Polydopamine-Based “Four-in-One” Versatile Nanoplatforms for Targeted Dual Chemo and Photothermal Synergistic Cancer Therapy

 ,
, 

Abstract
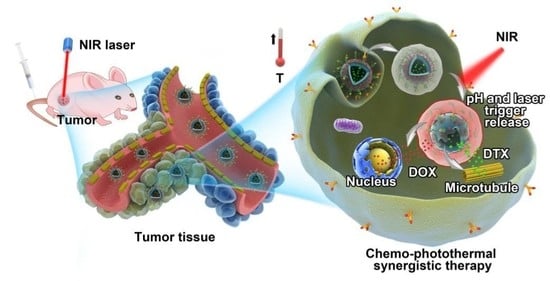
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Formulation of DTX-Loaded NPs
2.3. Surface Coating with PDA
2.4. Surface Conjugation of Ligands to PDA-coated NPs
2.5. Adsorption of DOX onto the PDA Layer
2.6. Characterization of NPs
2.7. Evaluation of Photothermal Effect
2.8. In Vitro Drug Release Study
2.9. Cellular Uptake of Fluorescent NPs
2.10. Cell Viability Study
2.11. Cellular Transport Mechanism Study
2.12. Pharmacokinetic Analysis
2.13. Animals and Tumor Model Establishment
2.14. In Vivo Imaging
2.15. In Vivo Antitumor Efficacy Study
2.16. Statistical Methodology
3. Results and Discussion
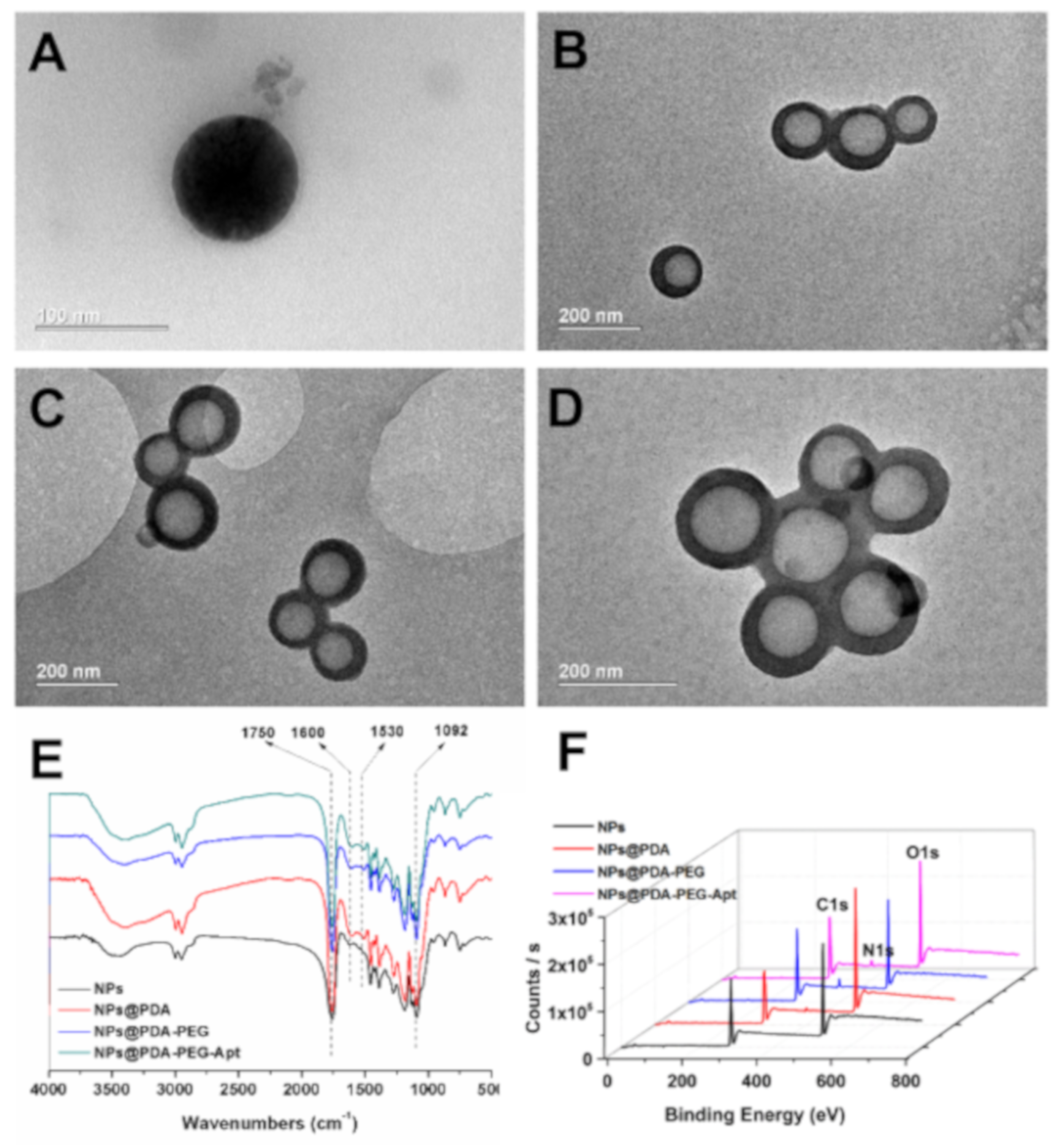
3.1. Preparation and Characterization of NPs
3.2. Photothermal Effect and Drug Release Profiles of NPs
3.3. Cellular Uptake of Fluorescent NPs
3.4. Effect of NPs on Cell Viability
3.5. Cellular Uptake Mechanism under NIR Irradiation
3.6. In Vivo Pharmacokinetics and Tumor Targeting
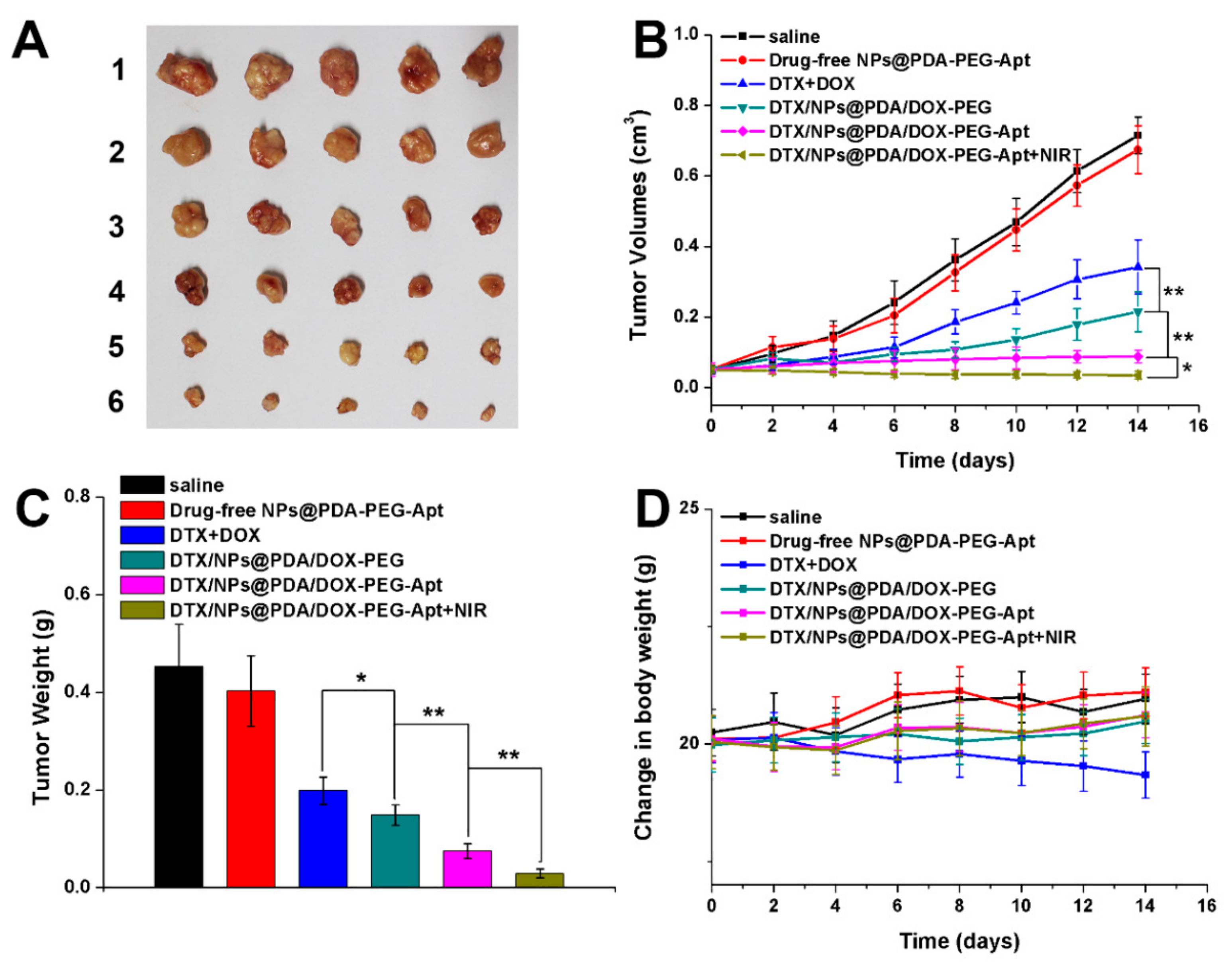
3.7. In Vivo Multimodal Antitumor Efficacy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2016, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.; Simon, P.; Loewer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schroers, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Grönroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Sengupta, S.; Eavarone, D.; Capila, I.; Zhao, G.; Watson, N.; Kiziltepe, T.; Sasisekharan, R. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature 2005, 436, 568–572. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Upadhyay, A.; Clark, S.L.; Fong, S.E.; Smith, V.; Koeppen, H.; Ross, S.; Polakis, P. Identification and immunotherapeutic targeting of antigens induced by chemotherapy. Nat. Biotechnol. 2006, 24, 205–209. [Google Scholar] [CrossRef]
- Greco, F.; Vicent, M.J. Combination therapy: Opportunities and challenges for polymer–drug conjugates as anticancer nanomedicines. Adv. Drug Deliv. Rev. 2009, 61, 1203–1213. [Google Scholar] [CrossRef]
- Dai, Y.; Yang, Z.; Cheng, S.; Wang, Z.; Zhang, R.; Zhu, G.; Wang, Z.; Yung, B.C.; Tian, R.; Jacobson, O.; et al. Toxic Reactive Oxygen Species Enhanced Synergistic Combination Therapy by Self-Assembled Metal-Phenolic Network Nanoparticles. Adv. Mater. 2018, 30, 1704877. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Cheng, W.; Zeng, X.; Mei, L.; Liu, G.; Deng, W. Folic Acid-Functionalized Black Phosphorus Quantum Dots for Targeted Chemo-Photothermal Combination Cancer Therapy. Pharmaceutics 2019, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Gao, H.; Zuo, Y.; Zeng, X.; Tao, W.; Tsai, H.; Mei, L. DACHPt-Loaded unimolecular micelles based on hydrophilic dendritic block copolymers for enhanced therapy of lung cancer. ACS Appl. Mater. Interfaces 2017, 9, 112–119. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, G.; Tao, W.; Ma, Y.; Zhang, X.; He, F.; Pan, J.; Mei, L.; Pan, G. A Drug-Self-Gated Mesoporous Antitumor Nanoplatform Based on pH-Sensitive Dynamic Covalent Bond. Adv. Funct. Mater. 2017, 27, 1605985. [Google Scholar] [CrossRef]
- Liu, G.; Tsai, H.-I.; Zeng, X.; Zuo, Y.; Tao, W.; Han, J.; Mei, L. Phosphorylcholine-based stealthy nanocapsules enabling tumor microenvironment-responsive doxorubicin release for tumor suppression. Theranostics 2017, 7, 1192–1203. [Google Scholar] [CrossRef]
- Liu, G.; Tsai, H.; Zeng, X.; Qi, J.; Luo, M.; Wang, X.; Mei, L.; Deng, W. Black phosphorus nanosheets-based stable drug delivery system via drug-self-stabilization for combined photothermal and chemo cancer therapy. Chem. Eng. J. 2019, 375, 121917. [Google Scholar] [CrossRef]
- Sun, X.; Pang, Z.; Ye, H.; Qiu, B.; Guo, L.; Li, J.; Ren, J.; Qian, Y.; Zhang, Q.; Chen, J.; et al. Co-delivery of pEGFP-hTRAIL and paclitaxel to brain glioma mediated by an angiopep-conjugated liposome. Biomaterials 2012, 33, 916–924. [Google Scholar] [CrossRef]
- Assanhou, A.G.; Li, W.; Zhang, L.; Xue, L.; Kong, L.; Sun, H.; Mo, R.; Zhang, C. Reversal of multidrug resistance by co-delivery of paclitaxel and lonidamine using a TPGS and hyaluronic acid dual-functionalized liposome for cancer treatment. Biomaterials 2015, 73, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Kolishetti, N.; Dhar, S.; Valencia, P.M.; Lin, L.Q.; Karnik, R.; Lippard, S.J.; Langer, R.; Farokhzad, O.C. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 17939–17944. [Google Scholar] [CrossRef] [PubMed]
- Elsabahy, M.; Wooley, K.L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012, 41, 2545–2561. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.E.; Carnes, E.C.; Phillips, G.K.; Padilla, D.; Durfee, P.N.; Brown, P.A.; Hanna, T.N.; Liu, J.W.; Phillips, B.; Carter, M.B.; et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat. Mater. 2011, 10, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Tarn, D.; Ashley, C.E.; Xue, M.; Carnes, E.C.; Zink, J.I.; Brinker, C.J. Mesoporous silica nanoparticle nanocarriers: Biofunctionality and biocompatibility. Acc. Chem. Res. 2013, 46, 792–801. [Google Scholar] [CrossRef]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ai, K.; Lu, L. Polydopamine and Its Derivative Materials: Synthesis and Promising Applications in Energy, Environmental, and Biomedical Fields. Chem. Rev. 2014, 114, 5057–5115. [Google Scholar] [CrossRef]
- Liu, S.; Pan, J.; Liu, J.; Ma, Y.; Qiu, F.; Mei, L.; Zeng, X.; Pan, G. Dynamically PEGylated and Borate-Coordination-Polymer-Coated Polydopamine Nanoparticles for Synergetic Tumor-Targeted, Chemo-Photothermal Combination Therapy. Small 2018, 14, 1703968. [Google Scholar] [CrossRef]
- Cheng, W.; Zeng, X.; Chen, H.; Li, Z.; Zeng, W.; Mei, L.; Zhao, Y. Versatile Polydopamine Platforms: Synthesis and Promising Applications for Surface Modification and Advanced Nanomedicine. ACS Nano 2019, 13, 8537–8565. [Google Scholar] [CrossRef]
- Park, J.; Brust, T.F.; Lee, H.J.; Lee, S.C.; Watts, V.J.; Yeo, Y. Polydopamine-based simple and versatile surface modification of polymeric nano drug carriers. ACS Nano 2014, 8, 3347–3356. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Luo, M.; Liu, G.; Wang, X.; Tao, W.; Lin, Y.; Ji, X.; Nie, L.; Mei, L. Polydopamine-Modified Black Phosphorous Nanocapsule with Enhanced Stability and Photothermal Performance for Tumor Multimodal Treatments. Adv. Sci. 2018, 5, 1800510. [Google Scholar] [CrossRef] [PubMed]
- Ci, L.-Q.; Huang, Z.-G.; Lv, F.-M.; Wang, J.; Feng, L.-L.; Sun, F.; Cao, S.-J.; Liu, Z.-P.; Liu, Y.; Wei, G.; et al. Enhanced Delivery of Imatinib into Vaginal Mucosa via a New Positively Charged Nanocrystal-Loaded in Situ Hydrogel Formulation for Treatment of Cervical Cancer. Pharmaceutics 2019, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Tao, W.; Zhang, H.; Liu, G.; Wang, T.; Zhang, L.; Zeng, X.; Mei, L. Docetaxel (DTX)-loaded polydopamine-modified TPGS-PLA nanoparticles as a targeted drug delivery system fore the treatment of liver cancer. Acta Biomater. 2016, 30, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Zeng, X.; Wu, J.; Zhu, X.; Yu, X.; Zhang, X.; Zhang, J.; Liu, G.; Mei, L. Polydopamine-Based Surface Modification of Novel Nanoparticle-Aptamer Bioconjugates for In Vivo Breast Cancer Targeting and Enhanced Therapeutic Effects. Theranostics 2016, 6, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Liang, C.; Xu, L.; Liu, G.; Gao, N.; Tao, W.; Luo, L.; Zuo, Y.; Wang, X.; Zhang, X.; et al. TPGS-Functionalized Polydopamine-Modified Mesoporous Silica as Drug Nanocarriers for Enhanced Lung Cancer Chemotherapy against Multidrug Resistance. Small 2017, 13, 1700623. [Google Scholar] [CrossRef]
- Chang, D.; Gao, Y.; Wang, L.; Liu, G.; Chen, Y.; Wang, T.; Tao, W.; Mei, L.; Huang, L.; Zeng, X. Polydopamine-based surface modification of mesoporous silica nanoparticles as pH-sensitive drug delivery vehicles for cancer therapy. J. Colloid Interface Sci. 2016, 463, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Nie, J.; Xu, L.; Liang, C.; Peng, Y.; Liu, G.; Wang, T.; Mei, L.; Huang, L.; Zeng, X. pH-Sensitive Delivery Vehicle Based on Folic Acid-Conjugated Polydopamine-Modified Mesoporous Silica Nanoparticles for Targeted Cancer Therapy. ACS Appl. Mater. Interfaces 2017, 9, 18462–18473. [Google Scholar] [CrossRef]
- Cheng, W.; Nie, J.; Gao, N.; Liu, G.; Tao, W.; Xiao, X.; Jiang, L.; Liu, Z.; Zeng, X.; Mei, L. A Multifunctional Nanoplatform against Multidrug Resistant Cancer: Merging the Best of Targeted Chemo/Gene/Photothermal Therapy. Adv. Funct. Mater. 2017, 27, 1704135. [Google Scholar] [CrossRef]
- Peng, Y.; Nie, J.; Cheng, W.; Liu, G.; Zhu, D.; Zhang, L.; Liang, C.; Mei, L.; Huang, L.; Zeng, X. A multifunctional nanoplatform for cancer chemo-photothermal synergistic therapy and overcoming multidrug resistance. Biomater. Sci. 2018, 6, 1084–1098. [Google Scholar] [CrossRef]
- Zeng, X.; Tao, W.; Mei, L.; Huang, L.; Tan, C.; Feng, S.-S. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials 2013, 34, 6058–6067. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Xu, D.; Liang, S.; Zhu, X.; Lu, Y.; Wang, H. Prolonging the plasma circulation of proteins by nano-encapsulation with phosphorylcholine-based polymer. Nano Res. 2016, 9, 2424–2432. [Google Scholar] [CrossRef]
- Liu, G.; Tsai, H.-I.; Zeng, X.; Cheng, W.; Jiang, L.; Chen, H.; Zhang, X.; Zhang, J.; Mei, L. Phosphorylcholine-Based Stealthy Nanocapsules Decorating TPGS for Combatting Multi-Drug-Resistant Cancer. ACS Biomater. Sci. Eng. 2018, 4, 1679–1686. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. Embo J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples (n = 3) | Size (d.nm) | PDI | ZP (mV) | DTX LC (%) | DOX LC (%) |
|---|---|---|---|---|---|
| DTX/NPs | 105.3 ± 2.3 | 0.128 | −21.5 ± 4.7 | 9.88 ± 0.41 | N.A. |
| DTX/NPs@PDA | 161.5 ± 3.6 | 0.151 | −17.3 ± 5.1 | 9.67 ± 0.39 | N.A. |
| DTX/NPs@PDA-PEG | 166.3 ± 4.1 | 0.139 | −13.6 ± 3.8 | 9.43 ± 0.43 | N.A. |
| DTX/NPs@PDA-PEG-Apt | 167.1 ± 3.9 | 0.145 | −13.4 ± 4.1 | 9.39 ± 0.32 | N.A. |
| DTX/NPs@PDA/DOX-PEG-Apt | 170.3 ± 4.6 | 0.160 | −10.6 ± 2.9 | 8.47 ± 0.27 | 8.61 ± 0.34 |
| Incubation Time (h) | IC50 (μg/mL) | |||||
|---|---|---|---|---|---|---|
| DTX | DTX/NPs@ PDA-PEG | DTX/NPs@ PDA-PEG-Apt | NPs@PDA/DOX-PEG-Apt | DTX/NPs@PDA/DOX-PEG-Apt | DTX/NPs@PDA/DOX-PEG-Apt+NIR | |
| 24 | 19.62 | 22.03 | 11.71 | 15.40 | 6.24 | 3.90 |
| 48 | 13.29 | 5.33 | 1.23 | 1.19 | 0.55 | 0.38 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Gao, N.; Zhou, Y.; Nie, J.; Cheng, W.; Luo, M.; Mei, L.; Zeng, X.; Deng, W. Polydopamine-Based “Four-in-One” Versatile Nanoplatforms for Targeted Dual Chemo and Photothermal Synergistic Cancer Therapy. Pharmaceutics 2019, 11, 507. https://doi.org/10.3390/pharmaceutics11100507
Liu G, Gao N, Zhou Y, Nie J, Cheng W, Luo M, Mei L, Zeng X, Deng W. Polydopamine-Based “Four-in-One” Versatile Nanoplatforms for Targeted Dual Chemo and Photothermal Synergistic Cancer Therapy. Pharmaceutics. 2019; 11(10):507. https://doi.org/10.3390/pharmaceutics11100507
Chicago/Turabian StyleLiu, Gan, Nansha Gao, Yun Zhou, Junpeng Nie, Wei Cheng, Miaomiao Luo, Lin Mei, Xiaowei Zeng, and Wenbin Deng. 2019. "Polydopamine-Based “Four-in-One” Versatile Nanoplatforms for Targeted Dual Chemo and Photothermal Synergistic Cancer Therapy" Pharmaceutics 11, no. 10: 507. https://doi.org/10.3390/pharmaceutics11100507
APA StyleLiu, G., Gao, N., Zhou, Y., Nie, J., Cheng, W., Luo, M., Mei, L., Zeng, X., & Deng, W. (2019). Polydopamine-Based “Four-in-One” Versatile Nanoplatforms for Targeted Dual Chemo and Photothermal Synergistic Cancer Therapy. Pharmaceutics, 11(10), 507. https://doi.org/10.3390/pharmaceutics11100507