Design of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles for Vaginal Co-Delivery of Griffithsin and Dapivirine and Their Synergistic Effect for HIV Prophylaxis


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
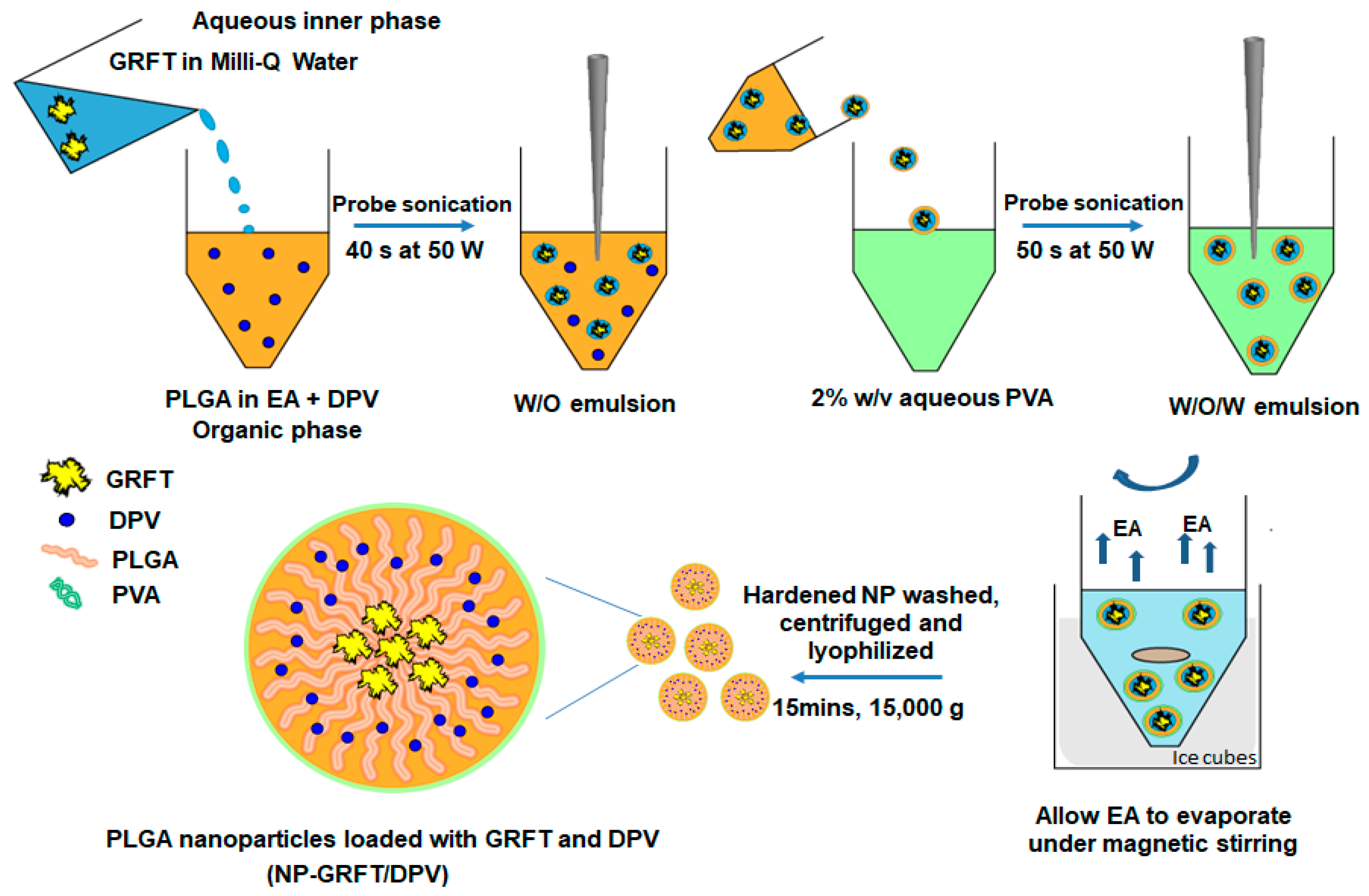
2.2.1. Fabrication of ARV-Loaded Nanoparticles
2.2.2. Characterization of Nanoparticles
2.2.3. Drug Loading
2.2.4. In Vitro Release of ARVs from PLGA Nanoparticles
2.2.5. Anti-HIV-1 Activity and Cellular Viability Assay of ARVs
2.2.6. Combination Effects
2.2.7. Cellular Uptake Assay
2.2.8. Statistical Analyses
3. Results
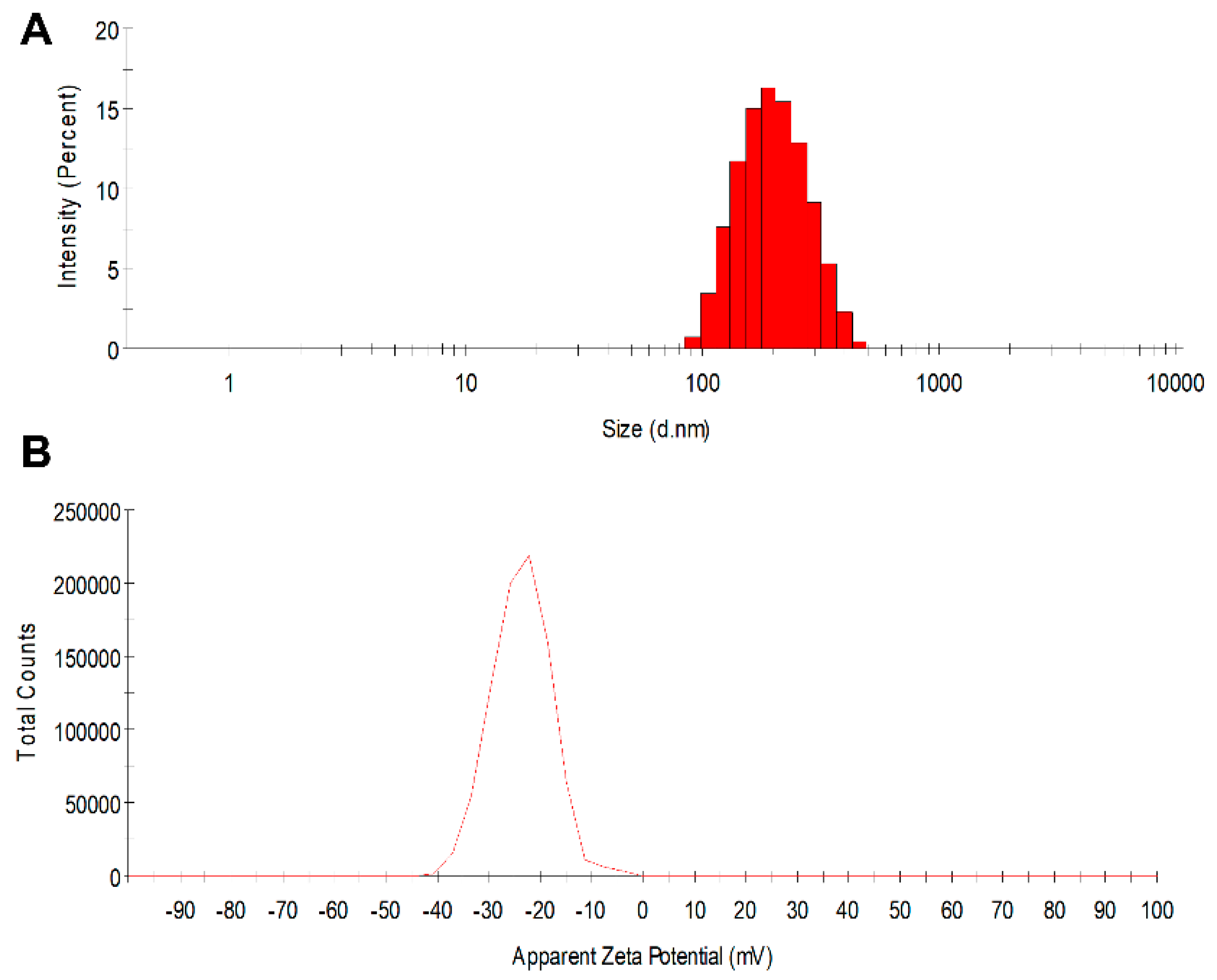
3.1. PLGA Nanoparticle Characterization
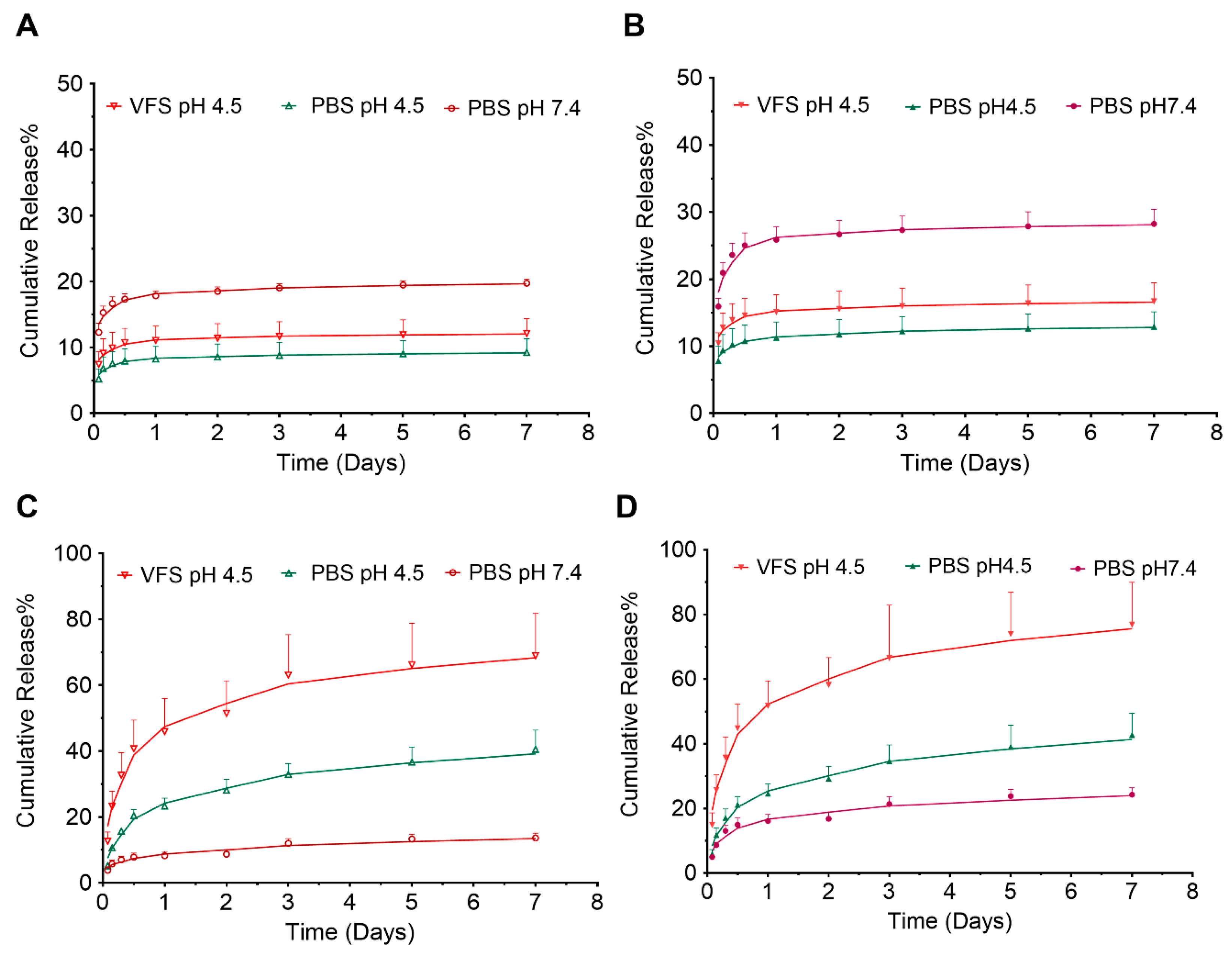
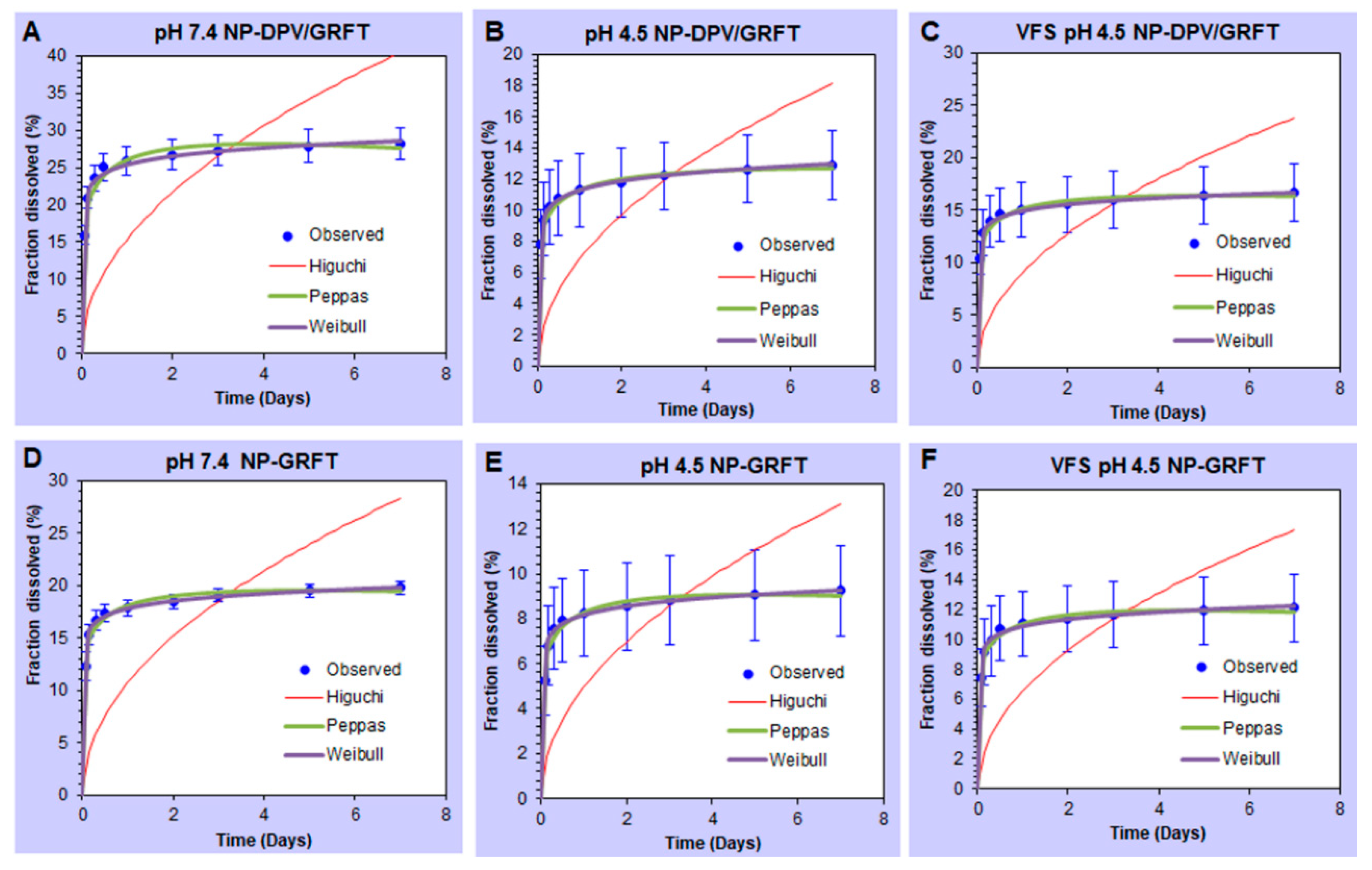
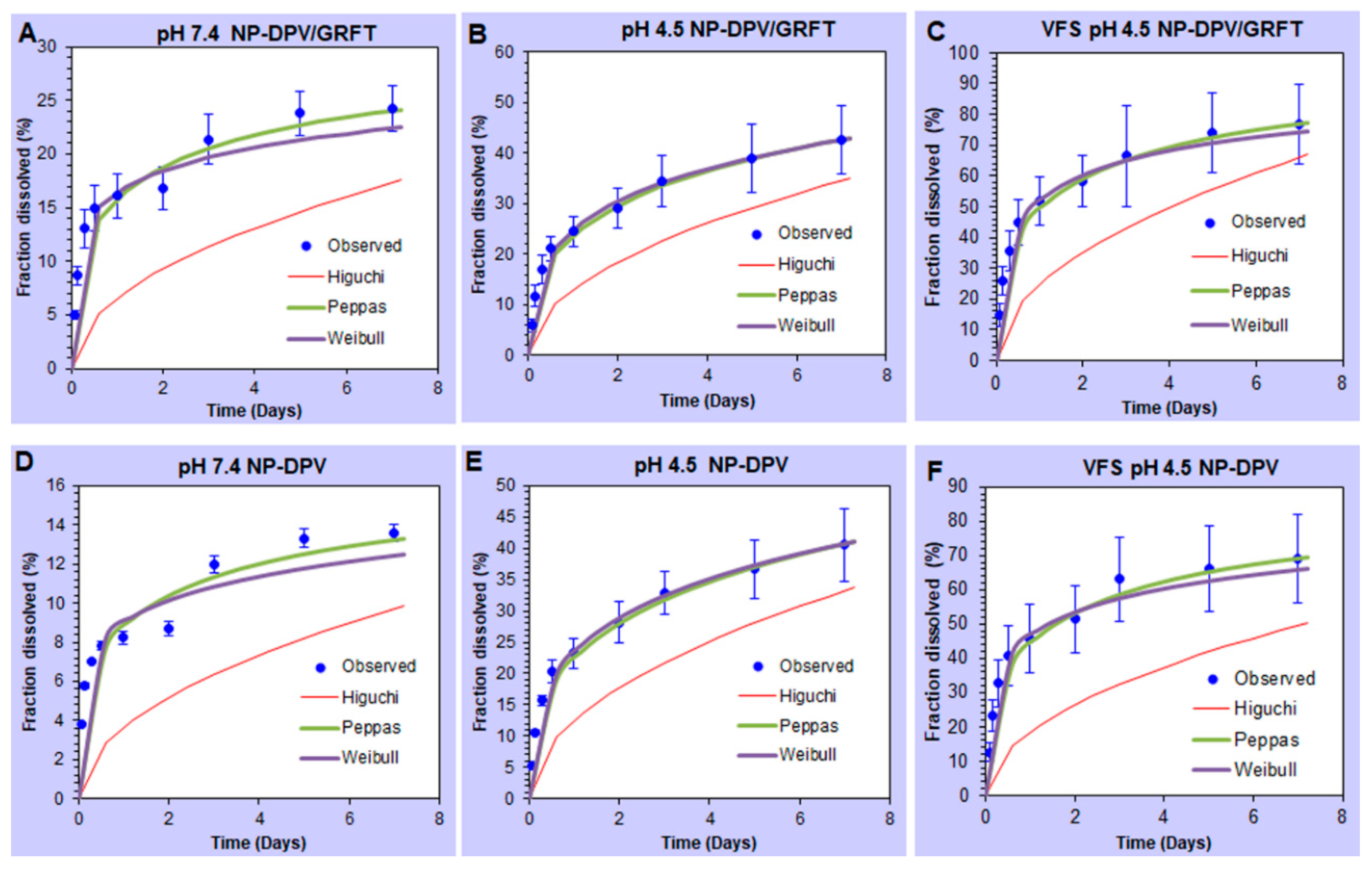
3.2. In Vitro Release Studies of ARVs from PLGA Nanoparticles Resulting in Sustained Drug Release
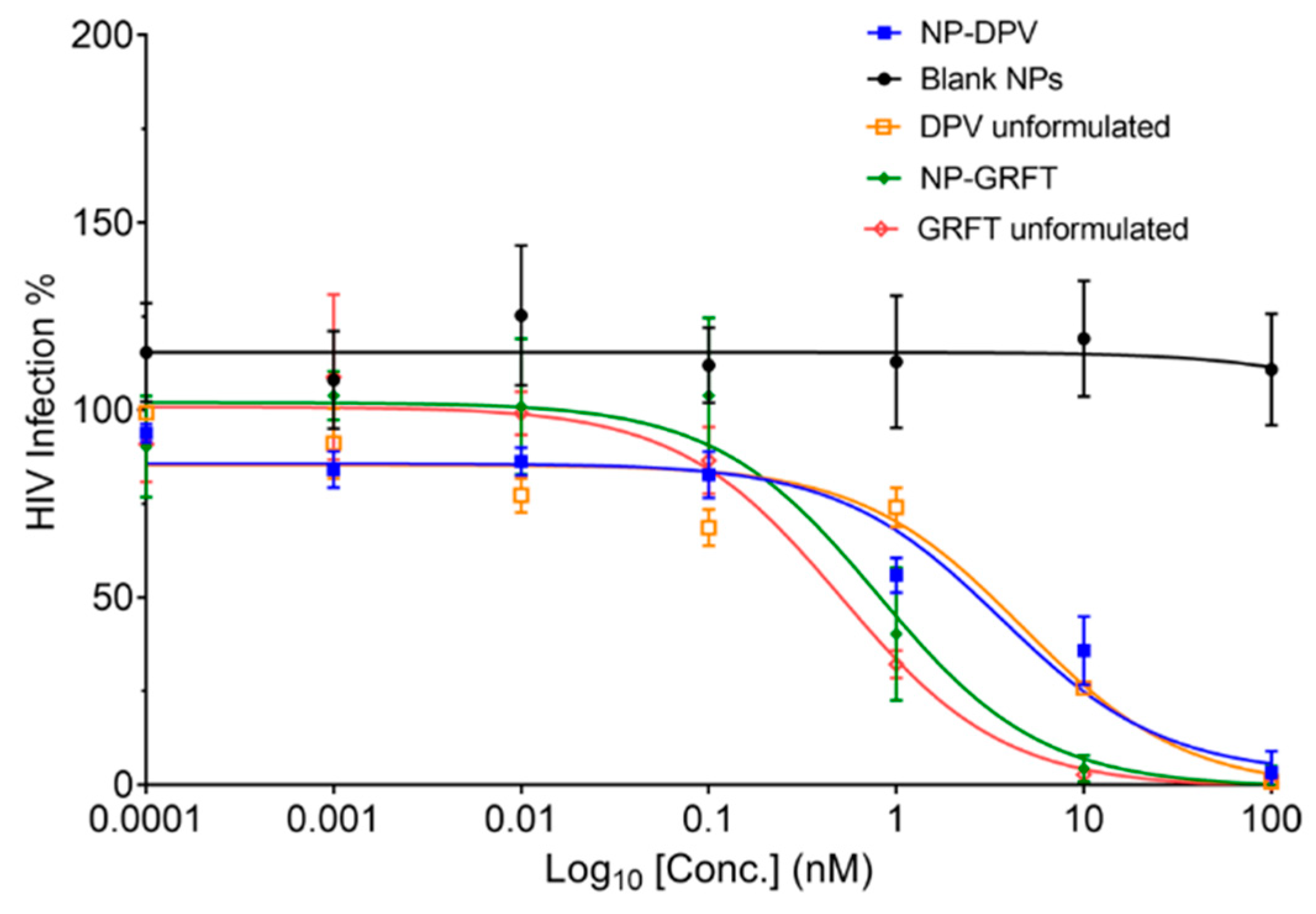
3.3. PLGA NP-ARVs Potently Inhibit HIV-1BaL Infection In Vitro
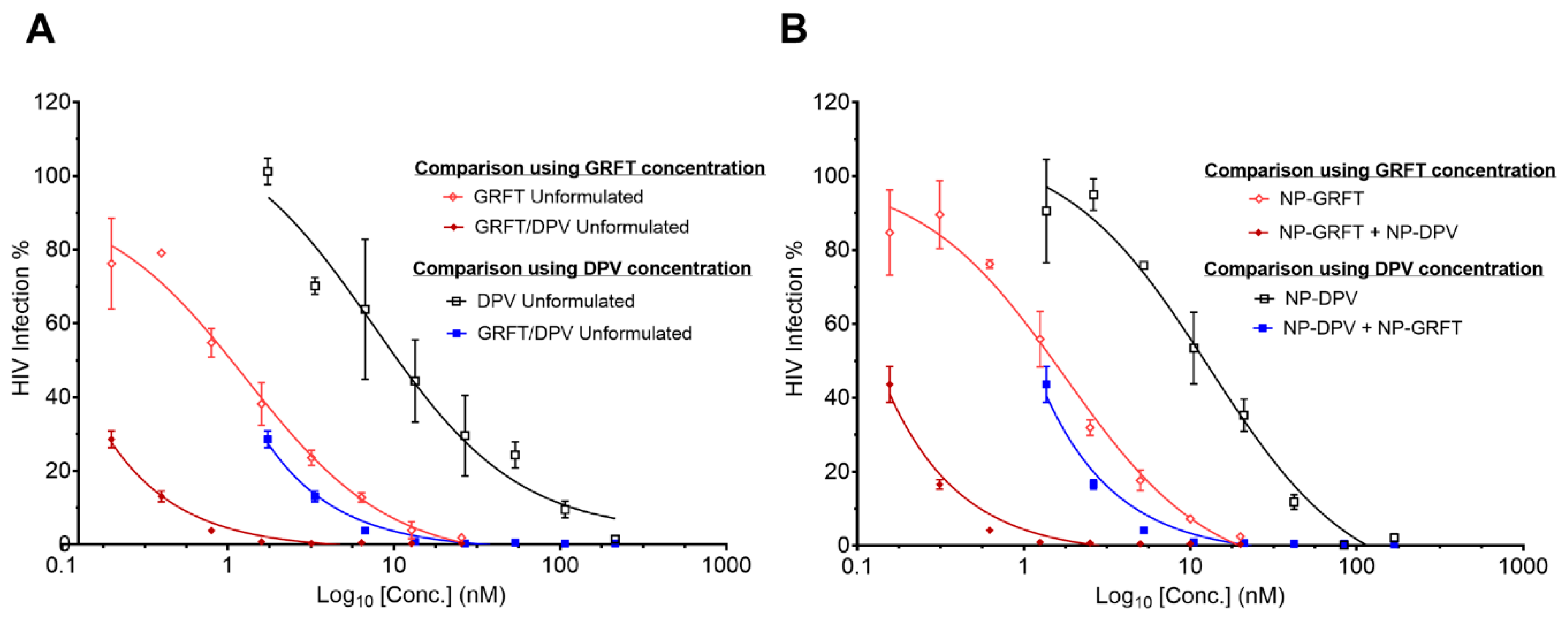
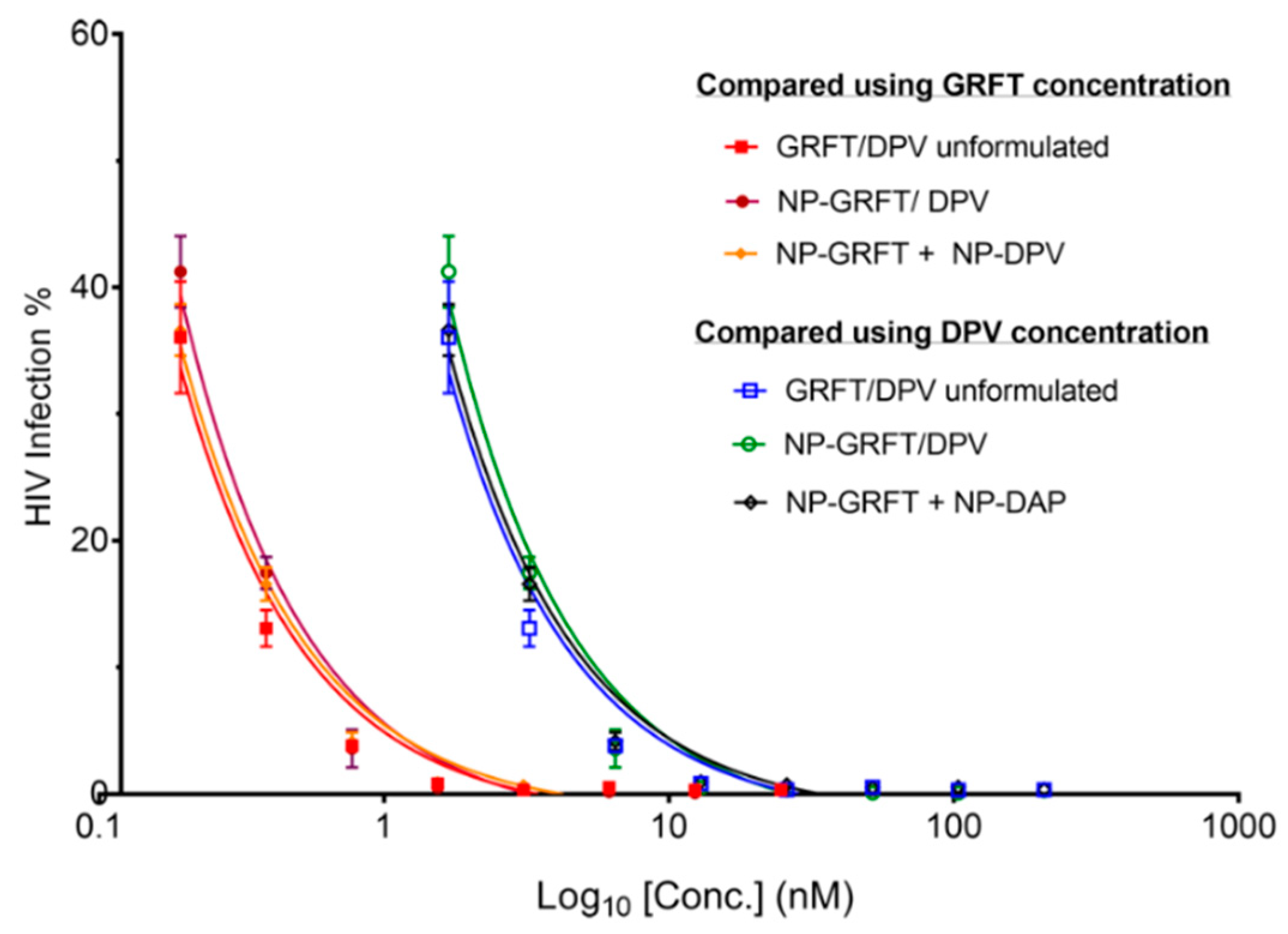
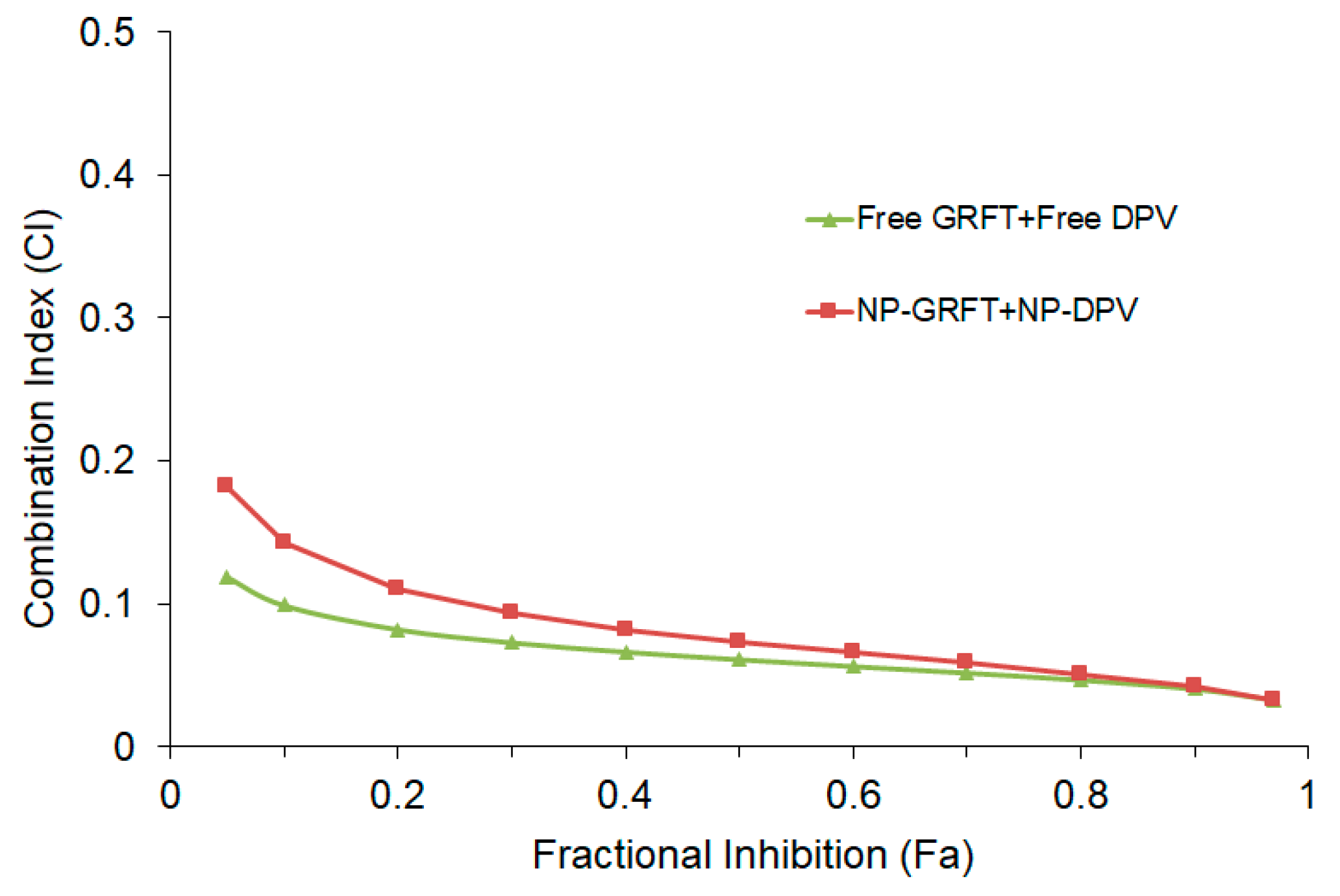
3.4. NP-ARV Combination Exhibits Strong Synergistic Bioactivity
3.5. NP-ARVs Are Nontoxic to In Vitro Cell Lines
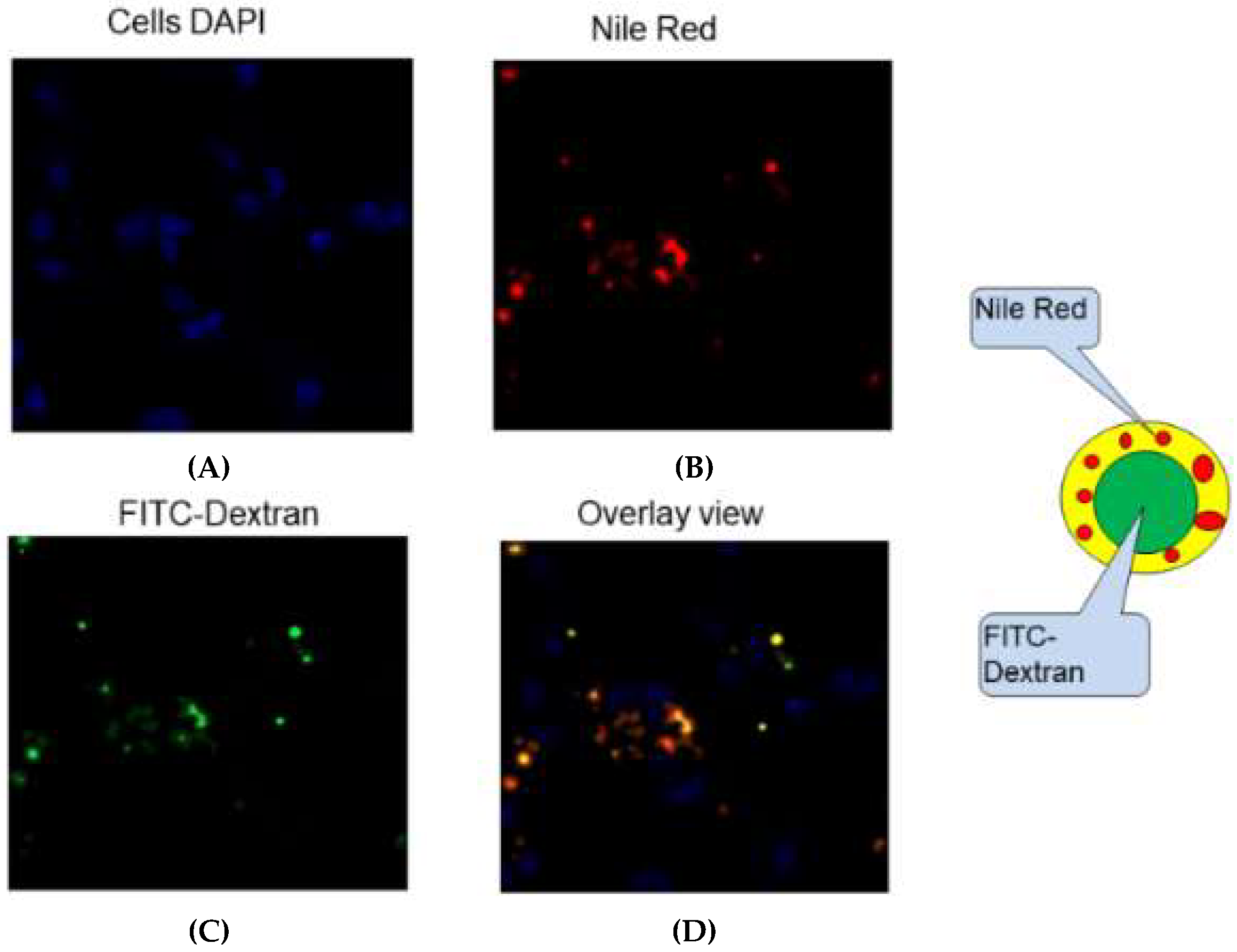
3.6. Cellular Uptake of NPs In Vitro
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- UNAIDS. Global HIV & AIDS Statistics—2018 Fact Sheet. Available online: http://www.unaids.org/en/resources/fact-sheet (accessed on 19 February 2019).
- AVERT. Women and Girls, HIV and AIDS. Available online: https://www.avert.org/professionals/hiv-social-issues/key-affected-populations/women (accessed on 19 February 2019).
- Centers for Disease Control and Prevention. HIV Among Women. Available online: https://www.cdc.gov/hiv/group/gender/women/index.html (accessed on 14 April 2019).
- Bedimo, A.L.; Bennett, M.; Kissinger, P.; Clark, R.A. Understanding barriers to condom usage among HIV-infected African American women. J. Assoc. Nurses AIDS Care 1998, 9, 48–58. [Google Scholar] [CrossRef]
- Santelli, J.S.; Kouzis, A.C.; Hoover, D.R.; Polacsek, M.; Burwell, L.G.; Celentano, D.D. Stage of behavior change for condom use: the influence of partner type, relationship and pregnancy factors. Fam. Plan. Perspect. 1996, 28, 101–107. [Google Scholar] [CrossRef]
- Moench, T.R.; Chipato, T.; Padian, N.S. Preventing disease by protecting the cervix: the unexplored promise of internal vaginal barrier devices. AIDS 2001, 15, 1595–1602. [Google Scholar] [CrossRef]
- Minnis, A.M.; Padian, N.S. Effectiveness of female controlled barrier methods in preventing sexually transmitted infections and HIV: current evidence and future research directions. Sex. Transm. Infect. 2005, 81, 193–200. [Google Scholar] [CrossRef]
- Matthews, J.; Harrison, T. An update on female-controlled methods for HIV prevention: Female condom, microbicides and cervical barriers. S. Afr. J. HIV Med. 2006, 7, 7–11. [Google Scholar]
- Padian, N.S.; Buve, A.; Balkus, J.; Serwadda, D.; Cates, W., Jr. Biomedical interventions to prevent HIV infection: evidence, challenges, and way forward. Lancet 2008, 372, 585–599. [Google Scholar] [CrossRef]
- Maenza, J.; Flexner, C. Combination antiretroviral therapy for HIV infection. Am. Fam. Physician 1998, 57, 2789–2798. [Google Scholar] [PubMed]
- Ludovici, D.W.; De Corte, B.L.; Kukla, M.J.; Ye, H.; Ho, C.Y.; Lichtenstein, M.A.; Kavash, R.W.; Andries, K.; de Bethune, M.P.; Azijn, H.; et al. Evolution of anti-HIV drug candidates. Part 3: Diarylpyrimidine (DAPY) analogues. Bioorg. Med. Chem. Lett. 2001, 11, 2235–2239. [Google Scholar] [CrossRef]
- Holt, J.D.; Cameron, D.; Dias, N.; Holding, J.; Muntendam, A.; Oostebring, F.; Dreier, P.; Rohan, L.; Nuttall, J. The Sheep as a Model for Preclinical Safety and Pharmacokinetic Evaluations of Candidate Microbicides. Antimicrob. Agents Chemother. 2015, 59, 3761–3770. [Google Scholar] [CrossRef] [PubMed]
- Baeten, J.M.; Palanee-Phillips, T.; Brown, E.R.; Schwartz, K.; Soto-Torres, L.E.; Govender, V.; Mgodi, N.M.; Matovu Kiweewa, F.; Nair, G.; Mhlanga, F.; et al. Use of a Vaginal Ring Containing Dapivirine for HIV-1 Prevention in Women. N. Engl. J. Med. 2016, 375, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.; van Niekerk, N.; Kapiga, S.; Bekker, L.G.; Gama, C.; Gill, K.; Kamali, A.; Kotze, P.; Louw, C.; Mabude, Z.; et al. Safety and Efficacy of a Dapivirine Vaginal Ring for HIV Prevention in Women. N. Engl. J. Med. 2016, 375, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; O’Keefe, B.R.; Sowder, R.C., 2nd; Bringans, S.; Gardella, R.; Berg, S.; Cochran, P.; Turpin, J.A.; Buckheit, R.W., Jr.; McMahon, J.B.; et al. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J. Biol. Chem. 2005, 280, 9345–9353. [Google Scholar] [CrossRef]
- Kagiampakis, I.; Gharibi, A.; Mankowski, M.K.; Snyder, B.A.; Ptak, R.G.; Alatas, K.; LiWang, P.J. Potent strategy to inhibit HIV-1 by binding both gp120 and gp41. Antimicrob. Agents Chemother. 2011, 55, 264–275. [Google Scholar] [CrossRef]
- Alexandre, K.B.; Gray, E.S.; Pantophlet, R.; Moore, P.L.; McMahon, J.B.; Chakauya, E.; O’Keefe, B.R.; Chikwamba, R.; Morris, L. Binding of the mannose-specific lectin, griffithsin, to HIV-1 gp120 exposes the CD4-binding site. J. Virol. 2011, 85, 9039–9050. [Google Scholar] [CrossRef] [PubMed]
- Emau, P.; Tian, B.; O’Keefe, B.R.; Mori, T.; McMahon, J.B.; Palmer, K.E.; Jiang, Y.; Bekele, G.; Tsai, C.C. Griffithsin, a potent HIV entry inhibitor, is an excellent candidate for anti-HIV microbicide. J. Med. Primatol. 2007, 36, 244–253. [Google Scholar] [CrossRef]
- Barton, C.L. Evaluation of the Safety and Pharmacokinetic Profile of the Broad Spectrum Antiviral Lectin Griffithsin. Ph.D. Thesis, University of Louisville, Louisville, KY, USA, 2014. [Google Scholar]
- O’Keefe, B.R.; Vojdani, F.; Buffa, V.; Shattock, R.J.; Montefiori, D.C.; Bakke, J.; Mirsalis, J.; d’Andrea, A.L.; Hume, S.D.; Bratcher, B.; et al. Scaleable manufacture of HIV-1 entry inhibitor griffithsin and validation of its safety and efficacy as a topical microbicide component. Proc. Natl. Acad. Sci. USA 2009, 106, 6099–6104. [Google Scholar] [CrossRef]
- Girard, L.; Birse, K.; Holm, J.B.; Gajer, P.; Humphrys, M.S.; Garber, D.; Guenthner, P.; Noel-Romas, L.; Abou, M.; McCorrister, S.; et al. Impact of the griffithsin anti-HIV microbicide and placebo gels on the rectal mucosal proteome and microbiome in non-human primates. Sci. Rep. 2018, 8, 8059. [Google Scholar] [CrossRef]
- Ferir, G.; Palmer, K.E.; Schols, D. Synergistic activity profile of griffithsin in combination with tenofovir, maraviroc and enfuvirtide against HIV-1 clade C. Virology 2011, 417, 253–258. [Google Scholar] [CrossRef]
- Kiser, P.F.; Mesquita, P.M.; Herold, B.C. A perspective on progress and gaps in HIV prevention science. AIDS Res. Hum. Retroviruses 2012, 28, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Nutan. Clinical use of vaginal or rectally applied microbicides in patients suffering from HIV/AIDS. HIV AIDS (Auckl) 2013, 5, 295–307. [Google Scholar] [CrossRef][Green Version]
- Thurman, A.R.; Clark, M.R.; Hurlburt, J.A.; Doncel, G.F. Intravaginal rings as delivery systems for microbicides and multipurpose prevention technologies. Int. J. Womens Health 2013, 5, 695–708. [Google Scholar] [CrossRef]
- das Neves, J.; Nunes, R.; Rodrigues, F.; Sarmento, B. Nanomedicine in the development of anti-HIV microbicides. Adv. Drug Deliv. Rev. 2016, 103, 57–75. [Google Scholar] [CrossRef]
- Cunha-Reis, C.; Machado, A.; Barreiros, L.; Araujo, F.; Nunes, R.; Seabra, V.; Ferreira, D.; Segundo, M.A.; Sarmento, B.; das Neves, J. Nanoparticles-in-film for the combined vaginal delivery of anti-HIV microbicide drugs. J. Control. Release 2016, 243, 43–53. [Google Scholar] [CrossRef]
- Malik, T.; Chauhan, G.; Rath, G.; Kesarkar, R.N.; Chowdhary, A.S.; Goyal, A.K. Efaverinz and nano-gold-loaded mannosylated niosomes: a host cell-targeted topical HIV-1 prophylaxis via thermogel system. Artif. Cells Nanomed. Biotechnol. 2017. [Google Scholar] [CrossRef]
- das Neves, J.; Araujo, F.; Andrade, F.; Amiji, M.; Bahia, M.F.; Sarmento, B. Biodistribution and pharmacokinetics of dapivirine-loaded nanoparticles after vaginal delivery in mice. Pharm. Res. 2014, 31, 1834–1845. [Google Scholar] [CrossRef]
- das Neves, J.; Araujo, F.; Andrade, F.; Michiels, J.; Arien, K.K.; Vanham, G.; Amiji, M.; Bahia, M.F.; Sarmento, B. In vitro and ex vivo evaluation of polymeric nanoparticles for vaginal and rectal delivery of the anti-HIV drug dapivirine. Mol. Pharm. 2013, 10, 2793–2807. [Google Scholar] [CrossRef]
- das Neves, J.; Michiels, J.; Arien, K.K.; Vanham, G.; Amiji, M.; Bahia, M.F.; Sarmento, B. Polymeric nanoparticles affect the intracellular delivery, antiretroviral activity and cytotoxicity of the microbicide drug candidate dapivirine. Pharm. Res. 2012, 29, 1468–1484. [Google Scholar] [CrossRef]
- Kovarova, M.; Council, O.D.; Date, A.A.; Long, J.M.; Nochi, T.; Belshan, M.; Shibata, A.; Vincent, H.; Baker, C.E.; Thayer, W.O.; et al. Correction: Nanoformulations of Rilpivirine for Topical Pericoital and Systemic Coitus-Independent Administration Efficiently Prevent HIV Transmission. PLoS Pathog. 2015, 11, e1005170. [Google Scholar] [CrossRef]
- das Neves, J.; Sarmento, B. Precise engineering of dapivirine-loaded nanoparticles for the development of anti-HIV vaginal microbicides. Acta Biomater. 2015, 18, 77–87. [Google Scholar] [CrossRef]
- Bala, I.; Hariharan, S.; Kumar, M.N. PLGA nanoparticles in drug delivery: The state of the art. Crit. Rev. Ther. Drug Carrier Syst. 2004, 21, 387–422. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Bouissou, C.; Rouse, J.J.; Price, R.; van der Walle, C.F. The influence of surfactant on PLGA microsphere glass transition and water sorption: remodeling the surface morphology to attenuate the burst release. Pharm. Res. 2006, 23, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Ham, A.S.; Cost, M.R.; Sassi, A.B.; Dezzutti, C.S.; Rohan, L.C. Targeted delivery of PSC-RANTES for HIV-1 prevention using biodegradable nanoparticles. Pharm. Res. 2009, 26, 502–511. [Google Scholar] [CrossRef]
- Menei, P.; Daniel, V.; Montero-Menei, C.; Brouillard, M.; Pouplard-Barthelaix, A.; Benoit, J.P. Biodegradation and brain tissue reaction to poly(D,L-lactide-co-glycolide) microspheres. Biomaterials 1993, 14, 470–478. [Google Scholar] [CrossRef]
- Shive, M.S.; Anderson, J.M. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar]
- McRae, A.; Ling, E.A.; Hjorth, S.; Dahlstrom, A.; Mason, D.; Tice, T. Catecholamine-containing biodegradable microsphere implants as a novel approach in the treatment of CNS neurodegenerative disease. A review of experimental studies in DA-lesioned rats. Mol. Neurobiol. 1994, 9, 191–205. [Google Scholar] [CrossRef]
- Engman, C.; Wen, Y.; Meng, W.S.; Bottino, R.; Trucco, M.; Giannoukakis, N. Generation of antigen-specific Foxp3+ regulatory T-cells in vivo following administration of diabetes-reversing tolerogenic microspheres does not require provision of antigen in the formulation. Clin. Immunol. 2015, 160, 103–123. [Google Scholar] [CrossRef]
- Chaowanachan, T.; Krogstad, E.; Ball, C.; Woodrow, K.A. Drug synergy of tenofovir and nanoparticle-based antiretrovirals for HIV prophylaxis. PLoS ONE 2013, 8, e61416. [Google Scholar] [CrossRef]
- Mallipeddi, R.; Rohan, L.C. Progress in antiretroviral drug delivery using nanotechnology. Int. J. Nanomed. 2010, 5, 533–547. [Google Scholar]
- das Neves, J.; Amiji, M.M.; Bahia, M.F.; Sarmento, B. Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv. Drug Deliv. Rev. 2010, 62, 458–477. [Google Scholar] [CrossRef]
- Destache, C.J.; Belgum, T.; Christensen, K.; Shibata, A.; Sharma, A.; Dash, A. Combination antiretroviral drugs in PLGA nanoparticle for HIV-1. BMC Infect. Dis. 2009, 9, 198. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Bikiaris, D. Novel self-assembled core-shell nanoparticles based on crystalline amorphous moieties of aliphatic copolyesters for efficient controlled drug release. J. Control. Release 2009, 138, 177–184. [Google Scholar] [CrossRef]
- Owen, D.H.; Katz, D.F. A vaginal fluid simulant. Contraception 1999, 59, 91–95. [Google Scholar] [CrossRef]
- Dezzutti, C.S.; Brown, E.R.; Moncla, B.; Russo, J.; Cost, M.; Wang, L.; Uranker, K.; Kunjara Na Ayudhya, R.P.; Pryke, K.; Pickett, J.; et al. Is wetter better? An evaluation of over-the-counter personal lubricants for safety and anti-HIV-1 activity. PLoS ONE 2012, 7, e48328. [Google Scholar] [CrossRef]
- Montefiori, D.C. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol. Biol. 2009, 485, 395–405. [Google Scholar] [CrossRef]
- Dezzutti, C.S.; Shetler, C.; Mahalingam, A.; Ugaonkar, S.R.; Gwozdz, G.; Buckheit, K.W.; Buckheit, R.W., Jr. Safety and efficacy of tenofovir/IQP-0528 combination gels - a dual compartment microbicide for HIV-1 prevention. Antiviral Res. 2012, 96, 221–225. [Google Scholar] [CrossRef]
- Kohli, A.; Islam, A.; Moyes, D.L.; Murciano, C.; Shen, C.; Challacombe, S.J.; Naglik, J.R. Oral and vaginal epithelial cell lines bind and transfer cell-free infectious HIV-1 to permissive cells but are not productively infected. PLoS ONE 2014, 9, e98077. [Google Scholar] [CrossRef] [PubMed]
- Montefiori, D.C. Evaluating neutralizing antibodies against HIV, SIV, and SHIV in luciferase reporter gene assays. Curr. Protoc. Immunol. 2005. [Google Scholar] [CrossRef]
- Dezzutti, C.S.; Rohan, L.C.; Wang, L.; Uranker, K.; Shetler, C.; Cost, M.; Lynam, J.D.; Friend, D. Reformulated tenofovir gel for use as a dual compartment microbicide. J. Antimicrob. Chemother. 2012, 67, 2139–2142. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef]
- Bilati, U.; Allemann, E.; Doelker, E. Poly(D,L-lactide-co-glycolide) protein-loaded nanoparticles prepared by the double emulsion method--processing and formulation issues for enhanced entrapment efficiency. J. Microencapsul. 2005, 22, 205–214. [Google Scholar] [CrossRef]
- Xu, B.; Dou, H.; Tao, K.; Sun, K.; Lu, R.; Shi, W. Influence of experimental parameters and the copolymer structure on the size control of nanospheres in double emulsion method. J. Polym. Res. 2011, 18, 131–137. [Google Scholar] [CrossRef]
- Liu, J.; Qiu, Z.; Wang, S.; Zhou, L.; Zhang, S. A modified double-emulsion method for the preparation of daunorubicin-loaded polymeric nanoparticle with enhanced in vitro anti-tumor activity. Biomed. Mater. 2010, 5, 065002. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.R.C.; Loebenberg, R.; Almukainzi, M. Simulated Biological Fluids with Possible Application in Dissolution Testing. Dissolution Technol. 2011, 18, 15–28. [Google Scholar] [CrossRef]
- Destache, C.J.; Belgum, T.; Goede, M.; Shibata, A.; Belshan, M.A. Antiretroviral release from poly(DL-lactide-co-glycolide) nanoparticles in mice. J. Antimicrob. Chemother. 2010, 65, 2183–2187. [Google Scholar] [CrossRef] [PubMed]
- Friend, D.R.; Kiser, P.F. Assessment of topical microbicides to prevent HIV-1 transmission: concepts, testing, lessons learned. Antiviral. Res. 2013, 99, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Abdool Karim, Q.; Abdool Karim, S.S.; Frohlich, J.A.; Grobler, A.C.; Baxter, C.; Mansoor, L.E.; Kharsany, A.B.; Sibeko, S.; Mlisana, K.P.; Omar, Z.; et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010, 329, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Long-acting HIV drugs advanced to overcome adherence challenge. Nat. Med. 2014, 20, 323–324. [Google Scholar] [CrossRef]
- Andrews, C.D.; Spreen, W.R.; Mohri, H.; Moss, L.; Ford, S.; Gettie, A.; Russell-Lodrigue, K.; Bohm, R.P.; Cheng-Mayer, C.; Hong, Z.; et al. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science 2014, 343, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Kramzer, L. Preformulation and Biological Evaluations for the Intravaginal Delivery of Griffithsin for HIV Prevention. Ph.D. Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 2015. [Google Scholar]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Ding, A.G.; Schwendeman, S.P. Acidic microclimate pH distribution in PLGA microspheres monitored by confocal laser scanning microscopy. Pharm. Res. 2008, 25, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Smith, P. Estrogens and the urogenital tract. Studies on steroid hormone receptors and a clinical study on a new estradiol-releasing vaginal ring. Acta Obstet. Gynecol. Scand. 1993, 72, 5–26. [Google Scholar] [CrossRef]
- Boskey, E.R.; Cone, R.A.; Whaley, K.J.; Moench, T.R. Origins of vaginal acidity: high D/L lactate ratio is consistent with bacteria being the primary source. Hum. Reprod. 2001, 16, 1809–1813. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef]
- Secchi, M.; Xu, Q.; Lusso, P.; Vangelista, L. The superior folding of a RANTES analogue expressed in lactobacilli as compared to mammalian cells reveals a promising system to screen new RANTES mutants. Protein Expr. Purif. 2009, 68, 34–41. [Google Scholar] [CrossRef][Green Version]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Size (d.nm ± SD) | PDI | Zeta Potential (mV ± SD) | Encapsulation Efficiency (% ± SD) |
|---|---|---|---|---|
| Placebo | 182.8 ± 1.7 | 0.066 | −23.7 ± 0.6 | - |
| GRFT | 188.8 ± 1.7 | 0.069 | −23.5 ± 0.3 | 40.7 ± 5.9 |
| DPV | 186.6 ± 1.6 | 0.079 | −24.9 ±1.3 | 70.1 ± 4.4 |
| GRFT/DPV | 184.3 ± 1.0 | 0.063 | −23.4 ± 0.3 | 45.9 ± 13.7 (GRFT) 69.4 ± 5.1 (DPV) |
| Drug | Alone (nM) | |
|---|---|---|
| Unformulated | NP-GRFT/NP-DPV | |
| DPV | 4.7 ± 2.9 | 3.6 ± 2.9 |
| GRFT | 0.5 ± 0.3 | 0.8 ± 0.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Li, J.; Patel, S.K.; Palmer, K.E.; Devlin, B.; Rohan, L.C. Design of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles for Vaginal Co-Delivery of Griffithsin and Dapivirine and Their Synergistic Effect for HIV Prophylaxis. Pharmaceutics 2019, 11, 184. https://doi.org/10.3390/pharmaceutics11040184
Yang H, Li J, Patel SK, Palmer KE, Devlin B, Rohan LC. Design of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles for Vaginal Co-Delivery of Griffithsin and Dapivirine and Their Synergistic Effect for HIV Prophylaxis. Pharmaceutics. 2019; 11(4):184. https://doi.org/10.3390/pharmaceutics11040184
Chicago/Turabian StyleYang, Haitao, Jing Li, Sravan Kumar Patel, Kenneth E. Palmer, Brid Devlin, and Lisa C. Rohan. 2019. "Design of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles for Vaginal Co-Delivery of Griffithsin and Dapivirine and Their Synergistic Effect for HIV Prophylaxis" Pharmaceutics 11, no. 4: 184. https://doi.org/10.3390/pharmaceutics11040184
APA StyleYang, H., Li, J., Patel, S. K., Palmer, K. E., Devlin, B., & Rohan, L. C. (2019). Design of Poly(lactic-co-glycolic Acid) (PLGA) Nanoparticles for Vaginal Co-Delivery of Griffithsin and Dapivirine and Their Synergistic Effect for HIV Prophylaxis. Pharmaceutics, 11(4), 184. https://doi.org/10.3390/pharmaceutics11040184