I.F.; Vyas, S.P. Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int. J. Pharm. 1998, 172, 33–70. [Google Scholar] [CrossRef]
- Kaur, I.P.; Garg, A.; Singla, A.K.; Aggarwal, D. Vesicular systems in ocular drug delivery: An overview. Int. J. Pharm. 2004, 269, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shilpa, S.; Srinivasan, B.; Chauhan, M. Niosomes as vesicular carriers for delivery of proteins and biologicals. Int. J. Drug Deliv. 2011, 3, 14–24. [Google Scholar] [CrossRef]
- Verma, S.; Singh, S.; Syan, N.; Mathur, P.; Valecha, V. Nanoparticle vesicular systems: A versatile tool for drug delivery. J Chem. Pharm. Res. 2010, 2, 496–509. [Google Scholar]
- Abdelkader, H.; Ismail, S.; Kamal, A.; Alany, R.G. Design and evaluation of controlled-release niosomes and discomes for naltrexone hydrochloride ocular delivery. J. Pharm. Sci. 2011, 100, 1833–1846. [Google Scholar] [CrossRef]
- Alam, M.; Zubair, S.; Farazuddin, M.; Ahmad, E.; Khan, A.; Zia, Q.; Malik, A.; Mohammad, O. Development, characterization and efficacy of niosomal diallyl disulfide in treatment of disseminated murine candidiasis. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 247–256. [Google Scholar] [CrossRef]
- Mozafari, M.; Reed, C.; Rostron, C.; Hasirci, V. A review of scanning probe microscopy investigations of liposome-DNA complexes. J. Liposome Res. 2005, 15, 93–107. [Google Scholar] [CrossRef]
- Mokhtar, M.; Sammour, O.A.; Hammad, M.A.; Megrab, N.A. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int. J. Pharm. 2008, 361, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Biswal, S.; Murthy, P.; Sahu, J.; Sahoo, P.; Amir, F. Vesicles of non-ionic surfactants (niosomes) and drug delivery potential. Int. J. Pharm. Sci. Nanotechnol. 2008, 1, 1–8. [Google Scholar]
- Waddad, A.Y.; Abbad, S.; Yu, F.; Munyendo, W.L.; Wang, J.; Lv, H.; Zhou, J. Formulation, characterization and pharmacokinetics of Morin hydrate niosomes prepared from various non-ionic surfactants. Int. J. Pharm. 2013, 456, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, G.; Zhang, X.; Li, B.; Chen, Y.; Lu, T.; Lu, T.J.; Xu, F. Magnetic hydrogels and their potential biomedical applications. Adv. Funct. Mater. 2013, 23, 660–672. [Google Scholar] [CrossRef]
- Tabata, Y. Biomaterial technology for tissue engineering applications. J. R. Soc. Interface 2009, 6, S311–S324. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, Z.; Gupta, D. Biopolymer Drived Hydrogels and Their Diverse Applications: A Review. In Modified Biopolymers: Challenges and Opportunities, 1st ed.; Nova Science Publishers: New York, NY, USA, 2017; pp. 83–103. [Google Scholar]
- Rao, K.R.; Devi, K.P. Swelling controlled-release systems: Recent developments and applications. Int. J. Pharm. 1988, 48, 1–13. [Google Scholar]
- Liang-Chang, D.; Qi, Y.; Hoffman, A.S. Controlled release of amylase from a thermal and pH-sensitive, macroporous hydrogel. J. Control. Release 1992, 19, 171–177. [Google Scholar] [CrossRef]
- van der Linden, H.; Olthuis, W.; Bergveld, P. An efficient method for the fabrication of temperature-sensitive hydrogel microactuators. Lab. A Chip. 2004, 4, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Kiptoo, P.K.; Hamad, M.O.; Crooks, P.A.; Stinchcomb, A.L. Enhancement of transdermal delivery of 6-β-naltrexol via a codrug linked to hydroxybupropion. J. Control. Release 2006, 113, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Leppänen, J.; Huuskonen, J.; Nevalainen, T.; Gynther, J.; Taipale, H.; Järvinen, T. Design and Synthesis of a Novel L-Dopa—Entacapone Codrug. J. Med. Chem. 2002, 45, 1379–1382. [Google Scholar] [CrossRef] [PubMed]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Langer, R. Biomaterials in drug delivery and tissue engineering: One laboratory’s experience. Acc. Chem. Res. 2000, 33, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Pani, K.; Gladieux, G.; Brandes, G.; Kulkarni, R.; Leonard, F. The degradation of n-butyl alpha-cyanoacrylate tissue adhesive. II. Surgery 1968, 63, 481. [Google Scholar] [PubMed]
- Soppimath, K.S.; Aminabhavi, T.M.; Kulkarni, A.R.; Rudzinski, W.E. Biodegradable polymeric nanoparticles as drug delivery devices. J. Control. Release 2001, 70, 1–20. [Google Scholar] [CrossRef]
- Mishima, K.; Matsuyama, K.; Tanabe, D.; Yamauchi, S.; Young, T.J.; Johnston, K.P. Microencapsulation of proteins by rapid expansion of supercritical solution with a nonsolvent. Aiche J. 2000, 46, 857–865. [Google Scholar] [CrossRef]
- Lombardo, D.; Kiselev, M.A.; Caccamo, M.T. Smart Nanoparticles for Drug Delivery Application: Development of Versatile Nanocarrier Platforms in Biotechnology and Nanomedicine. J. Nanomater. 2019, 2019, 3702518. [Google Scholar] [CrossRef]
- Crommelin, D.; Schreier, H. Colloidal Drug Delivery Systems; Kreuter, J., Ed.; Marcel Dekker Inc.: New York, NY, USA; pp. 73–190.
- Tomalia, D.A.; Naylor, A.M.; Goddard III, W.A. Starburst dendrimers: Molecular-level control of size, shape, surface chemistry, topology, and flexibility from atoms to macroscopic matter. Angew. Chem. Int. Ed. Engl. 1990, 29, 138–175. [Google Scholar] [CrossRef]
- Ballauff, M.; Likos, C.N. Dendrimers in solution: Insight from theory and simulation. Angew. Chem. Int. Ed. 2004, 43, 2998–3020. [Google Scholar] [CrossRef]
- Kesharwani, P.; Jain, K.; Jain, N.K. Dendrimer as nanocarrier for drug delivery. Prog. Polym. Sci. 2014, 39, 268–307. [Google Scholar] [CrossRef]
- Lombardo, D. Liquid-like ordering of negatively charged poly (amidoamine) (PAMAM) dendrimers in solution. Langmuir 2009, 25, 3271–3275. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gray, W.D.; Davis, M.E.; Luo, Y. Peptide-and saccharide-conjugated dendrimers for targeted drug delivery: A concise review. Interface Focus 2012, 2, 307–324. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Vogensen, S.B.; Brewster, M.E.; Konráðsdóttir, F. Effects of cyclodextrins on drug delivery through biological membranes. J. Pharm. Sci. 2007, 96, 2532–2546. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Receptor | Microbial Ligand(s) | Microbial Ligand Source | Endogenous Ligand(s) | Adaptor | TLR Localization | Expression |
|---|---|---|---|---|---|---|
| TLR1/2/6 | Triacyl and diacyl lipopeptides, glycolipids, zymosan, lipoteichoic acid | Bacterial lipoprotein and peptidoglycan, fungi, mycoplasma, gram-positive bacteria | HMGB1, HSPs, HDLs, hyaluronan, | MyD88-MAL | Cell surface | DCs, Mon, Mac, B |
| TLR3 | Double-stranded RNA (dsRNA) | Viruses | Self dsRNA | TRIF-TRAM | Endosomes | DCs, L, Plt, B |
| TLR4 | Lipopolysaccharide | Gram-negative bacteria | HSPs, fibrinogen, HA, heparin sulfate, HMGB1, LDLs | MyD88-MAL/TRIF-TRAM | Cell surface | Mon, Mac, N, DCs, M, B, C |
| TLR5 | Flagellin, profilin | Bacteria, Toxoplasma gondii | HMGB1 | MyD88-MAL | Cell surface | DCs, Mon, Mac, C, IE |
| TLR7 | Single-stranded RNA (ssRNA) | Viruses | Self ssRNA | MyD88-MAL | Endosomes | DCs, Mon, Mc, Plt, B |
| TLR8 | ssRNA | Viruses | Self ssRNA | MyD88-MAL | Endosomes | DCs, Mon, Mac, M, IECs |
| TLR9 | DNA | Bacteria, viruses | Self DNA | MyD88-MAL | Endosomes | DCs, Mon, Mac, Plt, B |
| TLR10 | Triacylated lipopeptides | NA | NA | MyD88-MAL | Cell surface | Mon, N, B, LN, S |
| TLR | Ligand | Type | Disease | Mechanism | Phase |
|---|---|---|---|---|---|
| TLR2 (with TLR1 or -6) | OPN-305 and derivatives | Monoclonal antibody | Inflammatory disease, myelodysplastic syndrome, kidney transplant rejection, pancreatic tumor | Anti-inflammatory | Phase II |
| CBLB6I2 | Synthetic lipopeptide | Cancers (breast) | Blood cell recovery | Phase II | |
| ISA-20I | Peptide | Head and neck tumor | Maturation of DCs | Phase II | |
| TLR3 | Poly-ICLC | Synthetic dsRNA | Various cancers (e.g., colon, ovarian, breast, prostate) | Immune stimulation and modulation of tumor microenvironment | Phase I and II |
| PRV-300 | Antibody | Asthma | Anti-inflammatory | Phase I and II | |
| TLR4 | NI-0I0I | Antibody | Rheumatoid arthritis | Anti-inflammatory | Phase II |
| GLA and derivatives | Glycolipid | Melanoma, sarcoma, viral infection | Immune stimulator | Phase I and II | |
| LPS | Glycolipid | Asthma | Inflammation | Phase I | |
| GSKI79509I | Glycolipid | Cancer | Immune stimulator | Phase I | |
| Eritoran | Glycolipid | Insulin sensitivity | Anti-inflammatory | Phase II | |
| CX-0I | Polysaccharide | Leukemia | Microenvironment modulator | Phase I | |
| PEPA-I0 | Small molecule | Cancer | Immune stimulator | Phase II | |
| PET-lipid A | Glycolipid | Cancers | Immune stimulator | Phase I | |
| JKB and derivatives | Small molecule | Hepatitis | Anti-inflammatory | Phase II | |
| MN-I66 | Small molecule | Brain injury, glioblastoma | Anti-inflammatory | Phase II | |
| TLR5 | Entolimod | Recombinant protein | Cancers | Immune stimulator | Phase I |
| Mobilan | Recombinant protein | Cancers (prostate) | Immune stimulator | Phase I and II | |
| VAX and derivatives | Recombinant protein | Influenza | Immune stimulator | Phase I and II | |
| TLR7 | Imiquimod | Small molecule | Various cancers, actinic keratosis, and viral infections | Immune stimulator | Phase I to Phase IV, approved |
| GSK2245035 | Small molecule | Asthma and rhinitis | IFN production | Phase II | |
| GS-9620 | Small molecule | Hepatitis B | pDC activator | Phase II | |
| RO702053I | Small molecule | Hepatitis B | Immune stimulator | Phase II | |
| GSK2245035 | Small molecule | Asthma | IFN-α production and immune stimulation | Phase II | |
| TLR8 | VTX-2337 | Small molecule | Various cancers | Immune stimulator | Phase I and II |
| TLR9 | SD-I0I | CpG-C class oligonucleotide | Lymphoma | Antitumor immune response | Phase I and II |
| CYT003 | Oligonucleotide | Asthma | TH-I–mediated immune response | Phase II | |
| MGNI703 | DNA-based molecule | HIV and melanoma | Antiviral and antitumor response | Phase I and II | |
| CpG-7909 | Oligonucleotide | Lymphoma, malaria, HIV | Immune stimulator | Phase I and II | |
| CpG-I0I04 | Oligonucleotide | Hookworm infection | Immune stimulator | Phase I | |
| CpG-ODN | Nucleotide-based | Lung tumor | Immune stimulator | Phase I | |
| Hydroxychloroquine | Small molecule | Sjogren’s syndrome | Immune suppressor | Phase III | |
| AZDI4I9 | CpG oligonucleotide | Asthma | IFN production | Phase II |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javaid, N.; Yasmeen, F.; Choi, S. Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods. Pharmaceutics 2019, 11, 441. https://doi.org/10.3390/pharmaceutics11090441
Javaid N, Yasmeen F, Choi S. Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods. Pharmaceutics. 2019; 11(9):441. https://doi.org/10.3390/pharmaceutics11090441
Chicago/Turabian StyleJavaid, Nasir, Farzana Yasmeen, and Sangdun Choi. 2019. "Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods" Pharmaceutics 11, no. 9: 441. https://doi.org/10.3390/pharmaceutics11090441
APA StyleJavaid, N., Yasmeen, F., & Choi, S. (2019). Toll-Like Receptors and Relevant Emerging Therapeutics with Reference to Delivery Methods. Pharmaceutics, 11(9), 441. https://doi.org/10.3390/pharmaceutics11090441