Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies



Abstract
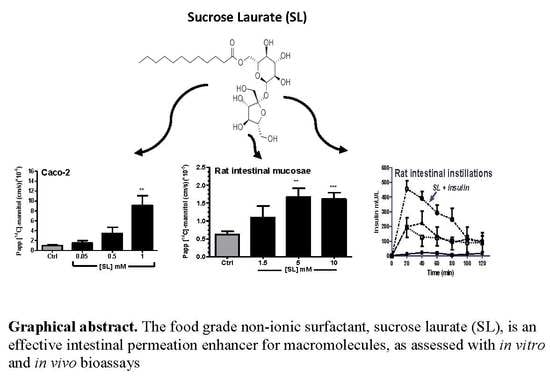
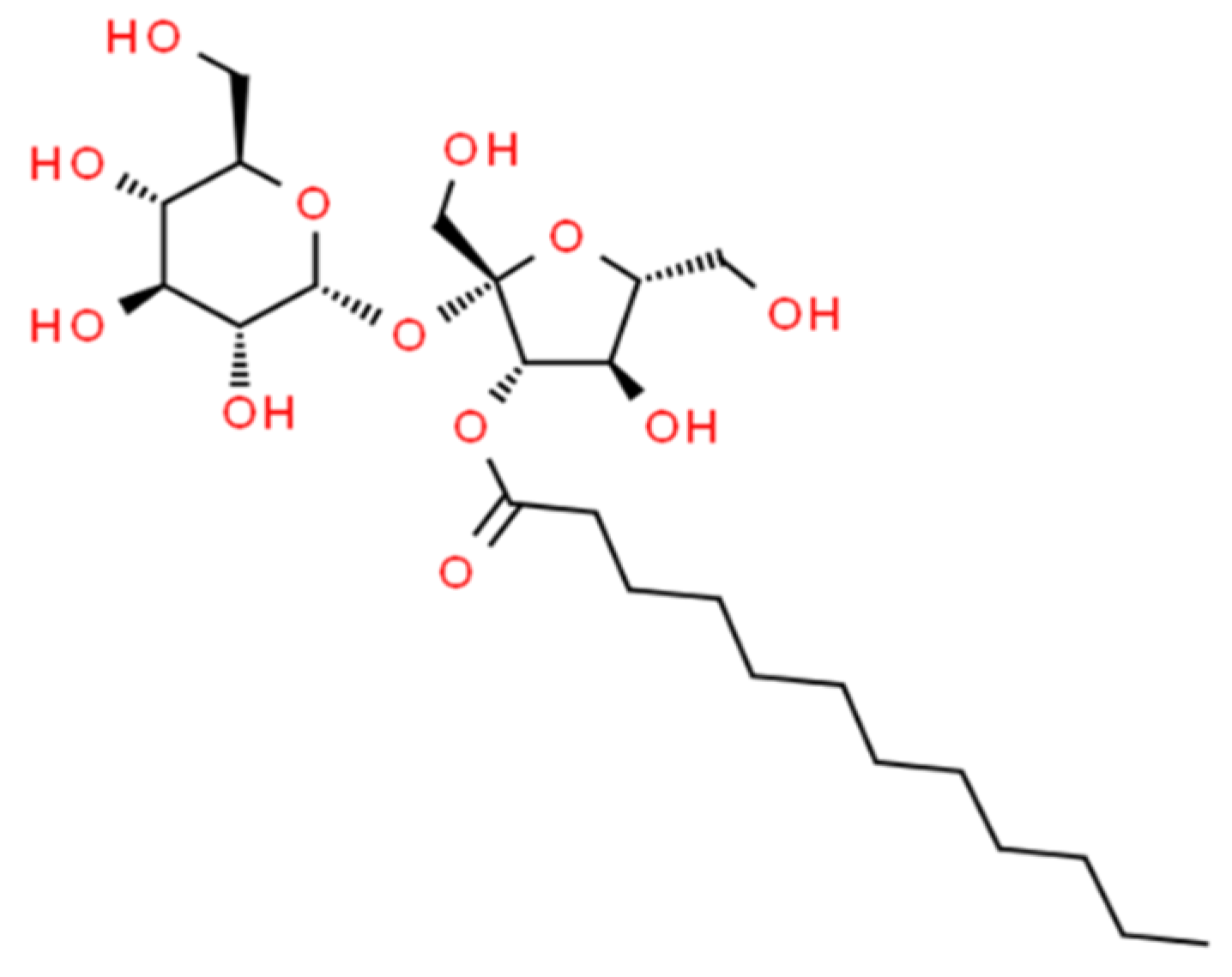
:
1. Introduction
2. Materials and Methods
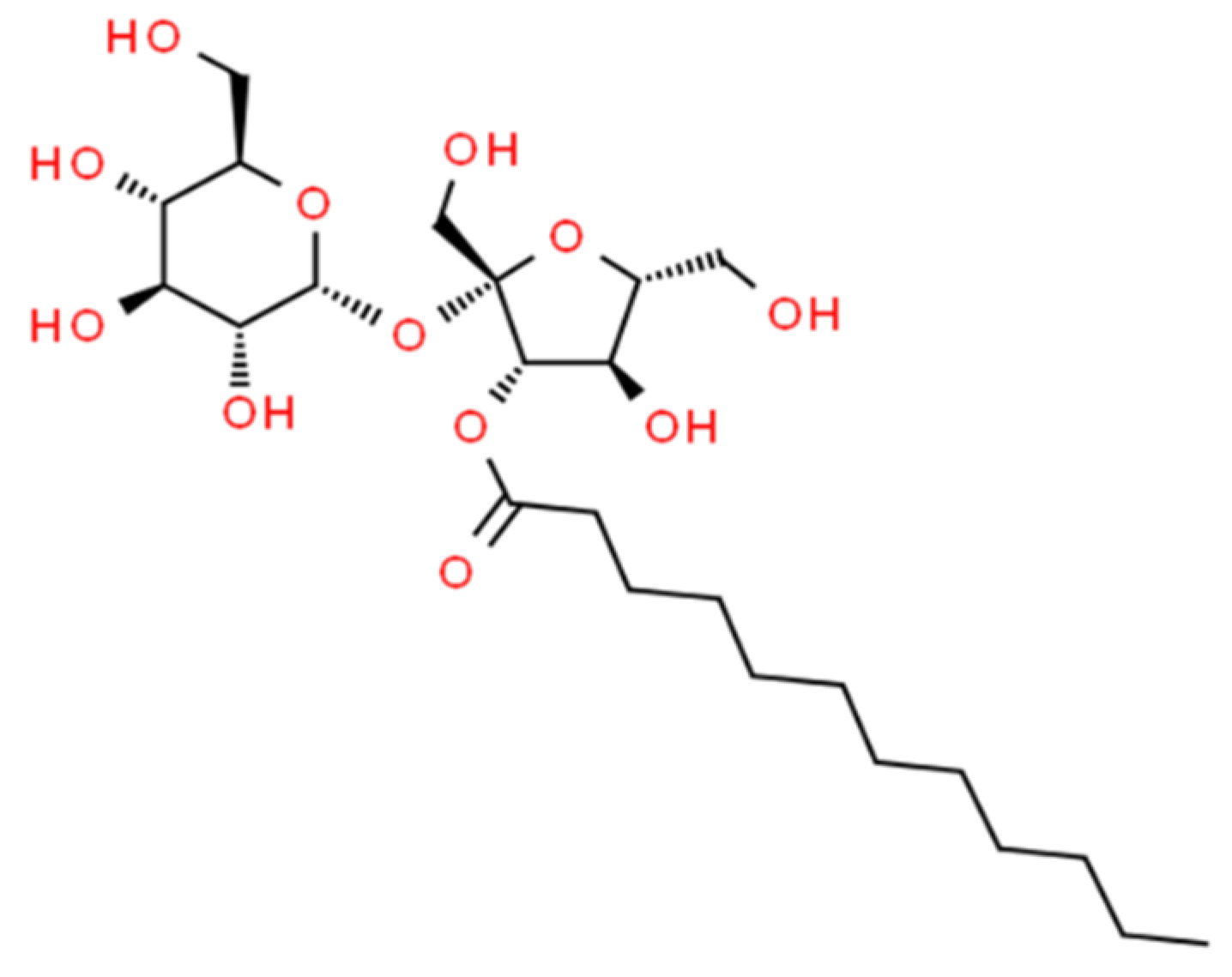
2.1. Materials
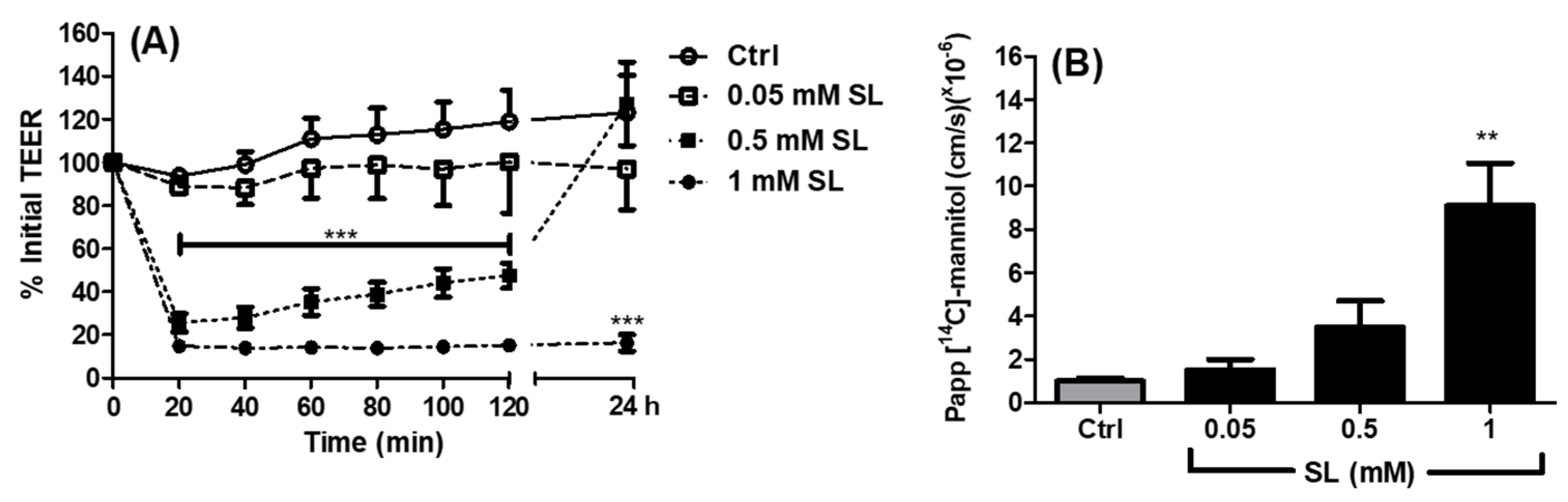
2.2. Apparent Permeability Coefficient (Papp) of 14C-Mannitol across Caco-2 Monolayers Exposed to SL
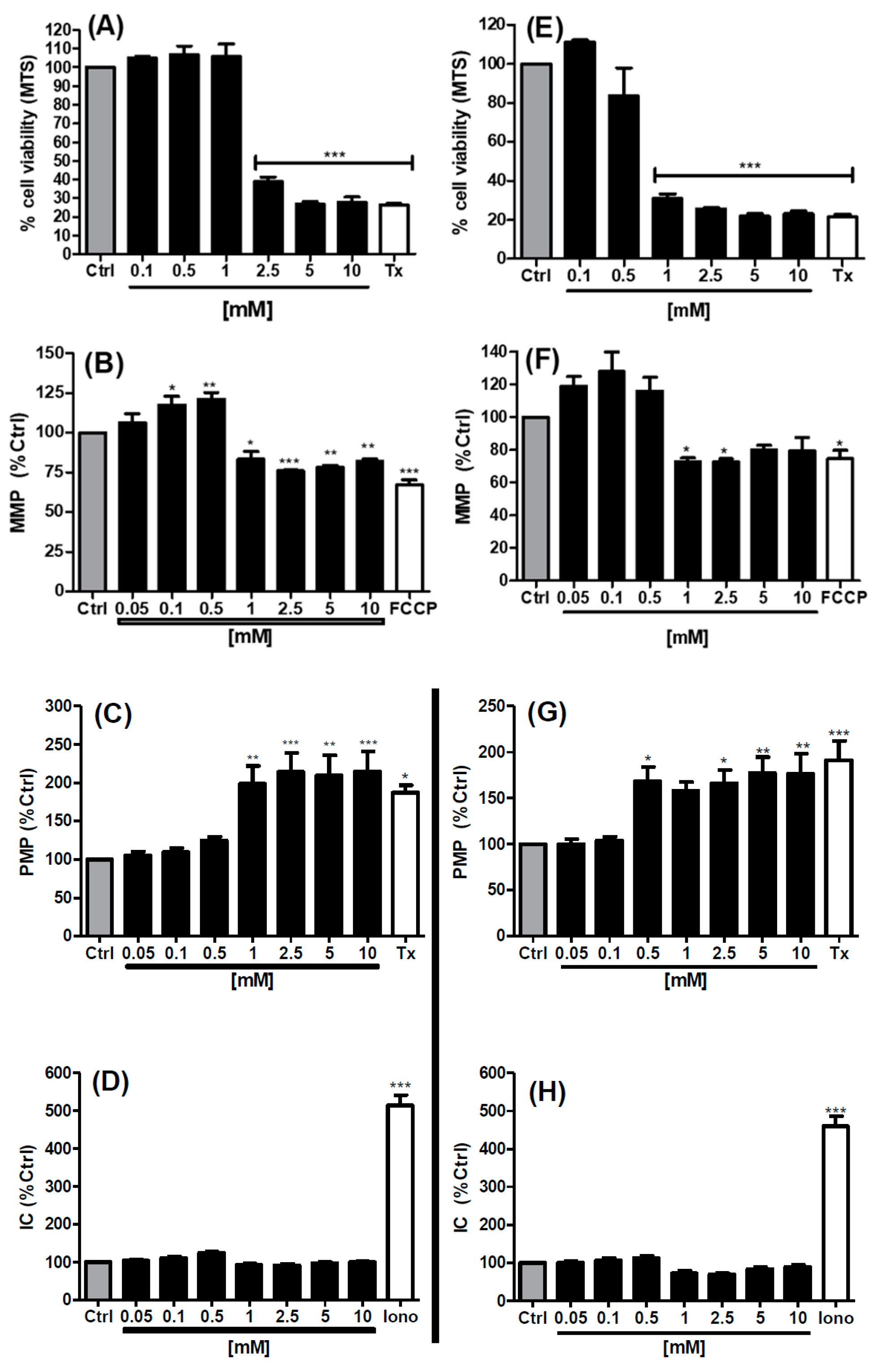
2.3. MTS Assay of Caco-2 Cells Exposed to SL
2.4. Immunofluorescence of Tight Junction Proteins in Caco-2 Cells Exposed to SL
2.5. High Content Analysis on Caco-2 Cells Exposed to SL
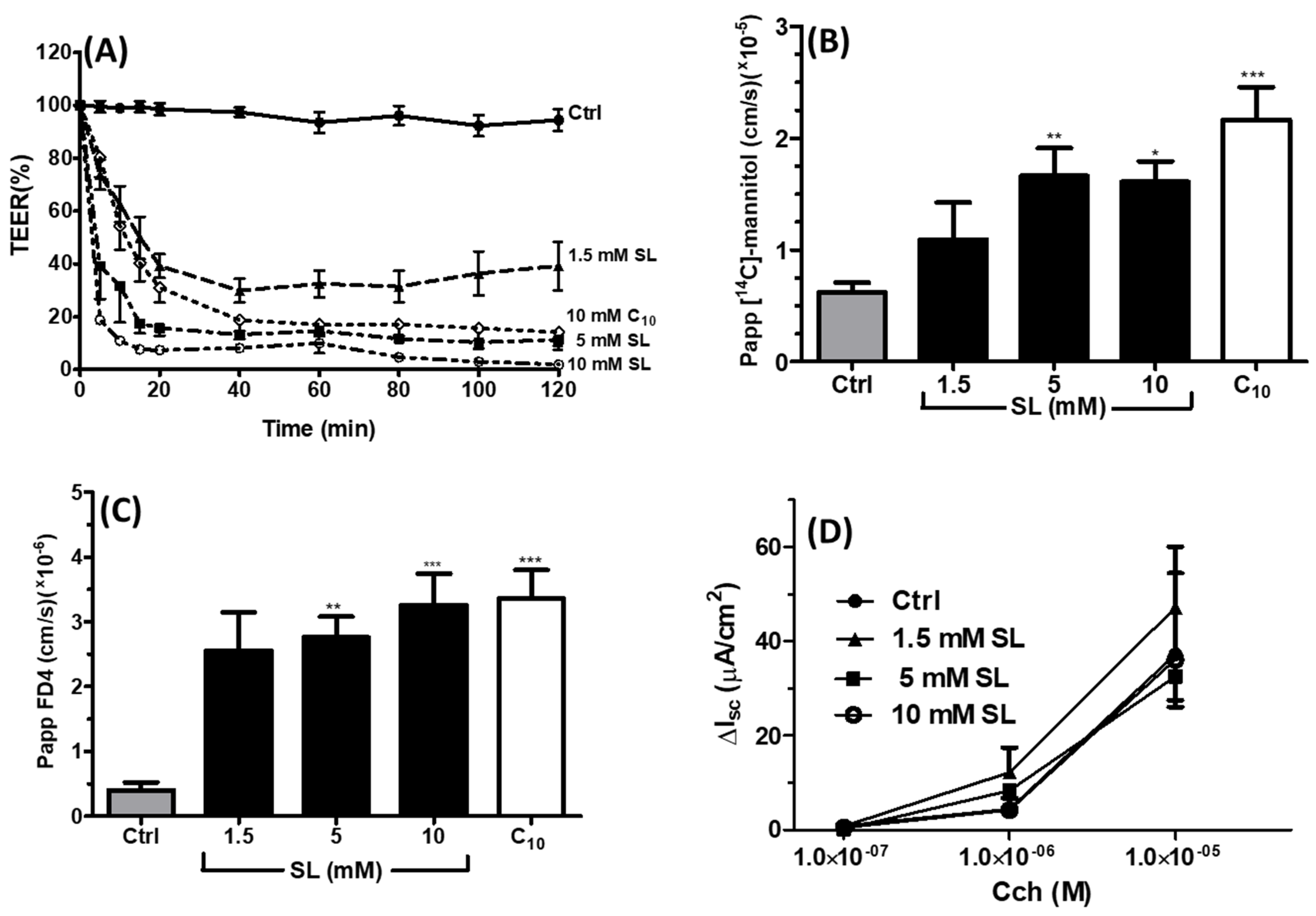
2.6. [14C]-Mannitol and FD4 Fluxes across Isolated Rat Colonic Mucosae Exposed to SL
2.7. Rat Jejunal and Colonic In Situ Instillations
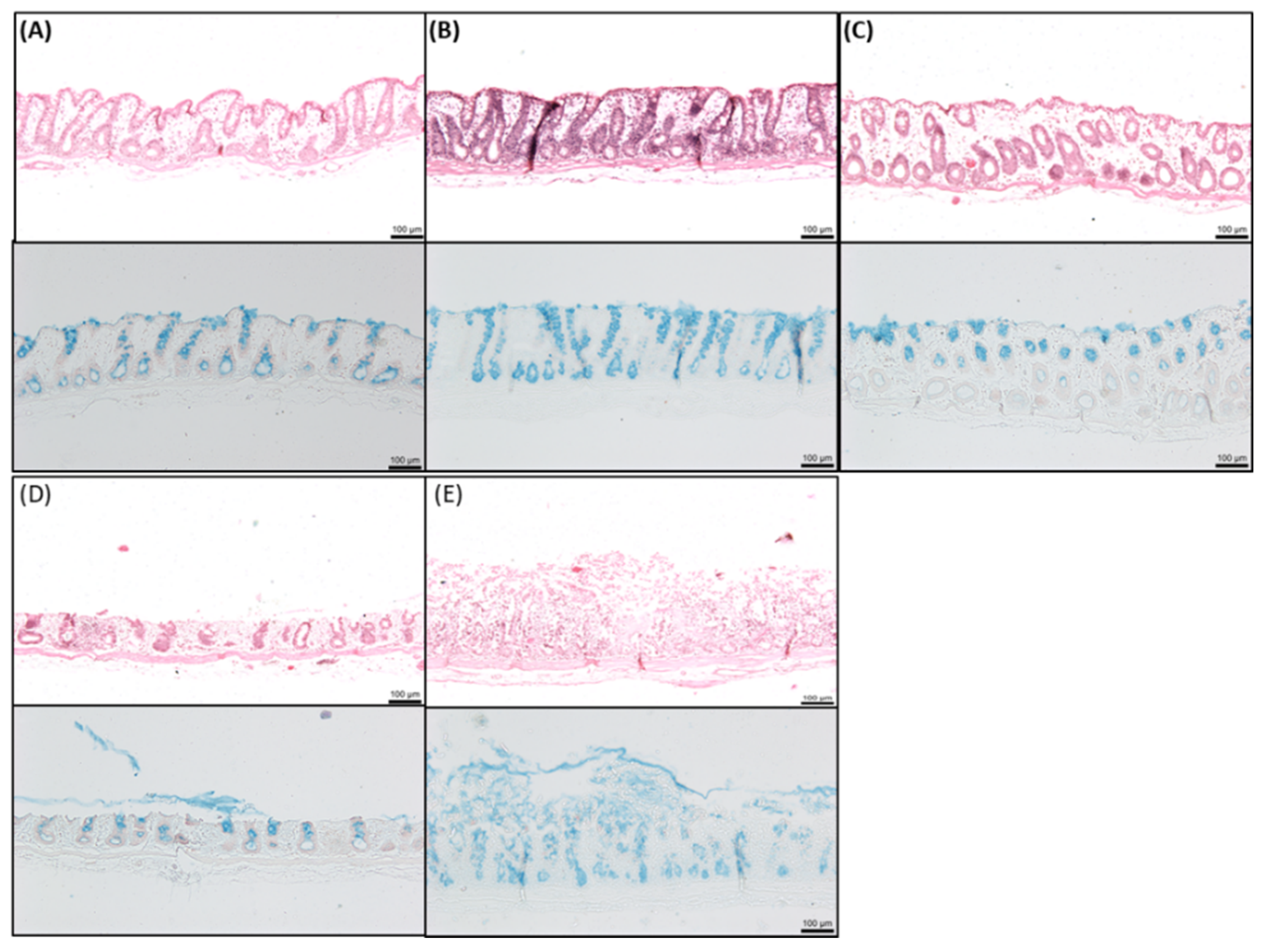
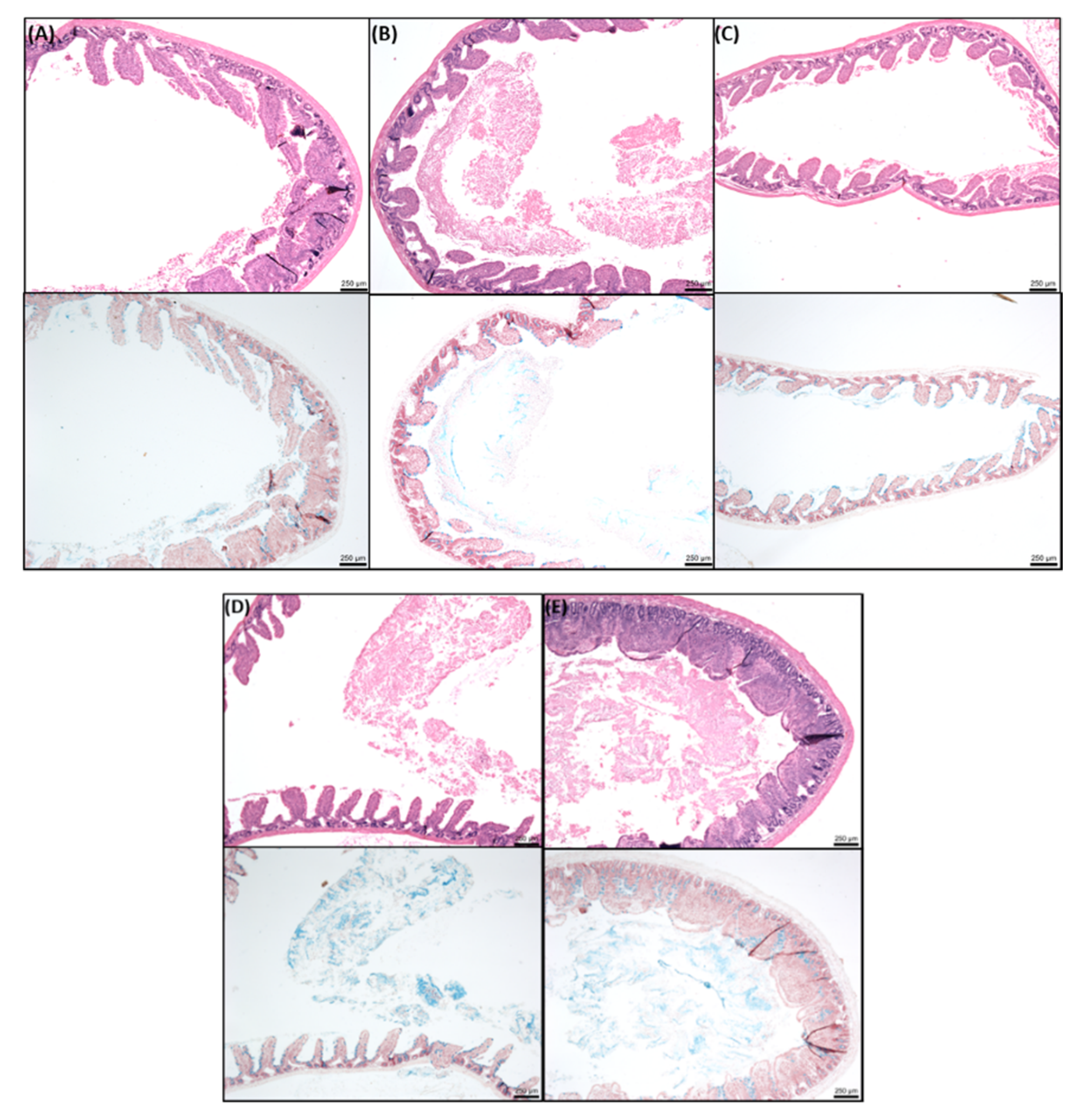
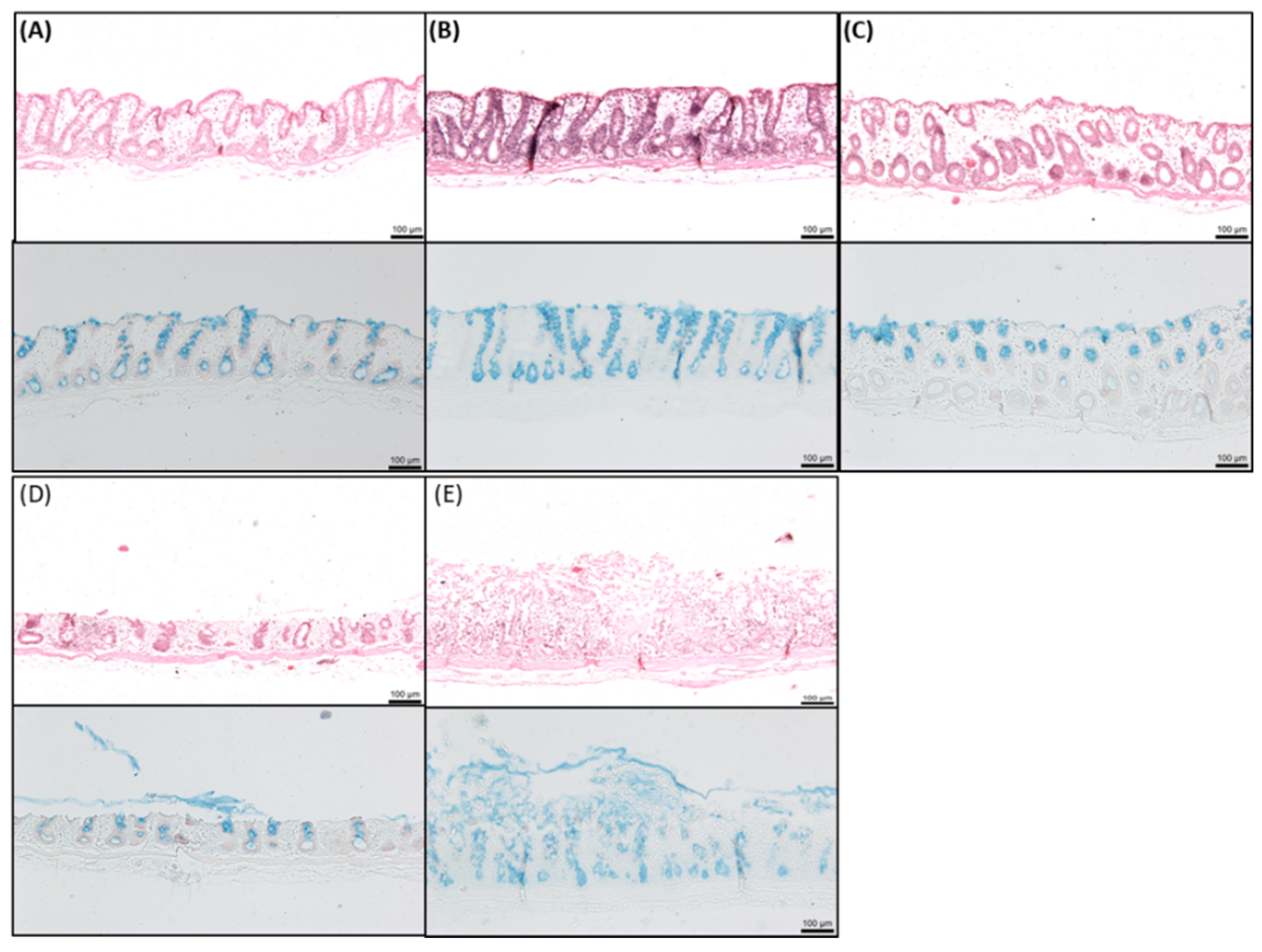
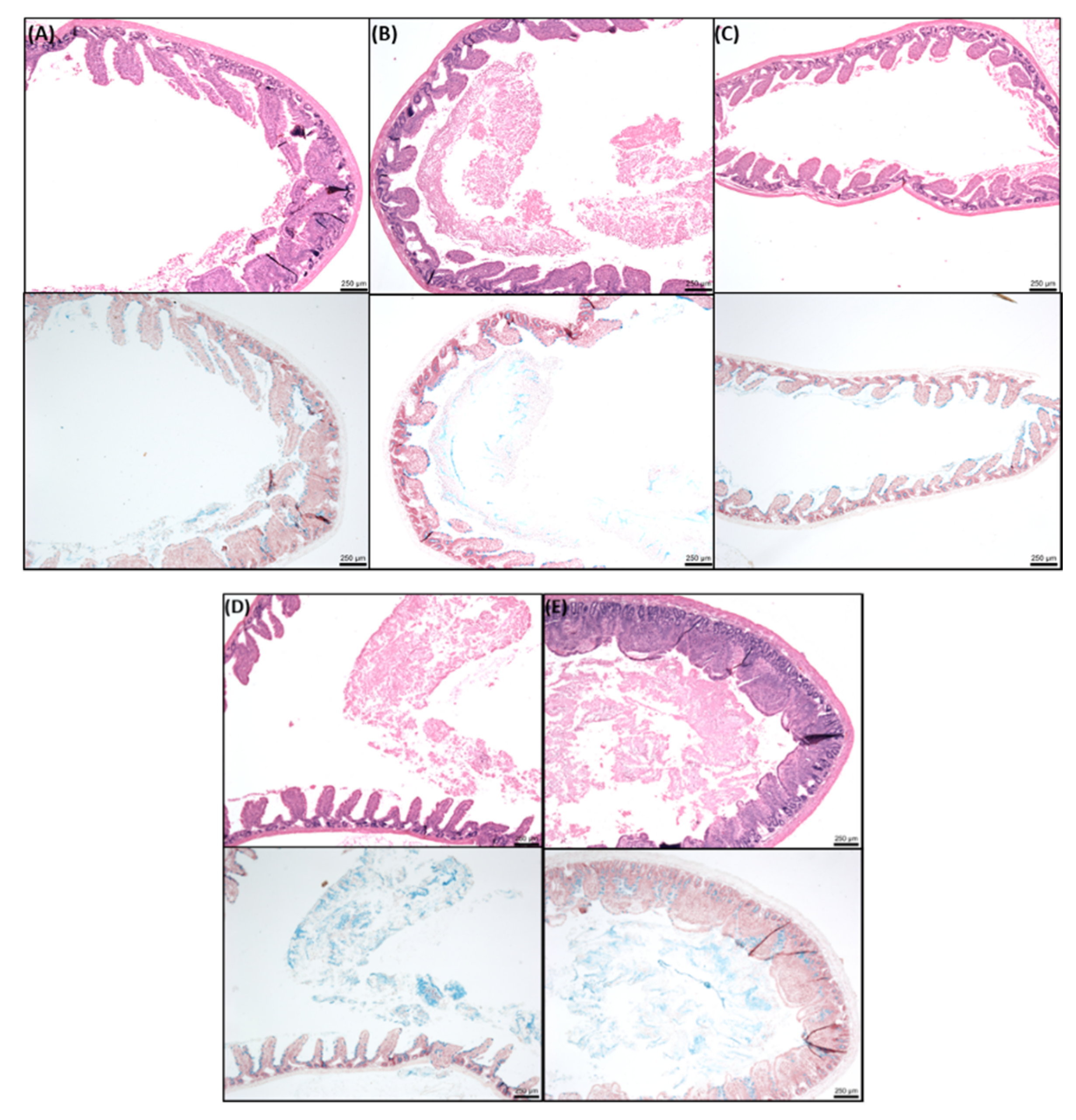
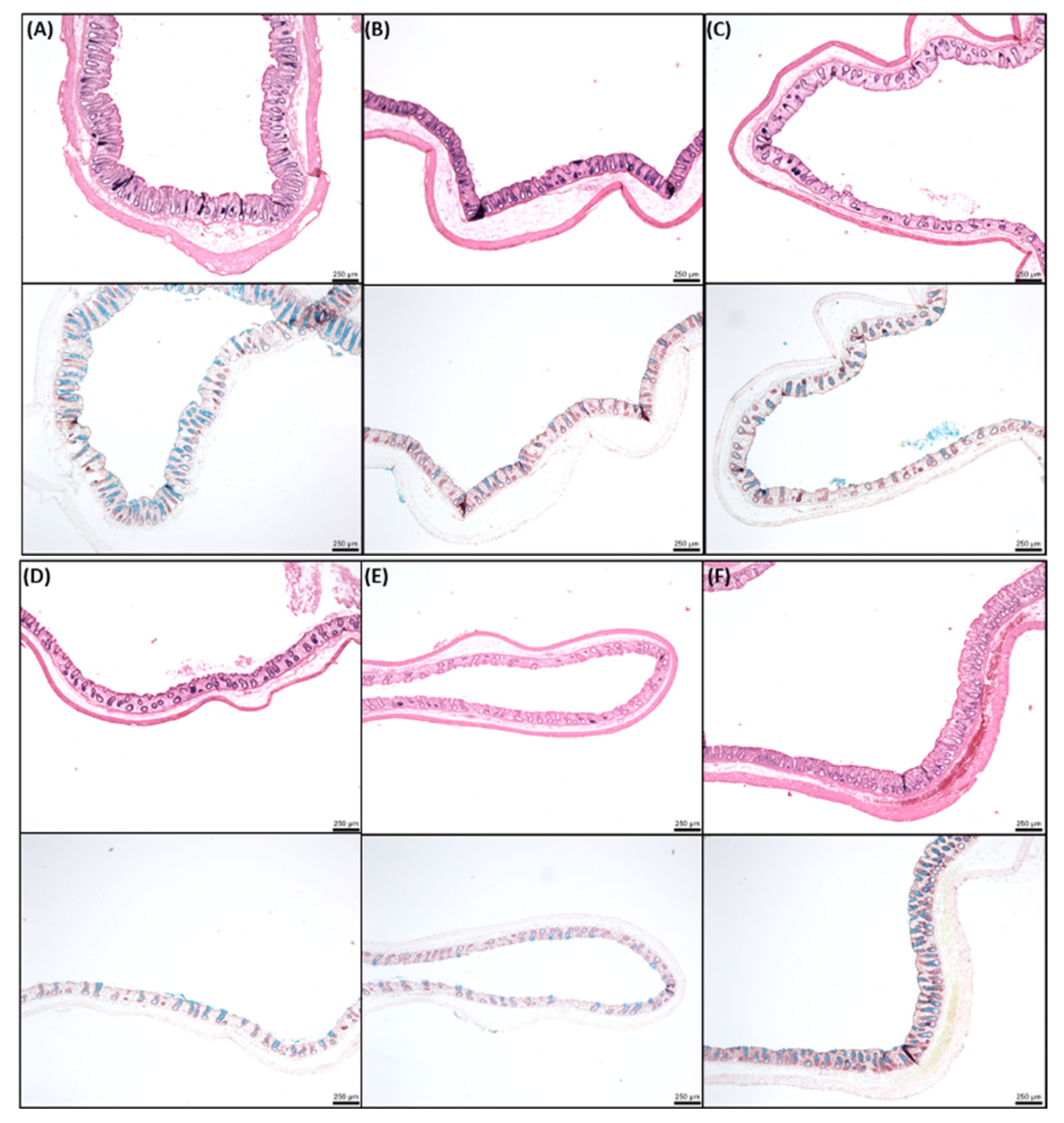
2.8. Histology of Rat Intestinal Tissue Mucosae Exposed to SL
2.9. Statistical Analysis
3. Results
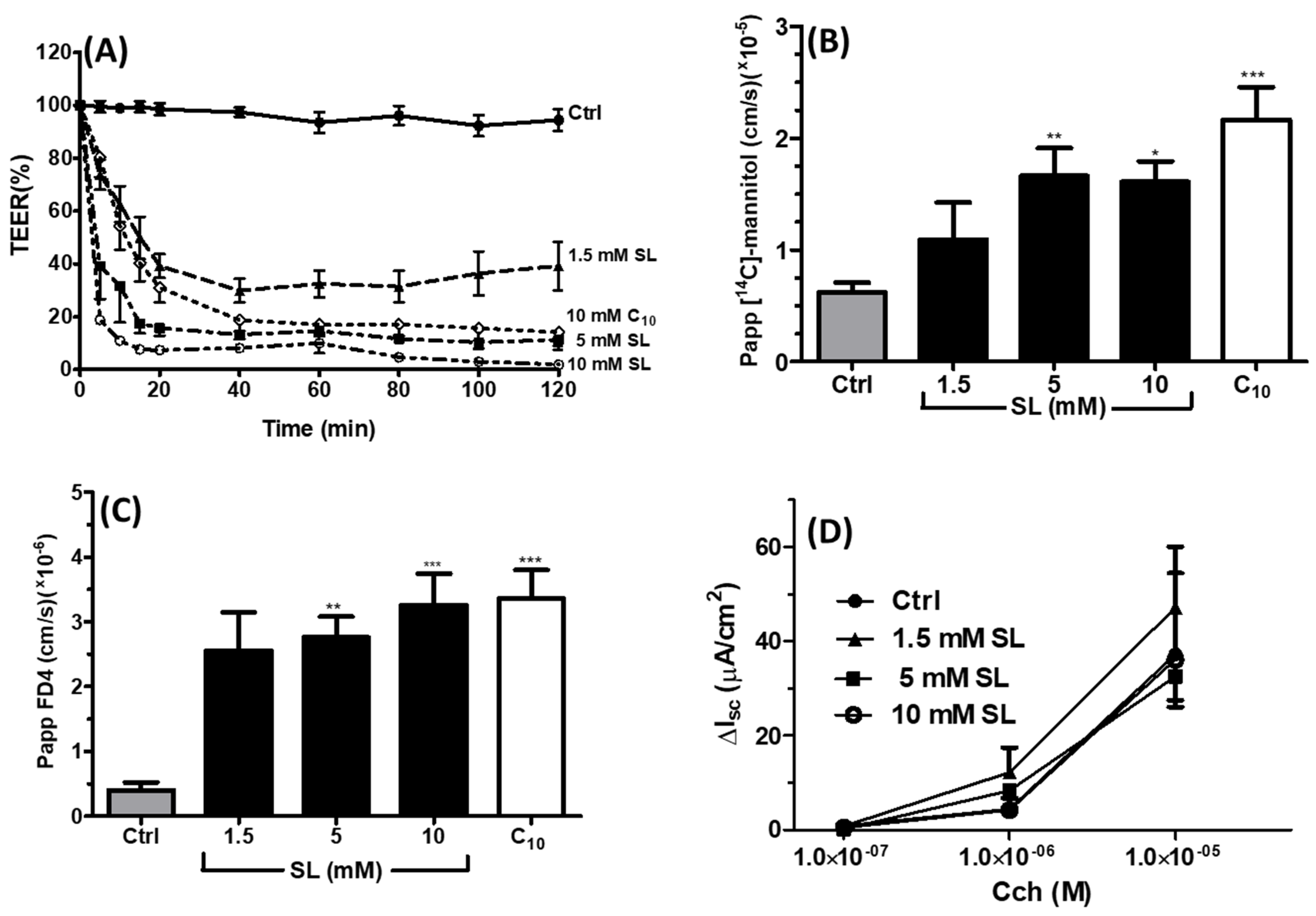
3.1. Effects of SL on TEER and Permeability across Caco-2 Monolayers
3.2. Effect of SL on ZO-1 Immunofluorescence in Caco-2 Cells
3.3. MTS and High Content Analysis (HCA) Studies in Caco-2 Cells
3.4. Effects of SL on Rat Colonic Tissue Mucosae Mounted in Ussing Chambers
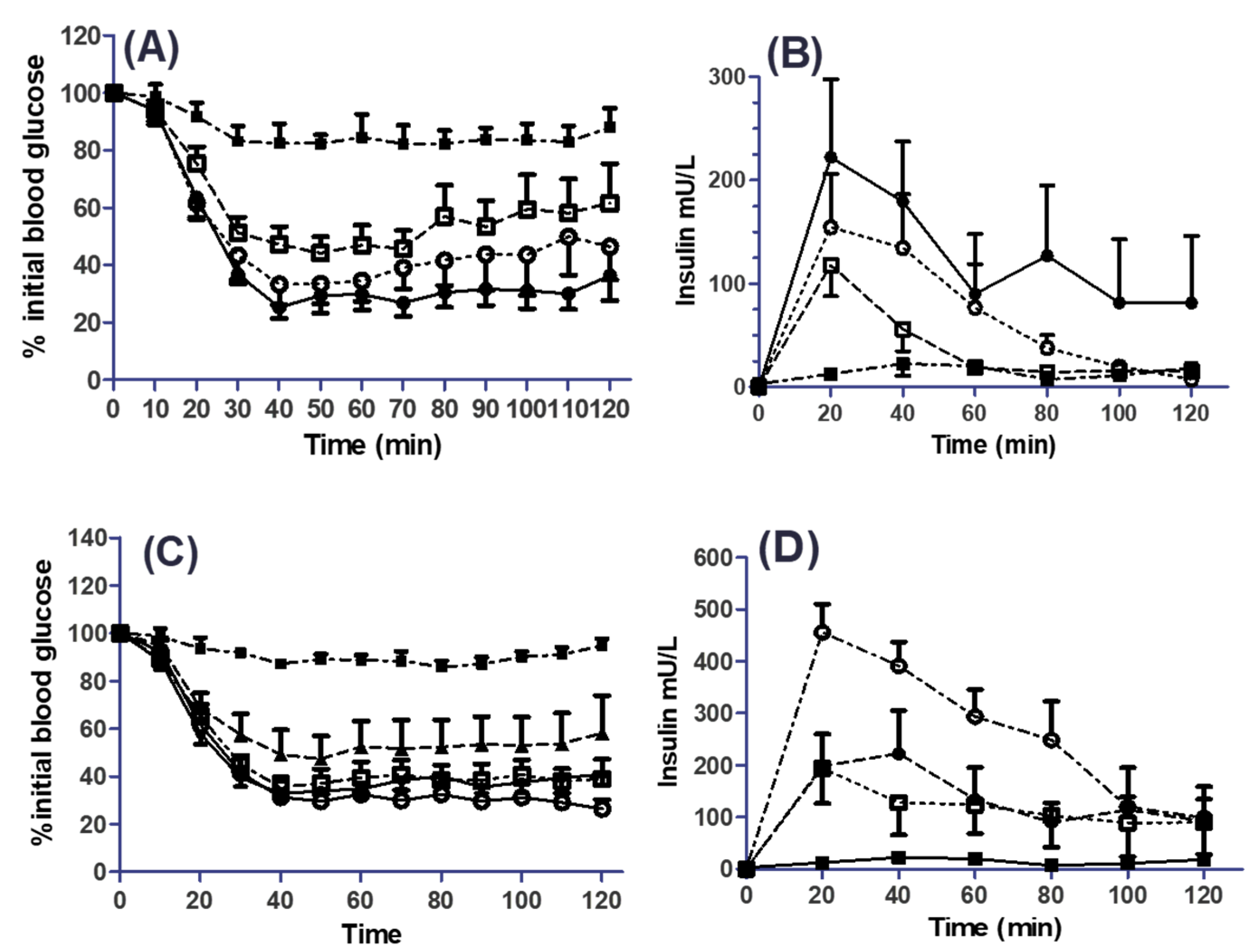
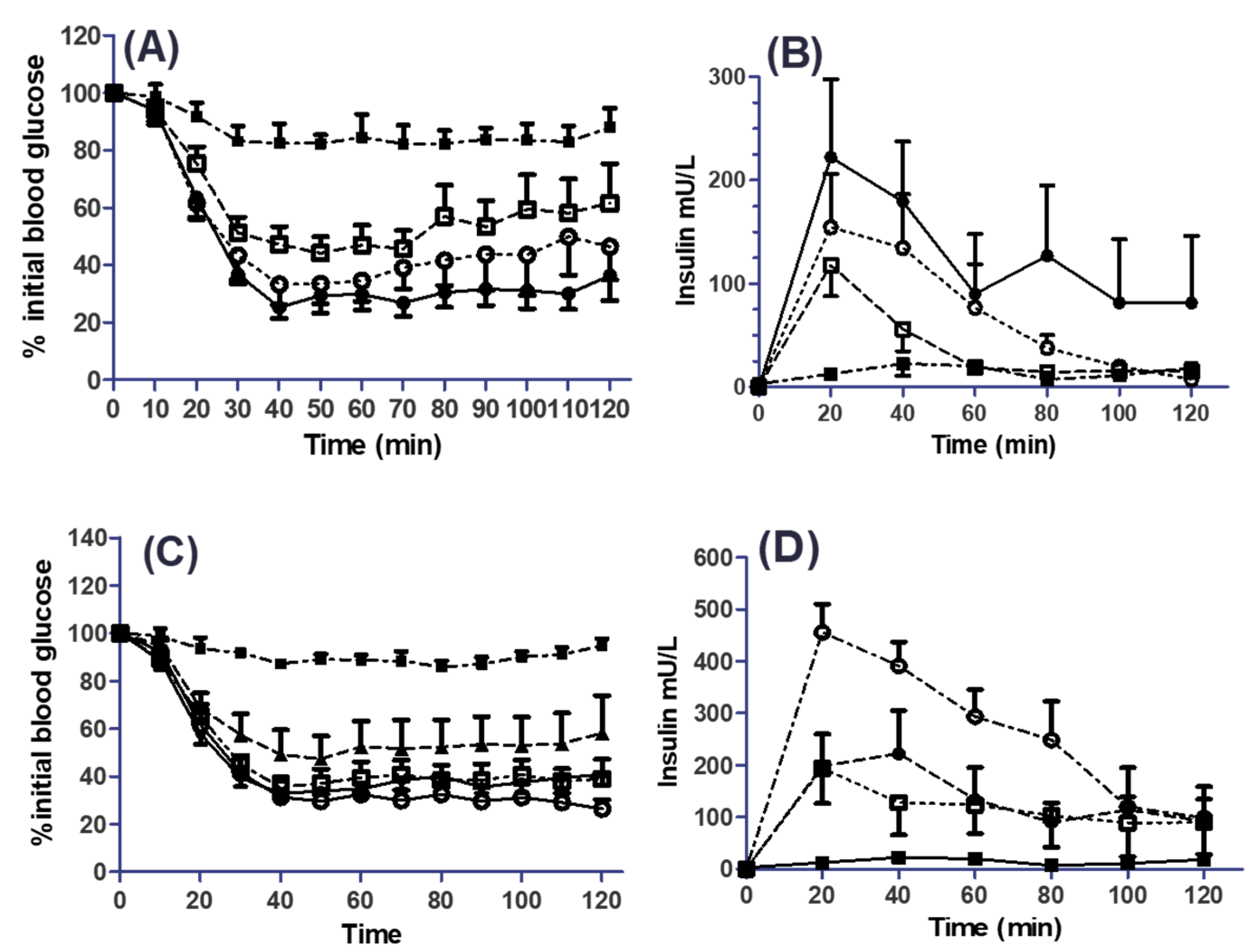
3.5. Effects of SL on the Pharmacodynamics (PD) and Pharmacokinetics (PK) of Co-Administered Insulin in Jejunal and Colonic Instillations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aguirre, T.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106 Pt B, 223–241. [Google Scholar] [CrossRef]
- Durán-Lobato, M.; Niu, Z.; Alonso, M.J. Oral delivery of biologics for precision medicine. Adv. Mater. 2019, 1901935. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106 Pt B, 277–319. [Google Scholar] [CrossRef]
- McCartney, F.; Gleeson, J.P.; Brayden, D.J. Safety concerns over the use of intestinal permeation enhancers: A mini-review. Tissue Barriers 2016, 4, e1176822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twarog, C.; Fattah, S.; Heade, J.; Maher, S.; Fattal, E.; Brayden, D.J. Intestinal permeation enhancers for oral delivery of macromolecules: A comparison between salcaprozate sodium (SNAC) and sodium caprate (C10). Pharmaceutics 2019, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Allison, D.; Birkenfeld, A.L.; Blicher, T.M.; Deenadayalan, S.; Jacobsen, J.B.; Serusclat, P.; Violante, R.; Watada, H.; Davies, M. Effect of additional oral semaglutide vs sitagliptin on glycated hemoglobin in adults with Type 2 diabetes uncontrolled with metformin alone or with sulfonylurea: The PIONEER 3 Randomized Clinical Trial. JAMA 2019, 321, 1466–1480. [Google Scholar] [CrossRef]
- Halberg, I.B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Mörschel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: A randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 179–188. [Google Scholar] [CrossRef]
- Almansour, K.; Taverner, A.; Turner, J.R.; Eggleston, I.M.; Mrsny, R.J. An intestinal paracellular pathway biased toward positively-charged macromolecules. J. Control. Release 2018, 288, 111–125. [Google Scholar] [CrossRef]
- Watari, A.; Kodaka, M.; Matsuhisa, K.; Sakamoto, Y.; Hisaie, K.; Kawashita, N.; Takagi, T.; Yamagishi, Y.; Suzuki, H.; Tsujino, H.; et al. Identification of claudin-4 binder that attenuates tight junction barrier function by TR-FRET-based screening assay. Sci. Rep. 2017, 7, 14514. [Google Scholar] [CrossRef] [Green Version]
- Maher, S.; Leonard, T.W.; Jacobsen, J.; Brayden, D.J. Safety and efficacy of sodium caprate in promoting oral drug absorption: From in vitro to the clinic. Adv. Drug Deliv. Rev. 2009, 61, 1427–1449. [Google Scholar] [CrossRef]
- Lindmark, T.; Kimura, Y.; Artursson, P. Absorption enhancement through intracellular regulation of tight junction permeability by medium chain fatty acids in Caco-2 cells. J. Pharmacol. Exp. Ther. 1998, 284, 362–369. [Google Scholar] [PubMed]
- Brayden, D.J.; Gleeson, J.; Walsh, E.G. A head-to-head multi-parametric high content analysis of a series of medium chain fatty acid intestinal permeation enhancers in Caco-2 cells. Eur. J. Pharm. Biopharm. 2014, 88, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Molinier, V.; Fenet, B.; Fitremann, J.; Bouchu, A.; Queneau, Y. PFGSE–NMR study of the self-diffusion of sucrose fatty acid monoesters in water. J. Colloid Interface Sci. 2005, 286, 360–368. [Google Scholar] [CrossRef] [PubMed]
- United States Food and Drug Administration (FDA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=172.859 (accessed on 3 July 2019).
- Chemspider. Available online: http://www.chemspider.com/Chemical-Structure.20121381.html (accessed on 29 October 2019).
- Szűts, A.; Szabó-Révész, P. Sucrose esters as natural surfactants in drug delivery systems—A mini-review. Int. J. Pharm. 2012, 433, 1–9. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. Scientific opinion on the exposure assessment of sucrose esters of fatty acids (E 473) from its use as food additive. EFSA J. 2012, 10, 2658. [Google Scholar] [CrossRef]
- Younes, M.; Aggett, P.; Aguilar, F.; Crebelli, R.; Dusemund, B.; Filipič, M.; Frutos, M.J.; Galtier, P.; Gott, D.; Gundert-Remy, U.; et al. Refined exposure assessment of sucrose esters of fatty acids (E 473) from its use as a food additive. EFSA J. 2018, 16, e05087. [Google Scholar] [Green Version]
- Marciello, M.; Mateo, C.; Guisán, J.M. Full enzymatic hydrolysis of commercial sucrose laurate by immobilized-stabilized derivatives of lipase from Thermomyces lanuginosa. Colloids Surf. B Biointerfaces 2011, 84, 556–560. [Google Scholar] [CrossRef]
- Christiansen, A.; Backensfeld, T.; Weitschies, W. Effects of non-ionic surfactants on in vitro triglyceride digestion and their susceptibility to digestion by pancreatic enzymes. Eur. J. Pharm. Sci. 2010, 41, 376–382. [Google Scholar] [CrossRef]
- Phan, T.N.Q.; Shahzadi, I.; Bernkop-Schnürch, A. Hydrophobic ion-pairs and lipid-based nanocarrier systems: The perfect match for delivery of BCS class 3 drugs. J. Control. Release 2019, 304, 146–155. [Google Scholar] [CrossRef]
- Paul, S.; Heng, P.W.S.; Chan, L.W. Improvement in dissolution rate and photodynamic efficacy of chlorin e6 by sucrose esters as drug carrier in nanosuspension formulation: Optimisation and in vitro characterisation. J. Pharm. Pharmacol. 2018, 70, 1152–1163. [Google Scholar] [CrossRef]
- Okamoto, H.; Sakai, T.; Tokuyama, C.; Danjo, K. Sugar Ester J-1216 enhances percutaneous permeation of ionized lidocaine. J. Pharm. Sci. 2011, 100, 4482–4490. [Google Scholar] [CrossRef] [PubMed]
- Kürti, L.; Veszelka, S.; Bocsik, A.; Dung, N.T.K.; Ózsvári, B.; Puskás, L.G.; Kittel, Á.; Szabó-Révész, P.; Deli, M.A. The effect of sucrose esters on a culture model of the nasal barrier. Toxicol. In Vitro 2012, 26, 445–454. [Google Scholar]
- Kiss, L.; Hellinger, É.; Pilbat, A.M.; Kittel, Á.; Török, Z.; Füredi, A.; Szakacs, G.; Veszelka, S.; Sipos, P.; Ózsvári, B.; et al. Sucrose esters increase drug penetration, but do noti P-glycoprotein in Caco-2 intestinal epithelial cells. J. Pharm. Sci. 2014, 103, 3107–3119. [Google Scholar] [CrossRef] [PubMed]
- Glynn, A.; Igra, A.M.; Sand, S.; Ilbäck, N.G.; Hellenäs, K.E.; Rosén, J.; Aspenstrom-Fagerlund, B. Are additive effects of dietary surfactants on intestinal tight junction integrity an overlooked human health risk? –A mixture study on Caco-2 monolayers. Food Chem. Toxicol. 2017, 106, 314–323. [Google Scholar] [CrossRef]
- Maher, S.; Heade, J.; McCartney, F.; Waters, S.; Bleiel, S.B.; Brayden, D.J. Effects of surfactant-based permeation enhancers on mannitol permeability, histology, and electrogenic ion transport responses in excised rat colonic mucosae. Int. J. Pharm. 2018, 539, 11–22. [Google Scholar] [CrossRef]
- Alama, T.; Katayama, H.; Hirai, S.; Ono, S.; Kajiyama, A.; Kusamori, K.; Katsumi, H.; Sakane, T.; Yamamoto, A. Enhanced oral delivery of alendronate by sucrose fatty acids esters in rats and their absorption-enhancing mechanisms. Int. J. Pharm. 2016, 515, 476–489. [Google Scholar] [CrossRef]
- Yamamoto, A.; Katsumi, H.; Kusamori, K.; Sakane, T. Improvement of intestinal absorption of poorly absorbable drugs by various sugar esters. YAKUGAKU ZASSHI 2014, 134, 47–53. [Google Scholar] [CrossRef]
- Onishi, H.; Imura, Y.; Uchida, M.; Machida, Y. Enhancement potential of sucrose laurate (L-1695) on intestinal absorption of water-soluble high molecular weight compounds. Curr. Drug Deliv. 2012, 9, 487–494. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ragnarsson, E.G.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef]
- Brayden, D.J.; Maher, S.; Bahar, B.; Walsh, E. Sodium caprate-induced increases in intestinal permeability and epithelial damage are prevented by misoprostol. Eur. J. Pharm. Biopharm. 2015, 94, 194–206. [Google Scholar] [CrossRef]
- Gleeson, J.P.; Heade, J.; Ryan, S.M.; Brayden, D.J. Stability, toxicity and intestinal permeation enhancement of two food-derived antihypertensive tripeptides, Ile-Pro-Pro and Leu-Lys-Pro. Peptides 2015, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Semba, S.; Khan, R.I.; Bochimoto, H.; Watanabe, T.; Fujiya, M.; Kohgo, Y.; Liu, Y.; Taniguchi, T. Focal adhesion kinase regulates intestinal epithelial barrier function via redistribution of tight junction. Biochim. Biophys. Acta 2013, 1832, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, S.; Kennelly, R.; Bzik, V.A.; Baird, A.W.; Wang, X.; Winter, D.; Brayden, D.J. Evaluation of intestinal absorption enhancement and local mucosal toxicity of two promoters. I. Studies in isolated rat and human colonic mucosae. Eur. J. Pharm. Sci. 2009, 38, 291–300. [Google Scholar] [CrossRef] [PubMed]
- McCartney, F.; Jannin, V.; Chevrier, S.; Boulghobra, H.; Hristov, D.R.; Ritter, N.; Miolane, C.; Chavant, Y.; Demarne, F.; Brayden, D.J. Labrasol® is an efficacious intestinal permeation enhancer across rat intestine: Ex vivo and in vivo rat studies. J. Control. Release 2019, 310, 115–126. [Google Scholar] [CrossRef]
- Brayden, D.J.; Bzik, V.A.; Lewis, A.L.; Illum, L. CriticalSorb™ promotes permeation of flux markers across isolated rat intestinal mucosae and Caco-2 monolayers. Pharm. Res. 2012, 29, 2543–2554. [Google Scholar] [CrossRef]
- Marusic, M.; Zupancic, T.; Hribar, G.; Komel, R.; Anderluh, G.; Caserman, S. The Caco-2 cell culture model enables sensitive detection of enhanced protein permeability in the presence of N-decyl-beta-d-maltopyranoside. New Biotechnol. 2013, 30, 507–515. [Google Scholar] [CrossRef]
- Petersen, S.B.; Nolan, G.; Maher, S.; Rahbek, U.L.; Guldbrandt, M.; Brayden, D.J. Evaluation of alkylmaltosides as intestinal permeation enhancers: Comparison between rat intestinal mucosal sheets and Caco-2 monolayers. Eur. J. Pharm. Sci. 2012, 47, 701–712. [Google Scholar] [CrossRef]
- Ungell, A.L.; Nylander, S.; Bergstrand, S.; Sjöberg, A.; Lennernäs, H. Membrane transport of drugs in different regions of the intestinal tract of the rat. J. Pharm. Sci. 1998, 87, 360–366. [Google Scholar] [CrossRef]
- Dharmsathaphorn, K.; Pandol, S.J. Mechanism of chloride secretion induced by carbachol in a colonic epithelial cell line. J. Clin. Investig. 1986, 77, 348–354. [Google Scholar] [CrossRef]
- Gurram, A.K.; Deshpande, P.B.; Kar, S.S.; Nayak, U.Y.; Udupa, N.; Reddy, M.S. Role of components in the formation of self-microemulsifying drug delivery Systems. Indian J. Pharm. Sci. 2015, 77, 249–257. [Google Scholar]
- Christiansen, A.; Backensfeld, T.; Denner, K.; Weitschies, W. Effects of non-ionic surfactants on cytochrome P450-mediated metabolism in vitro. Eur. J. Pharm. Biopharm. 2011, 78, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Lindmark, T.; Nikkilä, T.; Artursson, P. Mechanisms of absorption enhancement by medium chain fatty acids in intestinal epithelial Caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 1995, 275, 958–964. [Google Scholar] [PubMed]
- Ayala-Bravo, H.A.; Quintanar-Guerrero, D.; Naik, A.; Kalia, Y.N.; Cornejo-Bravo, J.M.; Ganem-Quintanar, A. Effects of sucrose oleate and sucrose laureate on in vivo human stratum corneum permeability. Pharm. Res. 2003, 20, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, T.A.; Rosa, M.; Guterres, S.S.; Pohlmann, A.R.; Coulter, I.; Brayden, D.J. Investigation of coco-glucoside as a novel intestinal permeation enhancer in rat models. Eur. J. Pharm. Biopharm. 2014, 88, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Walsh, E. Efficacious intestinal permeation enhancement induced by the sodium salt of 10-undecylenic acid, a medium chain fatty acid derivative. AAPS J. 2014, 16, 1064–1076. [Google Scholar] [CrossRef]
- Kimura, Y.; Hosoda, Y.; Shima, M.; Adachi, S.; Matsuno, R. Physico-chemical properties of fatty acids for assessing the threshold concentration to enhance the absorption of a hdrophilic substance. Biosci. Biotechnol. Biochem. 1998, 62, 443–447. [Google Scholar] [CrossRef]
- Mine, Y.; Zhang, J.W. Surfactants enhance the tight-junction permeability of food allergens in human intestinal epithelial Caco-2 cells. Int. Arch. Allergy Immunol. 2003, 130, 135–142. [Google Scholar] [CrossRef]
- Yalkowsky, S.H.; Zografi, G. Some micellar properties of long-chain acyl carnitines. J. Colloid Interface Sci. 1970, 34, 525–533. [Google Scholar] [CrossRef]
- McCartney, F. Use of Permeation Enhancers and Nanotechnology to Increase Intestinal Peptide Permeability; University College Dublin: Dublin, Ireland, 2016. [Google Scholar]
- O’Brien, P.J. High-content analysis in toxicology: Screening substances for human toxicity potential, elucidating subcellular mechanisms and in vivo use as translational safety biomarkers. Basic Clin. Pharmacol. Toxicol. 2014, 115, 4–17. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Boil. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Bernas, T.; Dobrucki, J. Mitochondrial and non-mitochondrial reduction of MTT: Interaction of MTT with TMRE, JC-1, and NAO mitochondrial fluorescent probes. Cytometry 2002, 47, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, R.J.; Smith, P.A.; Stolnik, S. Exposure to a non-ionic surfactant induces a response akin to heat-shock apoptosis in intestinal epithelial cells: Implications for excipients safety. Mol. Pharm. 2019, 16, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Anguissola, S.; Garry, D.; Salvati, A.; O’Brien, P.J.; Dawson, K.A. High content analysis provides mechanisti on the pathways of toxicity induced by amine-modified polystyrene nanoparticles. PLoS ONE 2014, 9, e108025. [Google Scholar] [CrossRef]
- Turner, J.R.; Buschmann, M.M.; Romero-Calvo, I.; Sailer, A.; Shen, L. The role of molecular re-modelling in differential regulation of tight junction permeability. Semin. Cell Dev. Boil. 2014, 36, 204–212. [Google Scholar] [CrossRef]
- Swenson, E.S.; Milisen, W.B.; Curatolo, W. Intestinal permeability enhancement: Eficacy, acute local toxicity, and reversibility. Pharm. Res. 1994, 11, 1132–1142. [Google Scholar] [CrossRef]
- Kararli, T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995, 16, 351–380. [Google Scholar] [CrossRef]
- Gradauer, K.; Nishiumi, A.; Unrinin, K.; Higashino, H.; Kataoka, M.; Pedersen, B.L.; Buckley, S.T.; Yamashita, S. Interaction with mixed micelles in the intestine attenuates the permeation-enhancing potential of alkyl maltosides. Mol. Pharm. 2015, 12, 2245–2253. [Google Scholar] [CrossRef]
- Muranushi, N.; Mack, E.; Kim, S.W. The effects of fatty acids and their derivatives on the intestinal absorption of insulin in rats. Drug Dev. Ind. Pharm. 1993, 19, 929–941. [Google Scholar] [CrossRef]
- Petersen, S.B.; Nielsen, L.G.; Rahbek, U.L.; Guldbrandt, M.; Brayden, D.J. Colonic absorption of salmon calcitonin using tetradecyl maltoside (TDM) as a permeation enhancer. Eur. J. Pharm. Sci. 2013, 48, 726–734. [Google Scholar] [CrossRef]
- Maroni, A.; Zema, L.; Del Curto, M.D.; Foppoli, A.; Gazzaniga, A. Oral colon delivery of insulin with the aid of functional adjuvants. Adv. Drug Deliv. Rev. 2012, 64, 540–556. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.L.; Liu, F.; Basit, A.W. Colonic treatments and targets: Issues and opportunities. J. Drug Target. 2009, 17, 335–363. [Google Scholar] [CrossRef] [PubMed]
- Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 2014, 15, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Leonard, T.W.; Lynch, J.; McKenna, M.J.; Brayden, D.J. Promoting absorption of drugs in humans using medium-chain fatty acid-based solid dosage forms: GIPET™. Expert Opin. Drug Deliv. 2006, 3, 685–692. [Google Scholar] [CrossRef]
- Tuvia, S.; Pelled, D.; Marom, K.; Salama, P.; Levin-Arama, M.; Karmeli, I.; Idelson, G.H.; Landau, I.; Mamluk, R. A novel suspension formulation enhances intestinal absorption of macromolecules via transient and reversible transport mechanisms. Pharm. Res. 2014, 31, 2010–2021. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Cmax (mU/L) | Tmax (min) | AUC(0–120) (mU/L·min) | % F | % PA |
|---|---|---|---|---|---|
| 1 IU/kg (s.c.) | 111 ± 26 | 20 ± 0 | 6936 ± 2436 | - | - |
| 50 mM SL (i.j.) | 118 ± 74 | 20 ± 16 | 4616 ± 1171 | 1.3 | 1.7 |
| 100 mM SL (i.j.) | 156 ± 80 | 30 ± 13 | 8782 ± 3007 | 2.5 | 2.4 |
| 50 mM C10 (i.j.) | 276 ± 54 | 43 ± 12 | 15,210 ± 5831 | 4.4 | 2.8 |
| 100 mM C10 (i.j.) | 215 ± 57 | 43 ± 13 | 11,348 ± 4489 | 3.3 | 2.5 |
| 1 mM SL (i.c.) | 179 ± 30 | 73 ± 13 | 9382 ± 2400 | 2.7 | 1.9 |
| 10 mM SL (i.c.) | 241 ± 59 | 37 ± 13 | 14,679 ± 6068 | 4.2 | 2.5 |
| 25 mM SL (i.c.) | 459 ± 52 | 23 ± 3 | 30,954 ± 4087 | 8.9 | 2.7 |
| 50 mM SL (i.c.) | 216 ± 2 | 33 ± 7 | 14,456 ± 2167 | 4.2 | 2.9 |
| 10 mM C10 (i.c.) | 293 ± 30 | 47 ± 0 | 21,395 ± 6966 | 6.2 | 2.1 |
| 25 mM C10 (i.c.) | 304 ± 52 | 37 ± 7 | 16,138 ± 5476 | 4.7 | 2.6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCartney, F.; Rosa, M.; Brayden, D.J. Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies. Pharmaceutics 2019, 11, 565. https://doi.org/10.3390/pharmaceutics11110565
McCartney F, Rosa M, Brayden DJ. Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies. Pharmaceutics. 2019; 11(11):565. https://doi.org/10.3390/pharmaceutics11110565
Chicago/Turabian StyleMcCartney, Fiona, Mónica Rosa, and David J. Brayden. 2019. "Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies" Pharmaceutics 11, no. 11: 565. https://doi.org/10.3390/pharmaceutics11110565
APA StyleMcCartney, F., Rosa, M., & Brayden, D. J. (2019). Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies. Pharmaceutics, 11(11), 565. https://doi.org/10.3390/pharmaceutics11110565