Vibrating Mesh Nebulisation of Pro-Antimicrobial Peptides for Use in Cystic Fibrosis

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis
2.3. HPLC Analysis
2.4. Aerosol Droplet Size Analysis
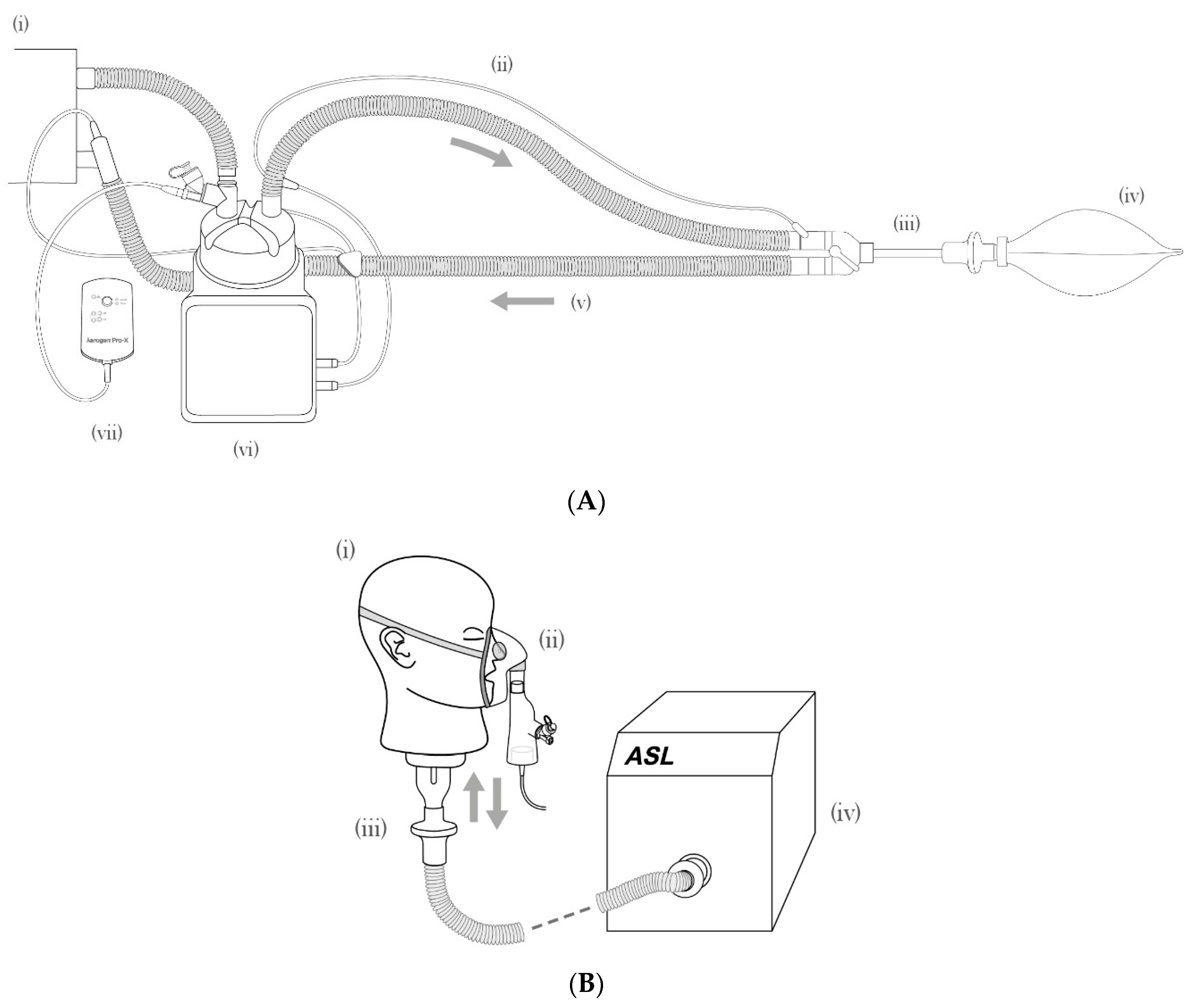
2.5. Breathing Apparatus
2.6. Bacterial Susceptibility Testing
3. Results
3.1. Characterisation of Pro- and AAG-WMR after Nebulisation
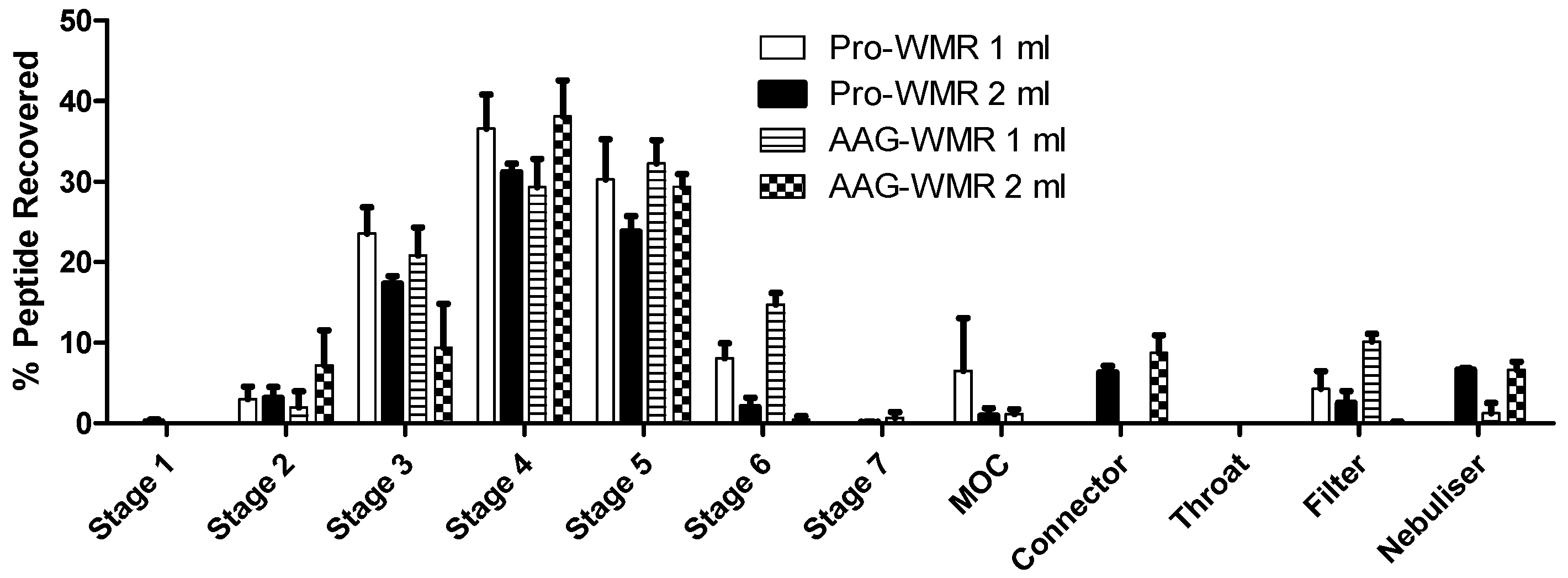
3.2. Aerodynamic Size Distribution of the Peptides
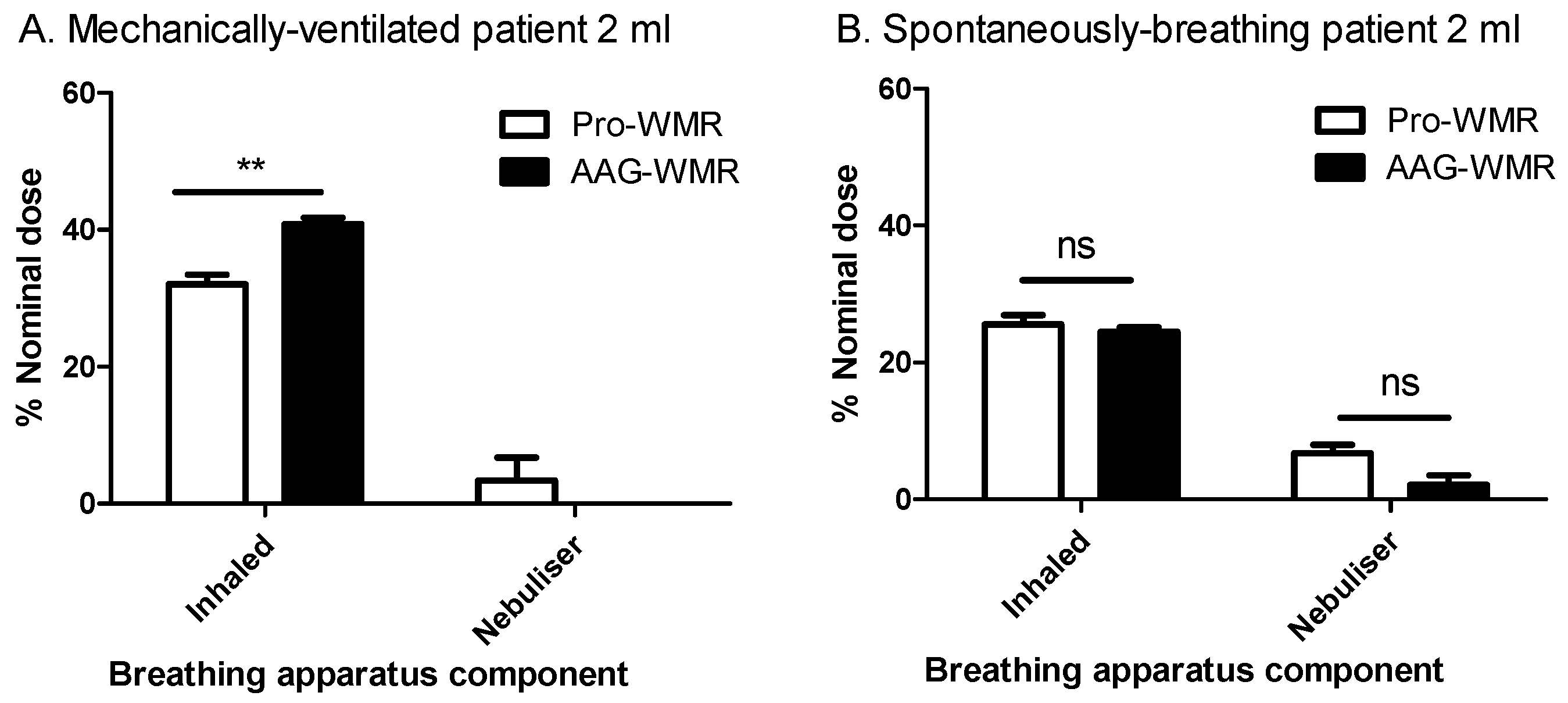
3.3. Aerosol Delivery during Simulated Breathing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Greally, P.; Whitaker, P.; Peckham, D. Challenges with current inhaled treatments for chronic Pseudomonas aeruginosa infection in patients with cystic fibrosis. Curr. Med. Res. Opin. 2012, 28, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Forde, E.; Humphreys, H.; Greene, C.M.; Fitzgerald-Hughes, D.; Devocelle, M. The potential of host defence peptide prodrugs as neutrophil elastase-dependent anti-infective agents for cystic fibrosis. Antimicrob. Agents Chemother. 2013, 58, 978. [Google Scholar] [CrossRef] [PubMed]
- Forde, E.; Schutte, A.; Reeves, E.; Greene, C.; Humphreys, H.; Mall, M.; Fitzgerald-Hughes, D.; Devocelle, M. Differential In Vitro and In Vivo Toxicities of Antimicrobial Peptide Prodrugs for Potential Use in Cystic Fibrosis. Antimicrob. Agents Chemother. 2016, 60, 2813–2821. [Google Scholar] [CrossRef]
- Doring, G.; Flume, P.; Heijerman, H.; Elborn, J.S. Treatment of lung infection in patients with cystic fibrosis: Current and future strategies. J. Cyst. Fibros. 2012, 11, 461–479. [Google Scholar] [CrossRef]
- Ratjen, F.; Brockhaus, F.; Angyalosi, G. Aminoglycoside therapy against Pseudomonas aeruginosa in cystic fibrosis: A review. J. Cyst. Fibros. 2009, 8, 361–369. [Google Scholar] [CrossRef]
- Lewis, A.L.; Richard, J. Challenges in the delivery of peptide drugs: An industry perspective. Ther. Deliv. 2015, 6, 149–163. [Google Scholar] [CrossRef]
- Greene, C.M.; Carroll, T.P.; Smith, S.G.; Taggart, C.C.; Devaney, J.; Griffin, S.; O’Neill, S.J.; McElvaney, N.G. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 2005, 174, 1638–1646. [Google Scholar] [CrossRef]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Exp. Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef]
- Hess, D.R. Aerosol delivery devices in the treatment of asthma. Respir. Care 2008, 53, 699–723; discussion 723–725. [Google Scholar]
- Agu, R.U.; Ugwoke, M.I.; Armand, M.; Kinget, R.; Verbeke, N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir. Res. 2001, 2, 198–209. [Google Scholar]
- Martin, A.R.; Finlay, W.H. Nebulizers for drug delivery to the lungs. Exp. Opin. Drug Deliv. 2014, 12, 889–900. [Google Scholar] [CrossRef]
- Khatri, L.; Taylor, K.M.; Craig, D.Q.; Palin, K. An assessment of jet and ultrasonic nebulisers for the delivery of lactate dehydrogenase solutions. Int. J. Pharm. 2001, 227, 121–131. [Google Scholar] [CrossRef]
- Hurt, K.; Bilton, D. Inhaled interventions in cystic fibrosis: Mucoactive and antibiotic therapies. Respiration 2014, 88, 441–448. [Google Scholar] [CrossRef]
- Daniels, T.; Mills, N.; Whitaker, P. Nebuliser systems for drug delivery in cystic fibrosis. Cochrane Database Syst. Rev. 2013, 4, CD007639. [Google Scholar] [CrossRef]
- Hertel, S.; Pohl, T.; Friess, W.; Winter, G. That’s cool!—Nebulization of thermolabile proteins with a cooled vibrating mesh nebulizer. Eur. J. Pharm. Biopharm. 2014, 87, 357–365. [Google Scholar] [CrossRef]
- Johnson, J.C.; Waldrep, J.C.; Guo, J.; Dhand, R. Aerosol delivery of recombinant human DNase I: In vitro comparison of a vibrating-mesh nebulizer with a jet nebulizer. Respir. Care 2008, 53, 1703–1708. [Google Scholar]
- Hubert, D.; Leroy, S.; Nove-Josserand, R.; Murris-Espin, M.; Mely, L.; Dominique, S.; Delaisi, B.; Kho, P.; Kovarik, J.M. Pharmacokinetics and safety of tobramycin administered by the PARI eFlow rapid nebulizer in cystic fibrosis. J. Cyst. Fibros. 2009, 8, 332–337. [Google Scholar] [CrossRef]
- Chan, J.G.; Kwok, P.C.; Young, P.M.; Chan, H.K.; Traini, D. Mannitol delivery by vibrating mesh nebulisation for enhancing mucociliary clearance. J. Pharm. Sci. 2011, 100, 2693–2702. [Google Scholar] [CrossRef]
- Röhm, M.; Carle, S.; Maigler, F.; Flamm, J.; Kramer, V.; Mavoungou, C.; Schmid, O.; Schindowski, K. A comprehensive screening platform for aerosolizable protein formulations for intranasal and pulmonary drug delivery. Int. J. Pharm. 2017, 532, 537–546. [Google Scholar] [CrossRef]
- Diot, P.; Gagnadoux, F.; Martin, C.; Ellataoui, H.; Furet, Y.; Breteau, M.; Boissinot, E.; Lemarie, E. Nebulization and anti-Pseudomonas aeruginosa activity of colistin. Eur. Respir. J. 1997, 10, 1995–1998. [Google Scholar] [CrossRef]
- Michotte, J.-B.; Staderini, E.; Aubriot, A.-S.; Jossen, E.; Dugernier, J.; Liistro, G.; Reychler, G. Pulmonary Drug Delivery Following Continuous Vibrating Mesh Nebulization and Inspiratory Synchronized Vibrating Mesh Nebulization During Noninvasive Ventilation in Healthy Volunteers. J. Aerosol Med. Pulm. Drug Deliv. 2017, 31, 33–41. [Google Scholar] [CrossRef]
- Réminiac, F.; Vecellio, L.; Loughlin, R.M.; Le Pennec, D.; Cabrera, M.; Vourc’h, N.H.; Fink, J.B.; Ehrmann, S. Nasal high flow nebulization in infants and toddlers: An in vitro and in vivo scintigraphic study. Pediatr. Pulmonol. 2017, 52, 337–344. [Google Scholar] [CrossRef]
- Dugernier, J.; Hesse, M.; Vanbever, R.; Depoortere, V.; Roeseler, J.; Michotte, J.-B.; Laterre, P.-F.; Jamar, F.; Reychler, G. SPECT-CT comparison of lung deposition using a system combining a vibrating-mesh nebulizer with a valved holding chamber and a conventional jet nebulizer: A randomized cross-over study. Pharm. Res. 2017, 34, 290–300. [Google Scholar] [CrossRef]
- Hibbitts, A.; O’mahony, A.; Forde, E.; Nolan, L.; Ogier, J.; Desgranges, S.; Darcy, R.; MacLoughlin, R.; O’driscoll, C.; Cryan, S.-A. Early-stage development of novel cyclodextrin-siRNA nanocomplexes allows for successful postnebulization transfection of bronchial epithelial cells. J. Aerosol Med. Pulm. Drug Deliv. 2014, 27, 466–477. [Google Scholar] [CrossRef]
- Wayne, P. Performance Standards for Antimicrobial Susceptibility Testing: 20th Informational Supplement; CLSI Document M100-S20; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2010. [Google Scholar]
- Wu, M.; Hancock, R.E. Interaction of the cyclic antimicrobial cationic peptide bactenecin with the outer and cytoplasmic membrane. J. Biol. Chem. 1999, 274, 29–35. [Google Scholar] [CrossRef]
- Sharma, K.; Somavarapu, S.; Colombani, A.; Govind, N.; Taylor, K.M. Nebulised siRNA encapsulated crosslinked chitosan nanoparticles for pulmonary delivery. Int. J. Pharm. 2013, 455, 241–247. [Google Scholar] [CrossRef]
- Lange, C.F.; Hancock, R.E.; Samuel, J.; Finlay, W.H. In vitro aerosol delivery and regional airway surface liquid concentration of a liposomal cationic peptide. J. Pharm. Sci. 2001, 90, 1647–1657. [Google Scholar] [CrossRef]
- Gibbons, A.M.; McElvaney, N.G.; Taggart, C.C.; Cryan, S.A. Delivery of rSLPI in a liposomal carrier for inhalation provides protection against cathepsin L degradation. J. Microencapsul. 2009, 26, 513–522. [Google Scholar] [CrossRef]
- Desai, T.R.; Tyrrell, G.J.; Ng, T.; Finlay, W.H. In vitro evaluation of nebulization properties, antimicrobial activity, and regional airway surface liquid concentration of liposomal polymyxin B sulfate. Pharm. Res. 2003, 20, 442–447. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kasiakou, S.K. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef]
- Coates, A.L.; Denk, O.; Leung, K.; Ribeiro, N.; Chan, J.; Green, M.; Martin, S.; Charron, M.; Edwardes, M.; Keller, M. Higher tobramycin concentration and vibrating mesh technology can shorten antibiotic treatment time in cystic fibrosis. Pediatr. Pulmonol. 2011, 46, 401–408. [Google Scholar] [CrossRef]
- Berlinski, A.; Willis, J.R. Effect of tidal volume and nebulizer type and position on albuterol delivery in a pediatric model of mechanical ventilation. Respir. Care 2015, 60, 1424–1430. [Google Scholar] [CrossRef]
- Ari, A.; Atalay, O.T.; Harwood, R.; Sheard, M.M.; Aljamhan, E.A.; Fink, J.B. Influence of nebulizer type, position, and bias flow on aerosol drug delivery in simulated pediatric and adult lung models during mechanical ventilation. Respir. Care 2010, 55, 845–851. [Google Scholar]
- Dhand, R.; Sohal, H. Pulmonary Drug Delivery System for inhalation therapy in mechanically ventilated patients. Exp. Rev. Med. Dev. 2008, 5, 9–18. [Google Scholar] [CrossRef]
- Kadrichu, N.; Boc, S.; Corkery, K.; Challoner, P. In vitro assessment of aerosolized amikacin in lung dose delivered by NKTR-061 PDDS clinical during on-ventilator and off-ventilator use. J. Aerosol Med. Pulm. Drug Deliv. 2018, 29, A39. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | MIC vs. P. aeruginosa Strains (μg/mL) | |||
|---|---|---|---|---|
| PAO1 | PABH01 | PABH02 | PABH03 | |
| AAG-WMR | 32 | 8 | 16 | 32 |
| AAG-WMR (nebulised) | 32 | 8 | 16 | 32 |
| Pro-WMR | >64 | >64 | >64 | >64 |
| Pro-WMR (nebulised) | >64 | >64 | >64 | >64 |
| Pro-WMR | AAG-WMR | |
|---|---|---|
| MMAD (GSD) 1 mL | 3.59 μm (1.67) | 3.14 μm (1.93) |
| MMAD (GSD) 2 mL | 3.92 μm (1.61) | 3.86 μm (1.51) |
| VMD (GSD) | 3.79 μm (1.76) | 3.80 μm (1.74) |
| FPF < 5 μm | 67.1% | 66.6% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forde, É.; Kelly, G.; Sweeney, L.; Fitzgerald-Hughes, D.; MacLoughlin, R.; Devocelle, M. Vibrating Mesh Nebulisation of Pro-Antimicrobial Peptides for Use in Cystic Fibrosis. Pharmaceutics 2019, 11, 239. https://doi.org/10.3390/pharmaceutics11050239
Forde É, Kelly G, Sweeney L, Fitzgerald-Hughes D, MacLoughlin R, Devocelle M. Vibrating Mesh Nebulisation of Pro-Antimicrobial Peptides for Use in Cystic Fibrosis. Pharmaceutics. 2019; 11(5):239. https://doi.org/10.3390/pharmaceutics11050239
Chicago/Turabian StyleForde, Éanna, Graeme Kelly, Louise Sweeney, Deirdre Fitzgerald-Hughes, Ronan MacLoughlin, and Marc Devocelle. 2019. "Vibrating Mesh Nebulisation of Pro-Antimicrobial Peptides for Use in Cystic Fibrosis" Pharmaceutics 11, no. 5: 239. https://doi.org/10.3390/pharmaceutics11050239
APA StyleForde, É., Kelly, G., Sweeney, L., Fitzgerald-Hughes, D., MacLoughlin, R., & Devocelle, M. (2019). Vibrating Mesh Nebulisation of Pro-Antimicrobial Peptides for Use in Cystic Fibrosis. Pharmaceutics, 11(5), 239. https://doi.org/10.3390/pharmaceutics11050239