
Changing Stem Cell Dynamics during Papillomavirus Infection: Potential Roles for Cellular Plasticity in the Viral Lifecycle and Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Target Cell of Papillomavirus Infection
3. A Cell Reservoir for Long-Term Viral Maintenance
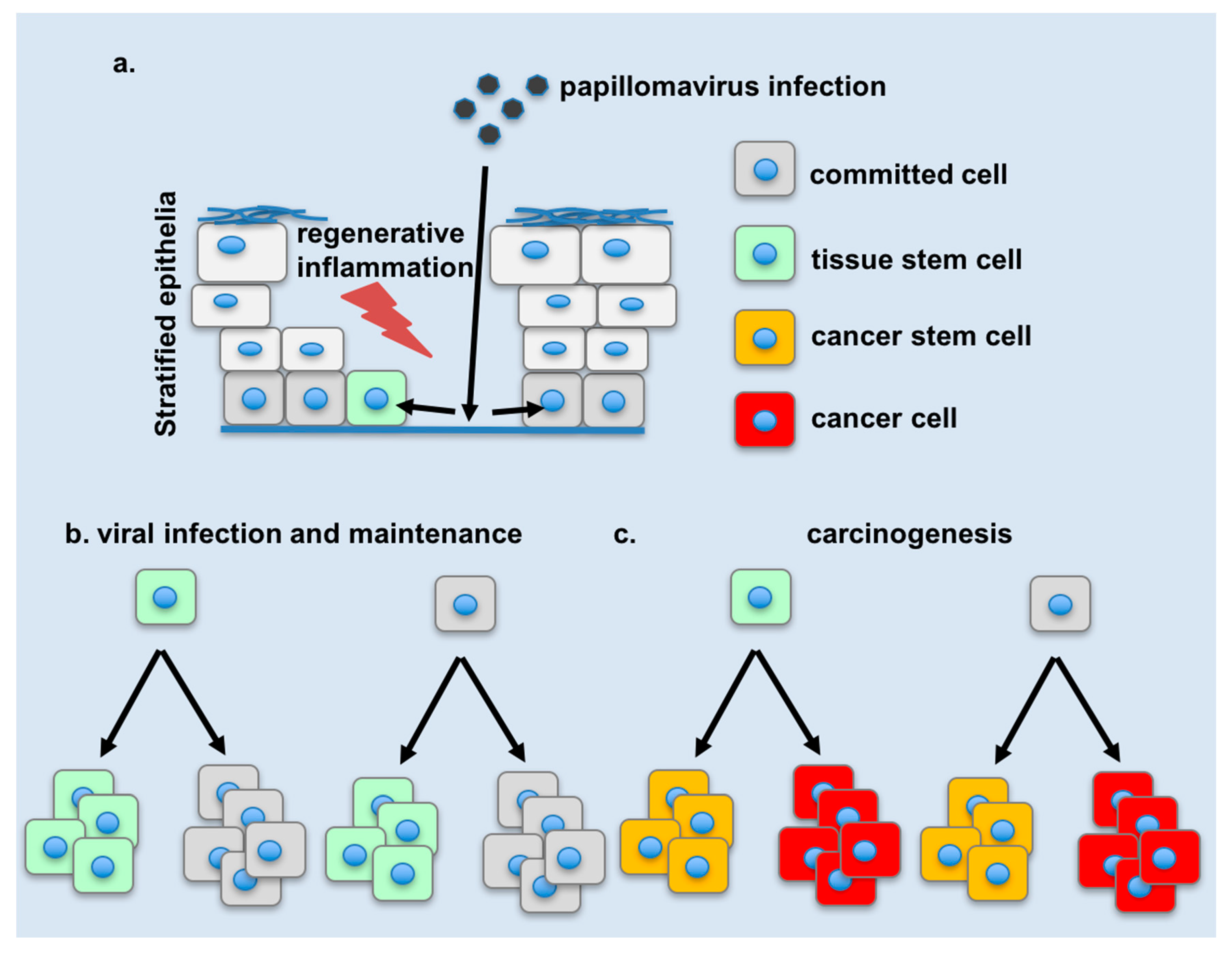
4. Changes in Tissue Stem Cell Dynamics during Infection
5. Changes in Cellular Plasticity during Infection
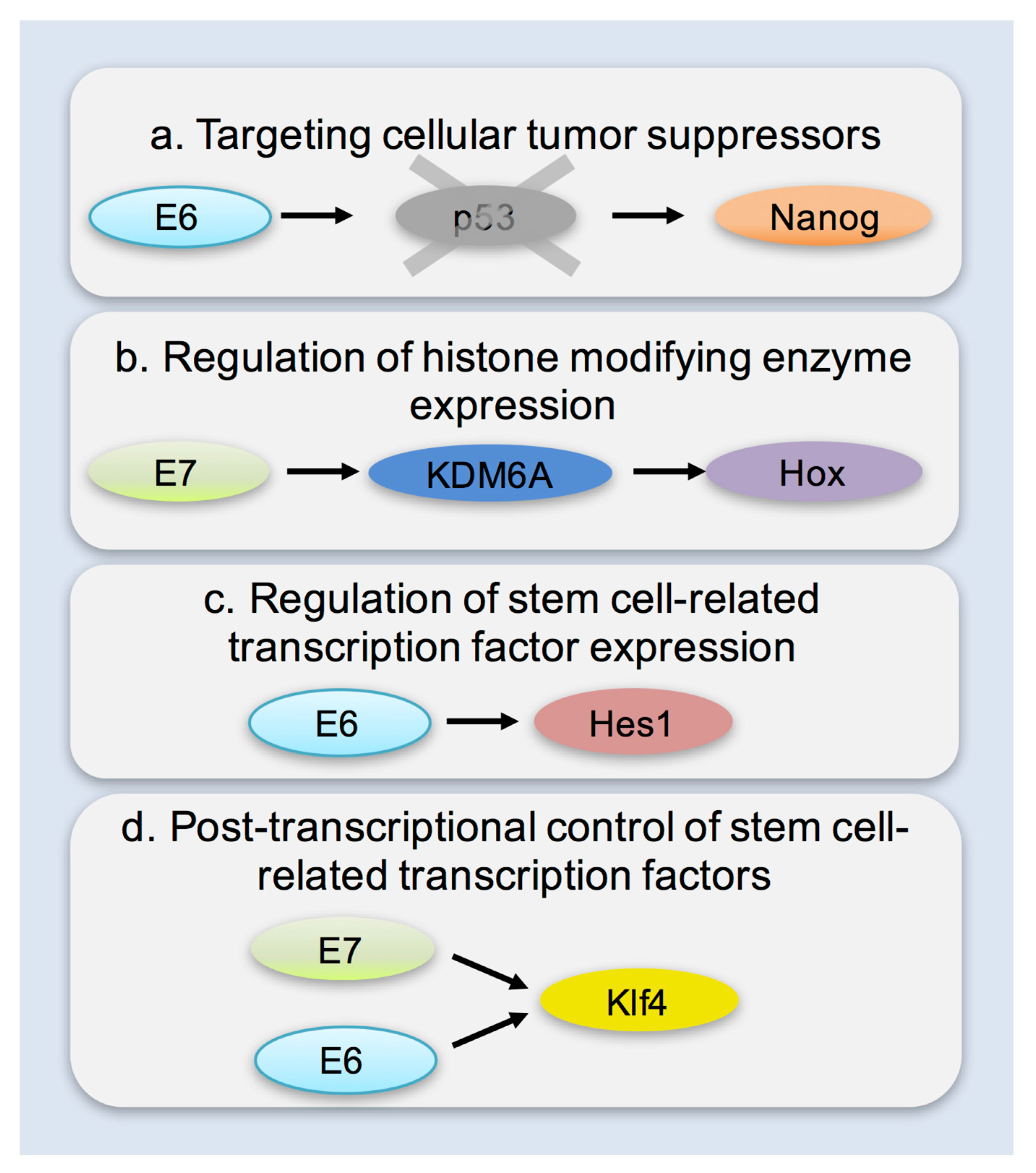
6. Mechanisms of Enhancing Cellular Plasticity
7. Potential Links of Cellular Plasticity to Disease
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Day, P.M.; Kines, R.C. Current understanding of the mechanism of HPV infection. Gynecol. Oncol. 2010, 118, S12–S17. [Google Scholar] [CrossRef] [PubMed]
- Mascre, G.; Dekoninck, S.; Drogat, B.; Youssef, K.K.; Brohee, S.; Sotiropoulou, P.A.; Simons, B.D.; Blanpain, C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012, 489, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Latent papillomavirus infections and their regulation. Curr. Opin. Virol. 2013, 3, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Diz, V.; Sole-Sanchez, S.; Valdes-Gutierrez, A.; Urpi, M.; Riba-Artes, D.; Penin, R.M.; Pascual, G.; Gonzalez-Suarez, E.; Casanovas, O.; Vinals, F.; et al. Progeny of Lgr5-expressing hair follicle stem cell contributes to papillomavirus-induced tumor development in epidermis. Oncogene 2013, 32, 3732–3743. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Doorbar, J. Human papillomavirus infection and induction of neoplasia: A matter of fitness. Curr. Opin. Virol. 2016, 20, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, F.; Laimins, L.A. Human papillomaviruses: Targeting differentiating epithelial cells for malignant transformation. Oncogene 2003, 22, 5201–5207. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of hpvs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Liu, Y.; Yang, Z.; Nguyen, J.; Liang, F.; Morris, R.J.; Cotsarelis, G. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 2005, 11, 1351–1354. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Fuchs, E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 2014, 344, 1242281. [Google Scholar] [CrossRef] [PubMed]
- Rompolas, P.; Mesa, K.R.; Greco, V. Spatial organization within a niche as a determinant of stem-cell fate. Nature 2013, 502, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Human papillomaviruses. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; WHO: Geneva, Switzerland, 2007; Volume 90, pp. 1–636.
- Brown, D.R.; Weaver, B. Human papillomavirus in older women: New infection or reactivation? J. Infect. Dis. 2013, 207, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Gravitt, P.E.; Rositch, A.F.; Silver, M.I.; Marks, M.A.; Chang, K.; Burke, A.E.; Viscidi, R.P. A cohort effect of the sexual revolution may be masking an increase in human papillomavirus detection at menopause in the united states. J. Infect. Dis. 2013, 207, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Rochat, A.; Zeltner, R.; Borenstein, L.; Barrandon, Y.; Wettstein, F.O.; Iftner, T. The primary target cells of the high-risk cottontail rabbit papillomavirus colocalize with hair follicle stem cells. J. Virol. 1996, 70, 1912–1922. [Google Scholar] [PubMed]
- Boxman, I.L.; Berkhout, R.J.; Mulder, L.H.; Wolkers, M.C.; Bouwes Bavinck, J.N.; Vermeer, B.J.; Ter Schegget, J. Detection of human papillomavirus dna in plucked hairs from renal transplant recipients and healthy volunteers. J. Investig. Dermatol. 1997, 108, 712–715. [Google Scholar] [CrossRef] [PubMed]
- De Koning, M.N.; Struijk, L.; Bavinck, J.N.; Kleter, B.; Ter Schegget, J.; Quint, W.G.; Feltkamp, M.C. β-papillomaviruses frequently persist in the skin of healthy individuals. J. Gen. Virol. 2007, 88, 1489–1495. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; Laimins, L.A. Human papillomaviruses: Shared and distinct pathways for pathogenesis. Curr. Opin. Virol. 2015, 14, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, M. The biology of β-human papillomaviruses. Virus Res. 2017, 231, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.E.; Arends, J.; Van der Linden, P.J.; De Boer, B.A.; Helmerhorst, T.J. Cytokeratin 17 and p63 are markers of the HPV target cell, the cervical stem cell. Anticancer Res. 2004, 24, 771–775. [Google Scholar] [PubMed]
- Herfs, M.; Yamamoto, Y.; Laury, A.; Wang, X.; Nucci, M.R.; McLaughlin-Drubin, M.E.; Munger, K.; Feldman, S.; McKeon, F.D.; Xian, W.; et al. A discrete population of squamocolumnar junction cells implicated in the pathogenesis of cervical cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 10516–10521. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Rossi, R.; Commere, P.H.; Jay, P.; Sansonetti, P.J. The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 2014, 15, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.D.; Sodhi, C.P.; Jia, H.; Dyer, M.; Egan, C.E.; Yazji, I.; Good, M.; Afrazi, A.; Marino, R.; Slagle, D.; et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J. Biol. Chem. 2012, 287, 37296–37308. [Google Scholar] [CrossRef] [PubMed]
- Apidianakis, Y.; Pitsouli, C.; Perrimon, N.; Rahme, L. Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 20883–20888. [Google Scholar] [CrossRef] [PubMed]
- Pitsouli, C.; Apidianakis, Y.; Perrimon, N. Homeostasis in infected epithelia: Stem cells take the lead. Cell Host Microbe 2009, 6, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Mysorekar, I.U.; Isaacson-Schmid, M.; Walker, J.N.; Mills, J.C.; Hultgren, S.J. Bone morphogenetic protein 4 signaling regulates epithelial renewal in the urinary tract in response to uropathogenic infection. Cell Host Microbe 2009, 5, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Michael, S.; Lambert, P.F.; Strati, K. The HPV16 oncogenes cause aberrant stem cell mobilization. Virology 2013, 443, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Lanfredini, S.; Olivero, C.; Borgogna, C.; Calati, F.; Powell, K.; Davies, K.J.; De Andrea, M.; Harries, S.; Tang, H.K.C.; Pfister, H.; et al. HPV8 field cancerization in a transgenic mouse model is due to Lrig1+ keratinocyte stem cell expansion. J. Investig. Dermatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- White, A.C.; Khuu, J.K.; Dang, C.Y.; Hu, J.; Tran, K.V.; Liu, A.; Gomez, S.; Zhang, Z.; Yi, R.; Scumpia, P.; et al. Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat. Cell. Biol. 2014, 16, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Jaks, V.; Barker, N.; Kasper, M.; Van Es, J.H.; Snippert, H.J.; Clevers, H.; Toftgård, R. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat. Genet. 2008, 40, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Andersen, B. Have hair follicle stem cells shed their tranquil image? Cell Stem Cell 2008, 3, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Canamero, M.; Rayon, T.; Ors, I.; Grana, O.; Megias, D.; Dominguez, O.; Martinez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Maherali, N.; Breault, D.T.; Hochedlinger, K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2008, 2, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Hochedlinger, K.; Plath, K. Epigenetic reprogramming and induced pluripotency. Development 2009, 136, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Kelaini, S.; Cochrane, A.; Margariti, A. Direct reprogramming of adult cells: Avoiding the pluripotent state. Stem Cells Cloning 2014, 7, 19–29. [Google Scholar] [PubMed]
- Margariti, A.; Winkler, B.; Karamariti, E.; Zampetaki, A.; Tsai, T.N.; Baban, D.; Ragoussis, J.; Huang, Y.; Han, J.D.; Zeng, L.; et al. Direct reprogramming of fibroblasts into endothelial cells capable of angiogenesis and reendothelialization in tissue-engineered vessels. Proc. Natl. Acad. Sci. USA 2012, 109, 13793–13798. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Qu, J.; Cholewa-Waclaw, J.; Burr, K.; Raaum, R.; Rambukkana, A. Reprogramming adult schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell 2013, 152, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; McGlinchey, A.; Tomlinson, S.R.; Qu, J.; Rambukkana, A. Reprogramming diminishes retention of mycobacterium leprae in schwann cells and elevates bacterial transfer property to fibroblasts. F1000Research 2013, 2, 198. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; McGlinchey, A.; Cholewa-Waclaw, J.; Qu, J.; Tomlinson, S.R.; Rambukkana, A. Innate immune response precedes Mycobacterium leprae-induced reprogramming of adult Schwann cells. Cell. Reprogram. 2014, 16, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Panayidou, S.; Apidianakis, Y. Regenerative inflammation: Lessons from Drosophila intestinal epithelium in health and disease. Pathogens 2013, 2, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; De Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Michael, S.; Achilleos, C.; Panayiotou, T.; Strati, K. Inflammation shapes stem cells and stemness during infection and beyond. Front. Cell Dev. Biol. 2016, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Ingle, A.; Ghim, S.; Joh, J.; Chepkoech, I.; Bennett Jenson, A.; Sundberg, J.P. Novel laboratory mouse papillomavirus (MusPV) infection. Vet. Pathol. 2011, 48, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Handisurya, A.; Day, P.M.; Thompson, C.D.; Buck, C.B.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Characterization of Mus musculus papillomavirus 1 infection in situ reveals an unusual pattern of late gene expression and capsid protein localization. J. Virol. 2013, 87, 13214–13225. [Google Scholar] [CrossRef] [PubMed]
- Uberoi, A.; Yoshida, S.; Frazer, I.H.; Pitot, H.C.; Lambert, P.F. Role of ultraviolet radiation in papillomavirus-induced disease. PLoS Pathog. 2016, 12, e1005664. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Izpisua Belmonte, J.C. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Collado, M.; Villasante, A.; Strati, K.; Ortega, S.; Canamero, M.; Blasco, M.A.; Serrano, M. The Ink4/Arf locus is a barrier for ips cell reprogramming. Nature 2009, 460, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Tejera, A.; Schoeftner, S.; Ortega, S.; Serrano, M.; Blasco, M.A. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 2009, 4, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Utikal, J.; Polo, J.M.; Stadtfeld, M.; Maherali, N.; Kulalert, W.; Walsh, R.M.; Khalil, A.; Rheinwald, J.G.; Hochedlinger, K. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 2009, 460, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. P53 induces differentiation of mouse embryonic stem cells by suppressing nanog expression. Nat. Cell. Biol. 2005, 7, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 2015, 16, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Chlon, T.M.; Hoskins, E.E.; Mayhew, C.N.; Wikenheiser-Brokamp, K.A.; Davies, S.M.; Mehta, P.; Myers, K.C.; Wells, J.M.; Wells, S.I. High-risk human papillomavirus E6 protein promotes reprogramming of fanconi anemia patient cells through repression of p53 but does not allow for sustained growth of induced pluripotent stem cells. J. Virol. 2014, 88, 11315–11326. [Google Scholar] [CrossRef] [PubMed]
- Gunasekharan, V.K.; Li, Y.; Andrade, J.; Laimins, L.A. Post-transcriptional regulation of Klf4 by high-risk human papillomaviruses is necessary for the differentiation-dependent viral life cycle. PLoS Pathog. 2016, 12, e1005747. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Ohbo, K.; Scholer, H. The carboxy-terminal transactivation domain of Oct-4 acquires cell specificity through the POU domain. Mol. Cell. Biol. 1997, 17, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Ohbo, K.; Zwerschke, W.; Botquin, V.; Jansen-Durr, P.; Scholer, H.R. Synergism with germ line transcription factor Oct-4: Viral oncoproteins share the ability to mimic a stem cell-specific activity. Mol. Cell. Biol. 1999, 19, 2635–2643. [Google Scholar] [CrossRef] [PubMed]
- Organista-Nava, J.; Gómez-Gómez, Y.; Ocadiz-Delgado, R.; García-Villa, E.; Bonilla-Delgado, J.; Lagunas-Martínez, A.; Tapia, J.S.; Lambert, P.F.; García-Carrancá, A.; Gariglio, P. The HPV16 E7 oncoprotein increases the expression of Oct3/4 and stemness-related genes and augments cell self-renewal. Virology 2016, 499, 230–242. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Munger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, M.; Biddle, A.; Borgogna, C.; Gariglio, M.; Doorbar, J.; Storey, A.; Pfister, H.; Mackenzie, I.; Akgül, B. Expression of β-papillomavirus oncogenes increases the number of keratinocytes with stem cell-like properties. J. Virol. 2013, 87, 12158–12165. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.M.; Uberoi, A.; Grace, M.; Lambert, P.F.; Munger, K. Cutaneous HPV8 and MmuPV1 E6 proteins target the Notch and TGF-β tumor suppressors to inhibit differentiation and sustain keratinocyte proliferation. PLoS Pathog. 2017, 13, e1006171. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Holleywood, C.; Libert, D.; Griffin, H.; Mahmood, R.; Isaacson, E.; Doorbar, J. Modulation of basal cell fate during productive and transforming HPV16 infection is mediated by progressive E6-driven depletion of Notch. J. Pathol. 2017, 242, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Vishnoi, K.; Mahata, S.; Verma, G.; Srivastava, Y.; Masaldan, S.; Roy, B.G.; Bharti, A.C.; Das, B.C. Cervical cancer stem cells selectively overexpress hpv oncoprotein E6 that controls stemness and self-renewal through upregulation of Hes1. Clin. Cancer Res. 2016, 22, 4170–4184. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.W.; Cho, H.; Choi, C.H.; Ylaya, K.; Chung, J.Y.; Kim, J.H.; Hewitt, S.M. Clinical significance of Oct4 and Sox2 protein expression in cervical cancer. BMC Cancer 2015, 15, 1015. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, D.; Näsman, A.; Tarján, M.; Henriksson, R.; Tot, T.; Dalianis, T.; Hedman, H. Expression of LRIG1 is associated with good prognosis and human papillomavirus status in oropharyngeal cancer. Br. J. Cancer 2014, 110, 1793–1800. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strati, K. Changing Stem Cell Dynamics during Papillomavirus Infection: Potential Roles for Cellular Plasticity in the Viral Lifecycle and Disease. Viruses 2017, 9, 221. https://doi.org/10.3390/v9080221
Strati K. Changing Stem Cell Dynamics during Papillomavirus Infection: Potential Roles for Cellular Plasticity in the Viral Lifecycle and Disease. Viruses. 2017; 9(8):221. https://doi.org/10.3390/v9080221
Chicago/Turabian StyleStrati, Katerina. 2017. "Changing Stem Cell Dynamics during Papillomavirus Infection: Potential Roles for Cellular Plasticity in the Viral Lifecycle and Disease" Viruses 9, no. 8: 221. https://doi.org/10.3390/v9080221
APA StyleStrati, K. (2017). Changing Stem Cell Dynamics during Papillomavirus Infection: Potential Roles for Cellular Plasticity in the Viral Lifecycle and Disease. Viruses, 9(8), 221. https://doi.org/10.3390/v9080221