
Characterization of Bacillus subtilis Viruses vB_BsuM-Goe2 and vB_BsuM-Goe3



Abstract
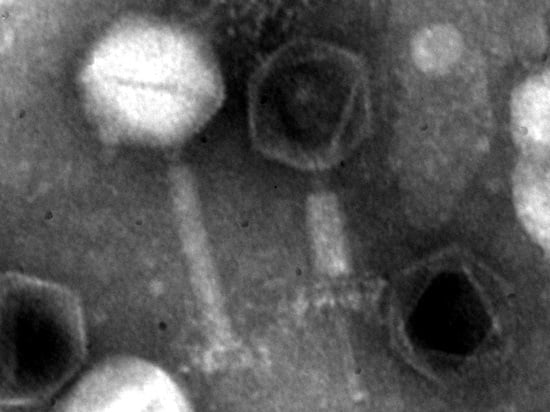
:
1. Introduction
2. Material and Methods
2.1. Naming
2.2. Media and Buffers
2.3. Virus Isolation and Preparation
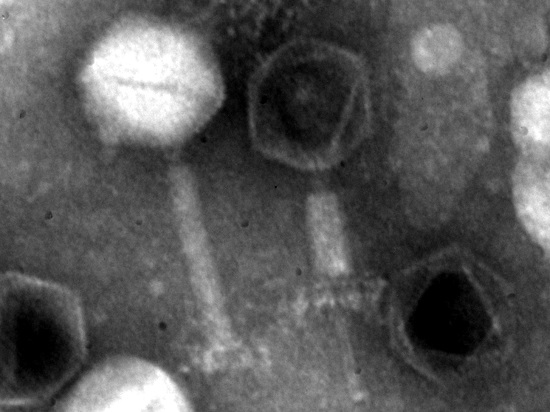
2.4. Transmission Electron Microscopy
2.5. Host Range and Sensitivity Determination
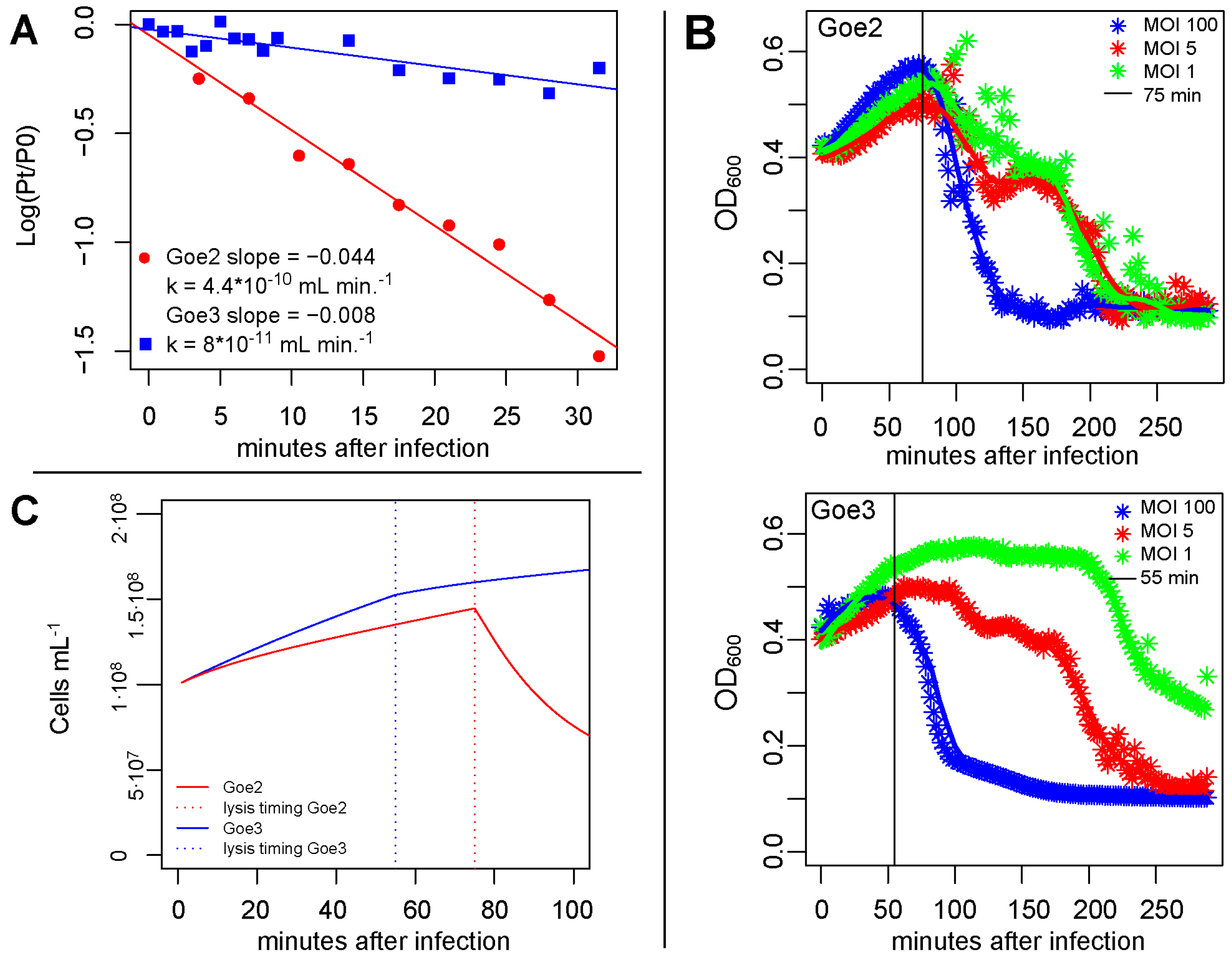
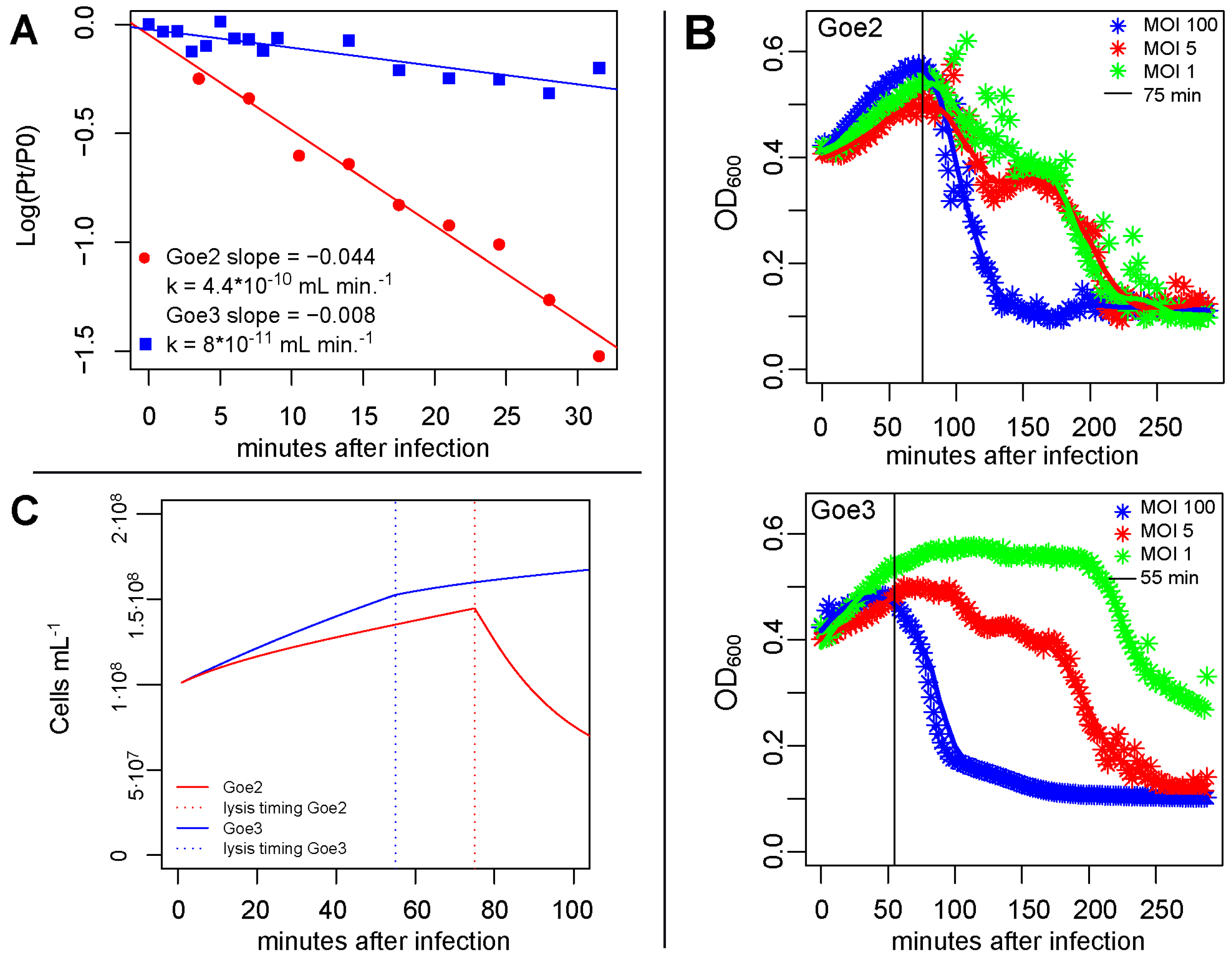
2.6. Adsorption Constant K
2.7. Latency Period
2.8. Burst Size
2.9. Simulation of a Virus Infection
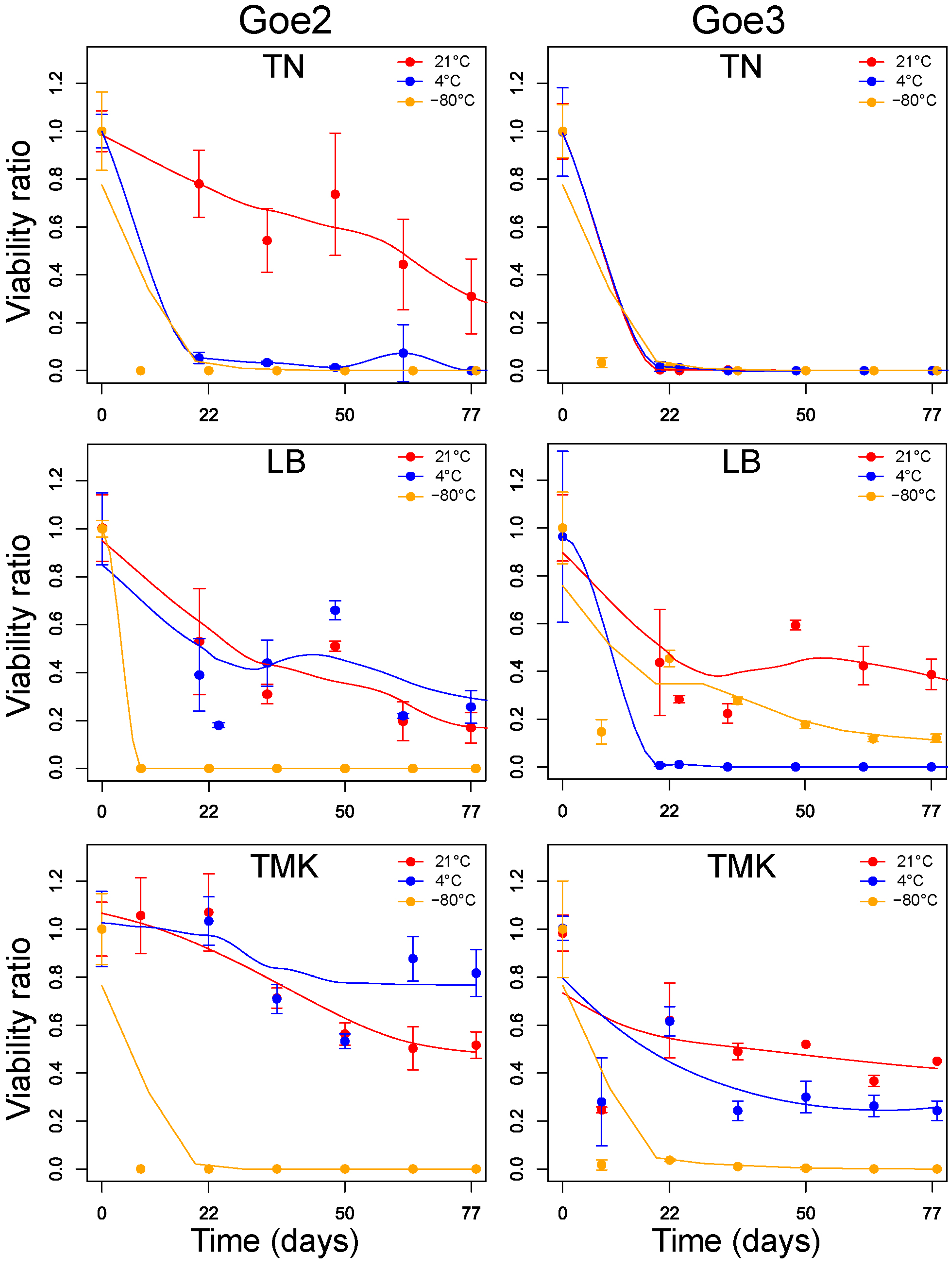
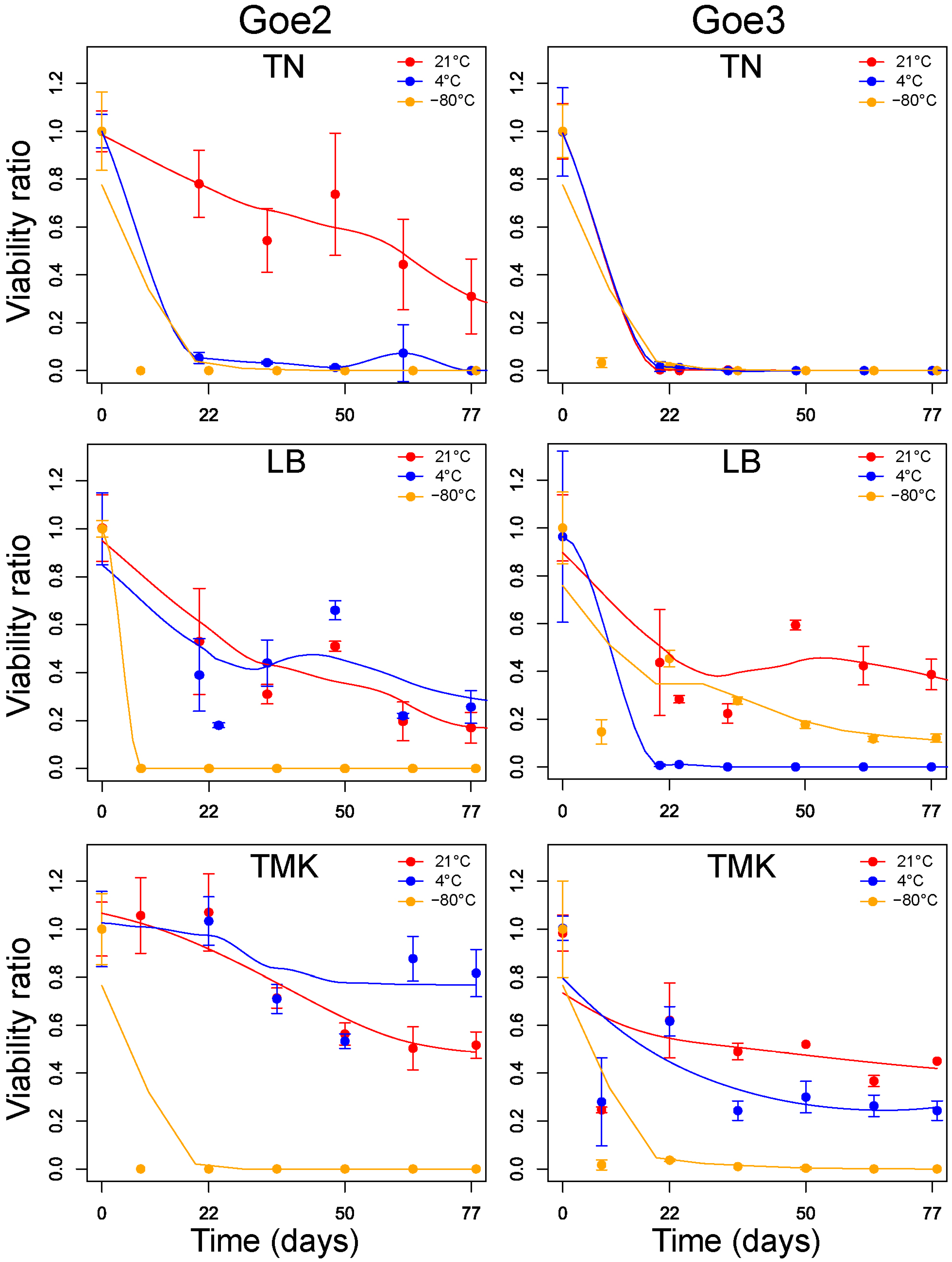
2.10. Viability Testing
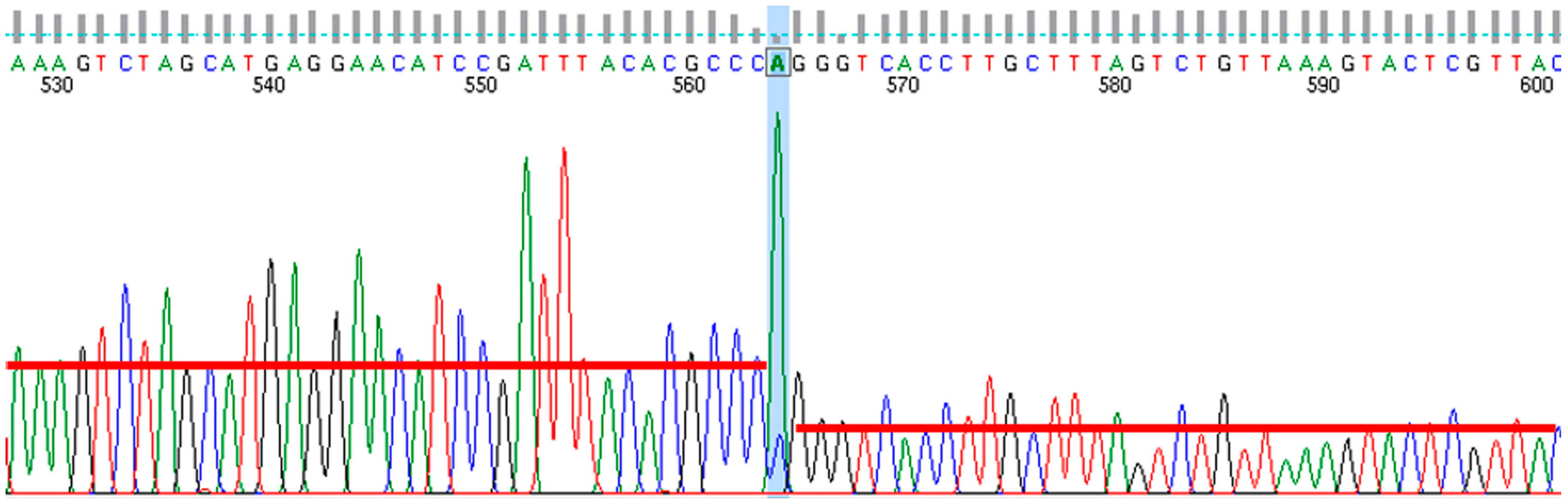
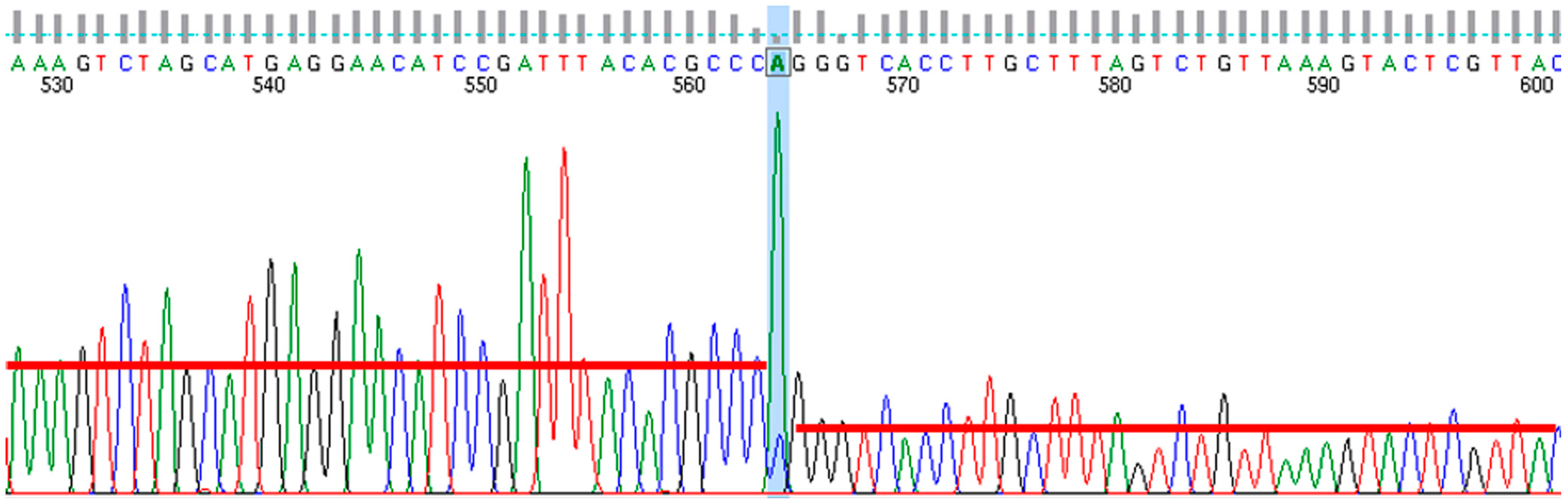
2.11. Genome Sequencing and Annotation
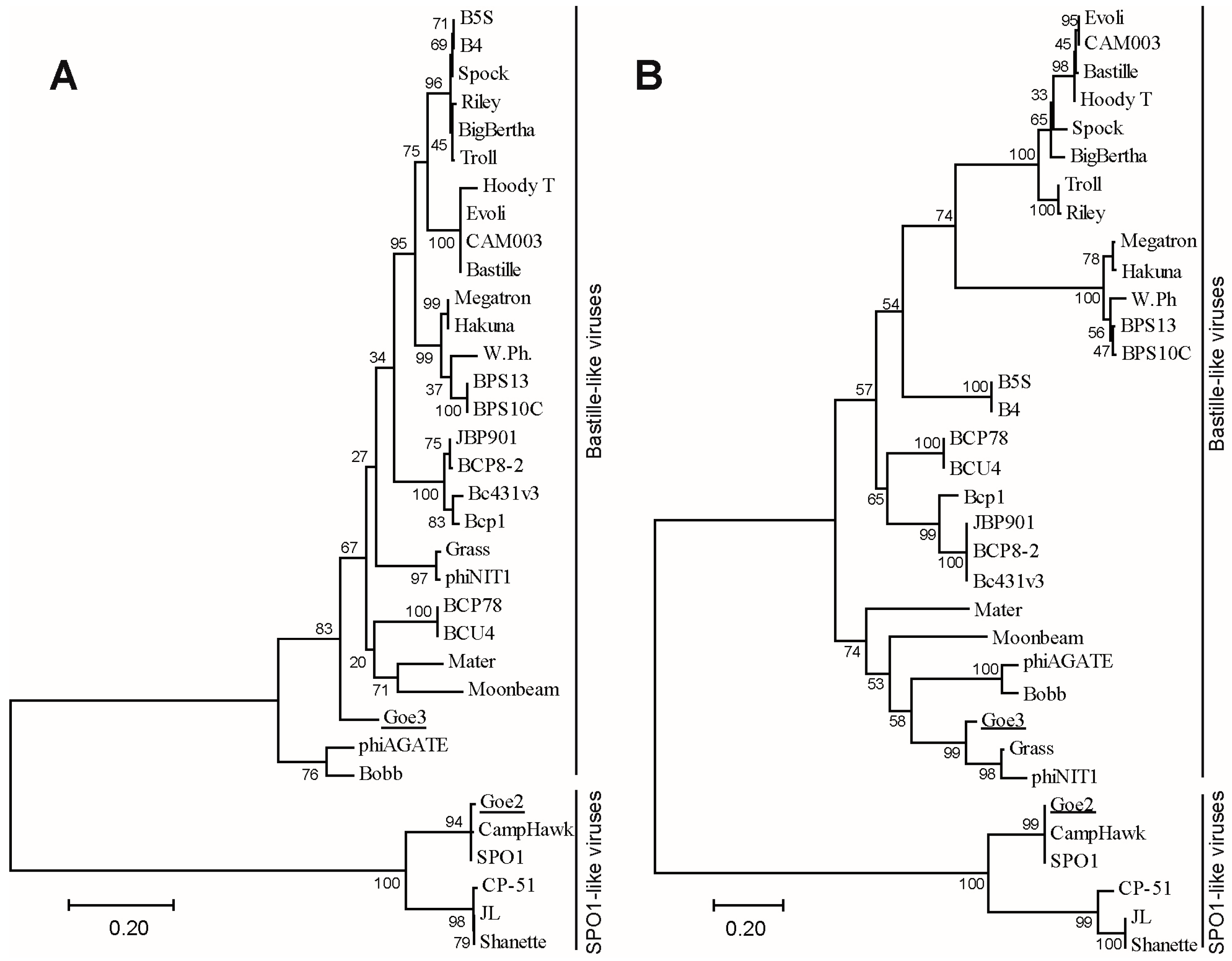
2.12. Phylogeny
3. Results
3.1. Virus Isolation
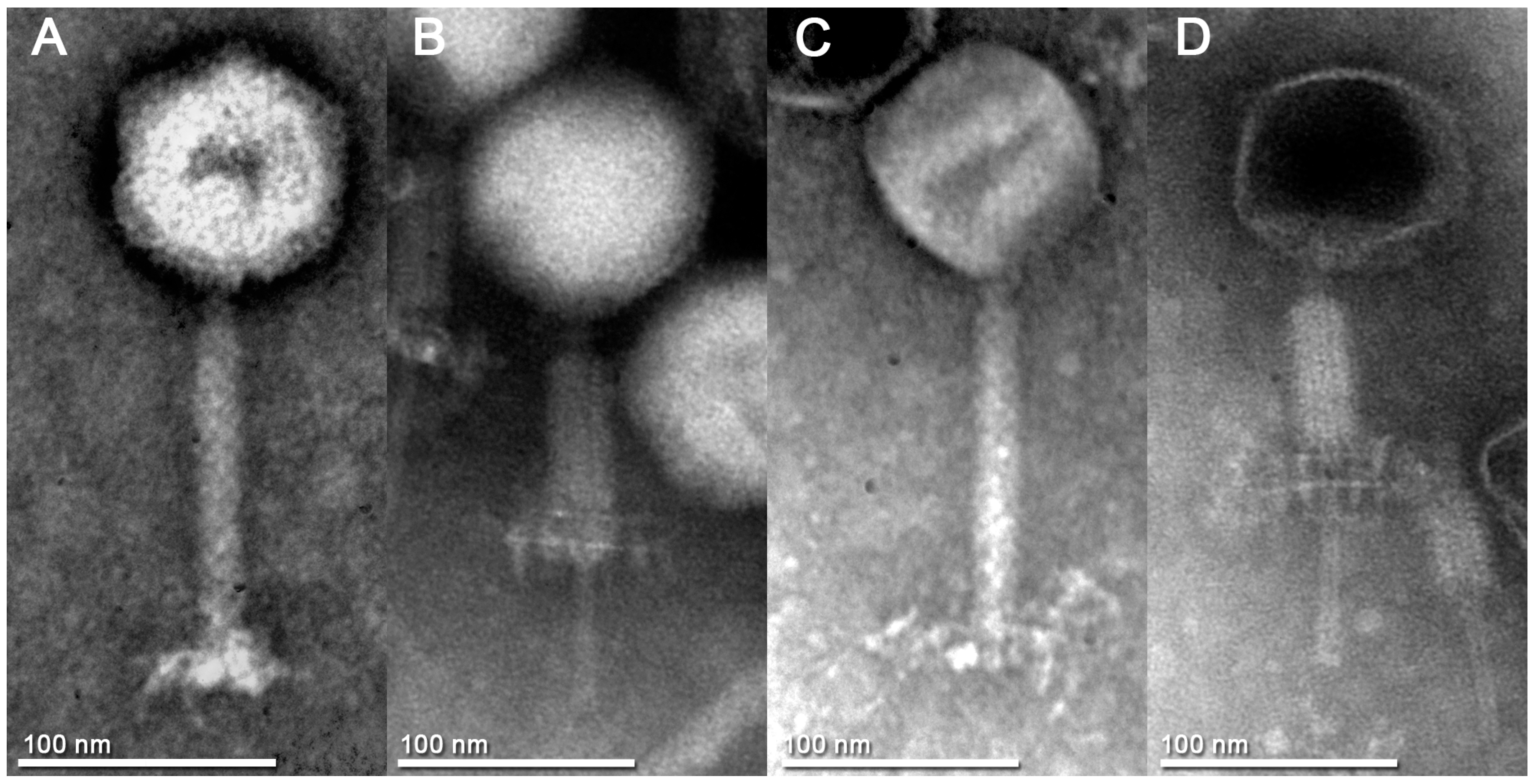
3.2. Phage Particle Morphology
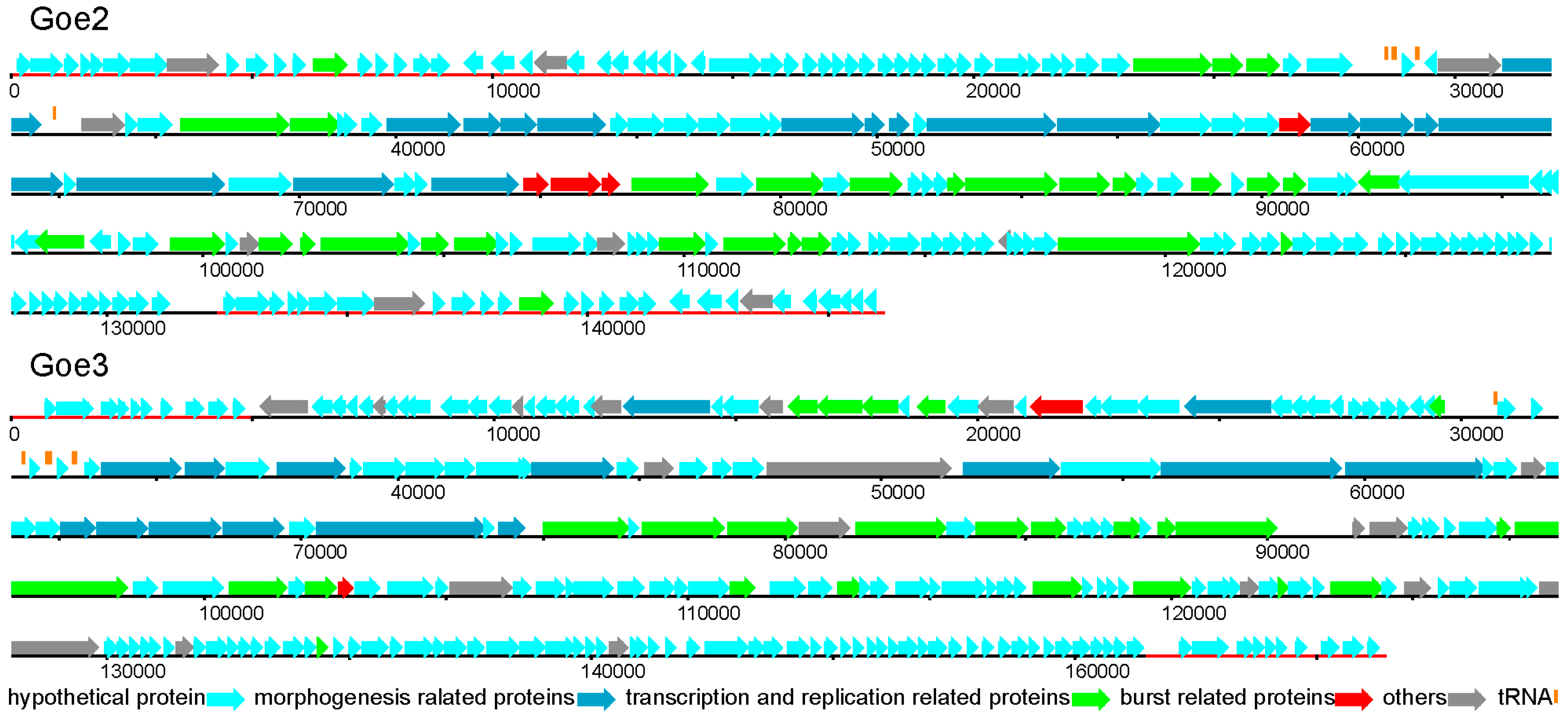
3.3. Genomic Structure and Content
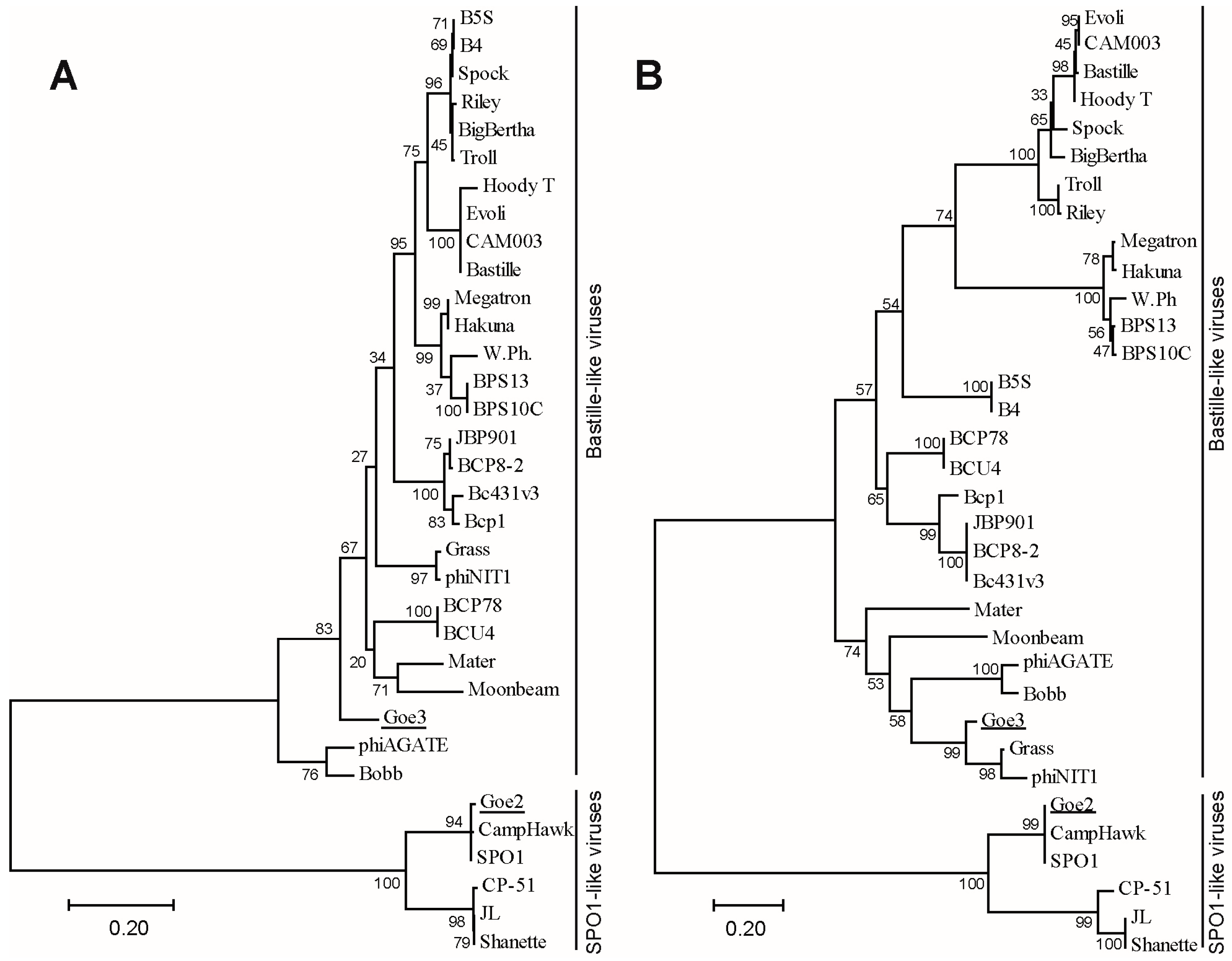
3.4. Phylogeny and Classification
3.5. Virus Host Interaction
3.6. Host Range and Sensitivity Determination
3.7. Viability Testing
4. Discussion
4.1. Comparability of Physiological Data
4.2. Know Your Phage
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of prokaryotic viruses: Update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2016, 161, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.F.; Brüssow, H. Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol. 2002, 10, 521–529. [Google Scholar] [CrossRef]
- Salmond, G.P.C.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Golkar, Z.; Bagasra, O.; Pace, D.G. Bacteriophage therapy: A potential solution for the antibiotic resistance crisis. J. Infect. Dev. Ctries. 2014, 8, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Łobocka, M.; Hejnowicz, M.S.; Dąbrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dąbrowska, B.; Kwiatek, M.; Parasion, S.; et al. Genomics of staphylococcal Twort-like phages—Potential therapeutics of the post-antibiotic era. Adv. Virus Res. 2012, 83, 143–216. [Google Scholar] [PubMed]
- Schoeni, J.L.; Wong, A.C.L. Bacillus cereus food poisoning and its toxins. J. Food Prot. 2005, 68, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Hanna, P.C.; Ireland, J.A.W. Understanding Bacillus anthracis pathogenesis. Trends Microbiol. 1999, 7, 180–182. [Google Scholar] [CrossRef]
- Ghose, C. Clostridium difficile infection in the twenty-first century. Emerg. Microbes Infect. 2013, 2, e62. [Google Scholar] [CrossRef] [PubMed]
- Kallen, A.J.; Mu, Y.; Bulens, S.; Reingold, A.; Petit, S.; Gershman, K.; Ray, S.M.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; et al. Health Care–Associated Invasive MRSA Infections, 2005–2008. JAMA 2010, 304, 641. [Google Scholar] [CrossRef] [PubMed]
- Hejnowicz, M.S.; Dąbrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dąbrowska, B.; Kwiatek, M.; Parasion, S.; Gawor, J.; et al. Chapter 5—Genomics of Staphylococcal Twort-like Phages—Potential Therapeutics of the Post-Antibiotic Era. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2012; Volume 83, pp. 143–216. [Google Scholar]
- Klumpp, J.; Lavigne, R.; Loessner, M.J.; Ackermann, H.W. The SPO1-related bacteriophages. Arch. Virol. 2010, 155, 1547–1561. [Google Scholar] [CrossRef] [PubMed]
- Asare, P.T.; Jeong, T.-Y.; Ryu, S.; Klumpp, J.; Loessner, M.J.; Merrill, B.D.; Kim, K.-P. Putative type 1 thymidylate synthase and dihydrofolate reductase as signature genes of a novel bastille-like group of phages in the subfamily Spounavirinae. BMC Genom. 2015, 16, 582. [Google Scholar] [CrossRef] [PubMed]
- Westers, H.; Dorenbos, R.; van Dijl, J.M.; Kabel, J.; Flanagan, T.; Devine, K.M.; Jude, F.; Seror, S.J.; Beekman, A.C.; Darmon, E.; et al. Genome engineering reveals large dispensable regions in Bacillus subtilis. Mol. Biol. Evol. 2003, 20, 2076–2090. [Google Scholar] [CrossRef] [PubMed]
- Kropinski, A.M.; Prangishvili, D.; Lavigne, R. Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 2009, 11, 2775–2777. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Marmur, J. Characterization of inducible bacteriophages in Bacillus licheniformis. J. Virol. 1970, 5, 237–246. [Google Scholar] [PubMed]
- Spizizen, J. Transformation of Biochemically Deficient Strains of Bacillus Subtilis by Deoxyribonucleate. Proc. Natl. Acad. Sci. USA 1958, 44, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Willms, I.M.; Hertel, R. Phage vB_BsuP-Goe1: The smallest identified lytic phage of Bacillus subtilis. FEMS Microbiol. Lett. 2016, 363, fnw208. [Google Scholar] [CrossRef] [PubMed]
- Hertel, R.; Pintor Rodríguez, D.; Hollensteiner, J.; Dietrich, S.; Leimbach, A.; Hoppert, M.; Liesegang, H.; Volland, S. Genome-Based Identification of Active Prophage Regions by Next Generation Sequencing in Bacillus licheniformis DSM13. PLoS ONE 2015, 10, e0120759. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Wang, I.-N. Bacteriophage adsorption rate and optimal lysis time. Genetics 2008, 180, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Practical methods for determining phage growth parameters. Methods Mol. Biol. 2009, 501, 175–202. [Google Scholar] [PubMed]
- RStudio Team. RStudio: Integrated Development for R. RStudio, Inc.: Boston, MA, USA, 2015; Available online: http://www.rstudio.com/ (accessed on 27 March 2017).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015; Available online: http://www.R-project.org/ (accessed on 27 March 2017).
- Bolger-Munro, M.; Cheung, K.; Fang, A.; Wang, L. T4 Bacteriophage Average Burst Size Varies with Escherichia coli B23 Cell Culture Age. J. Exp. Microbiol. Immunol. 2013, 17, 115–119. [Google Scholar]
- Stopar, D.; Abedon, S.T. Modeling bacteriophage population growth. In Bacteriophage Ecology; Abedon, S.T., Ed.; Advances in Molecular and Cellular Microbiology; Cambridge University Press: Cambridge, UK, 2008; pp. 389–414. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet—Next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, J.K.; Smith, K.F.; Staden, R. A new DNA sequence assembly program. Nucleic Acids Res. 1995, 23, 4992–4999. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualisation and analysis of high-throughput sequence-based experimental data. Bioinformatics 2011, 28, 464. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Itoh, Y. Characterization of poly-gamma-glutamate hydrolase encoded by a bacteriophage genome: Possible role in phage infection of Bacillus subtilis encapsulated with poly-gamma-glutamate. Appl. Environ. Microbiol. 2003, 69, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Ritz, M.P.; Perl, A.L.; Colquhoun, J.M.; Chamakura, K.R.; Kuty Everett, G.F. Complete Genome of Bacillus subtilis Myophage CampHawk. Genome Announc. 2013, 1, e00984-13. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Casjens, S.R.; Cresawn, S.G.; Houtz, J.M.; Smith, A.L.; Ford, M.E.; Peebles, C.L.; Hatfull, G.F.; Hendrix, R.W.; Huang, W.M.; et al. The Genome of Bacillus subtilis Bacteriophage SPO1. J. Mol. Biol. 2009, 388, 48–70. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.Y.; Colquhoun, J.M.; Perl, A.L.; Chamakura, K.R.; Everett, F.K. Complete Genome of Bacillus subtilis Myophage Grass. Genome Announc. 2013, 1, e00857-13. [Google Scholar] [CrossRef] [PubMed]
- Belda, E.; Sekowska, A.; Le Fèvre, F.; Morgat, A.; Mornico, D.; Ouzounis, C.; Vallenet, D.; Médigue, C.; Danchin, A. An updated metabolic view of the Bacillus subtilis 168 genome. Microbiology 2013, 159, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Zeigler, D.R.; Prágai, Z.; Rodriguez, S.; Chevreux, B.; Muffler, A.; Albert, T.; Bai, R.; Wyss, M.; Perkins, J.B. The origins of 168, W23, and other Bacillus subtilis legacy strains. J. Bacteriol. 2008, 190, 6983–6995. [Google Scholar] [CrossRef] [PubMed]
- Nishito, Y.; Osana, Y.; Hachiya, T.; Popendorf, K.; Toyoda, A.; Fujiyama, A.; Itaya, M.; Sakakibara, Y. Whole genome assembly of a natto production strain Bacillus subtilis natto from very short read data. BMC Genom. 2010, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- Veith, B.; Herzberg, C.; Steckel, S.; Feesche, J.; Maurer, K.H.; Ehrenreich, P.; Bäumer, S.; Henne, A.; Liesegang, H.; Merkl, R.; et al. The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J. Mol. Microbiol. Biotechnol. 2004, 7, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.W.; Ramaiya, P.; Nelson, B.A.; Brody-Karpin, S.D.; Zaretsky, E.J.; Tang, M.; Lopez de Leon, A.; Xiang, H.; Gusti, V.; Clausen, I.G.; et al. Complete genome sequence of the industrial bacterium Bacillus licheniformis and comparisons with closely related Bacillus species. Genome Biol. 2004, 5, R77. [Google Scholar] [CrossRef] [PubMed]
- Rachinger, M.; Volland, S.; Meinhardt, F.; Daniel, R.; Liesegang, H. First Insights into the Completely Annotated Genome Sequence of Bacillus licheniformis Strain 9945A. Genome Announc. 2013, 1, e00525-13. [Google Scholar] [CrossRef] [PubMed]
- DSMZ German Collection of Microorganisms and Cell Cultures. Available online: https://www.dsmz.de/catalogues/details/culture/DSM-12369.html (accessed on 27 March 2017).
- Chen, X.H.; Koumoutsi, A.; Scholz, R.; Eisenreich, A.; Schneider, K.; Heinemeyer, I.; Morgenstern, B.; Voss, B.; Hess, W.R.; Reva, O.; et al. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat. Biotechnol. 2007, 25, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Tirumalai, M.R.; Fox, G.E. An ICEBs1-like element may be associated with the extreme radiation and desiccation resistance of Bacillus pumilus SAFR-032 spores. Extremophiles 2013, 17, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Yin, J. Bacteriophage Plaques: Theory and Analysis. In Bacteriophages; Interscience Publishers: New York, NY, USA, 2009; pp. 161–174. [Google Scholar]
- Abedon, S.T.; Yin, J. Impact of spatial structure on phage population growth. In Bacteriophage Ecology; Abedon, S.T., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 94–113. [Google Scholar]
- Ashelford, K.E.; Day, M.J.; Bailey, M.J.; Lilley, A.K.; Fry, J.C. In situ population dynamics of bacterial viruses in a terrestrial environment. Appl. Environ. Microbiol. 1999, 65, 169–174. [Google Scholar] [PubMed]
- Wang, I.-N.; Dykhuizen, D.E.; Slobodkin, L.B. The evolution of phage lysis timing. Evol. Ecol. 1996, 10, 545–558. [Google Scholar] [CrossRef]
- Bandara, N.; Jo, J.; Ryu, S.; Kim, K.-P. Bacteriophages BCP1-1 and BCP8-2 require divalent cations for efficient control of Bacillus cereus in fermented foods. Food Microbiol. 2012, 31, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Stewart, C.R. A cytotoxic early gene of Bacillus subtilis bacteriophage SPO1. J. Bacteriol. 1993, 175, 7887–7900. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P.; Abedon, S.T. Practical Methods for Determining Phage Growth Parameters. In Bacteriophages; Interscience Publishers: New York, NY, USA, 2009; pp. 175–202. [Google Scholar]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis–lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Shin, H.; Son, B.; Heu, S.; Ryu, S. Characterization and complete genome sequence of a virulent bacteriophage B4 infecting food-borne pathogenic Bacillus cereus. Arch. Virol. 2013, 158, 2101–2108. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Wishart, D.S.; She, Y.-M.; Villegas, A.; Lingohr, E.J.; Kropinski, A.M.; Gavin, O.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Barylski, J.; Nowicki, G.; Goździcka-Józefiak, A. The Discovery of phiAGATE, A Novel Phage Infecting Bacillus pumilus, Leads to New Insights into the Phylogeny of the Subfamily Spounavirinae. PLoS ONE 2014, 9, e86632. [Google Scholar] [CrossRef] [PubMed]
- Klumpp, J.; Schmuki, M.; Sozhamannan, S.; Beyer, W.; Fouts, D.E.; Bernbach, V.; Calendar, R.; Loessner, M.J. The odd one out: Bacillus ACT bacteriophage CP-51 exhibits unusual properties compared to related Spounavirinae W.Ph. and Bastille. Virology 2014, 462–463, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W.; Tremblay, D.; Moineau, S. Long-Term Bacteriophage Preservation. WFCC Newsl. 2004, 38, 35–40. [Google Scholar]

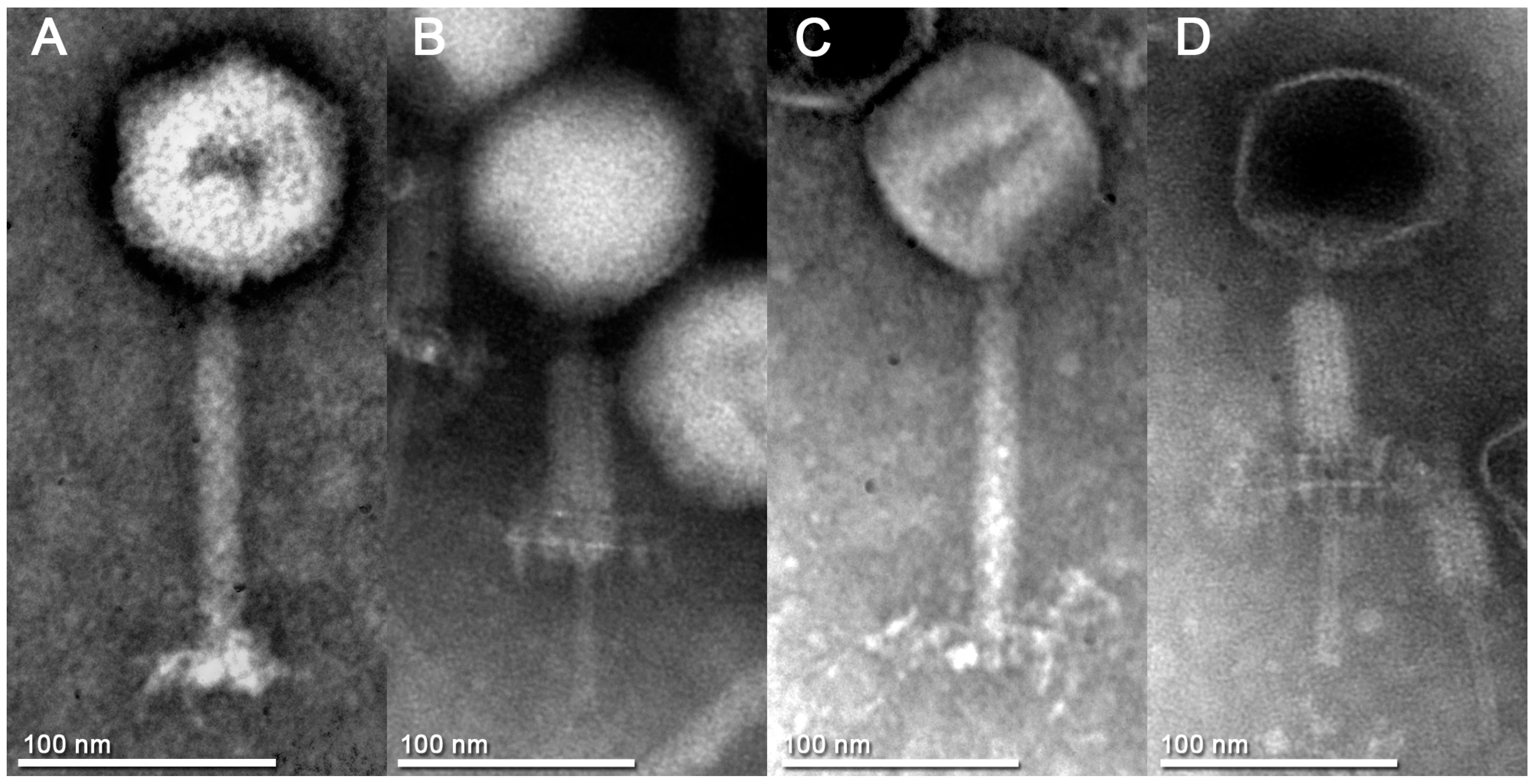
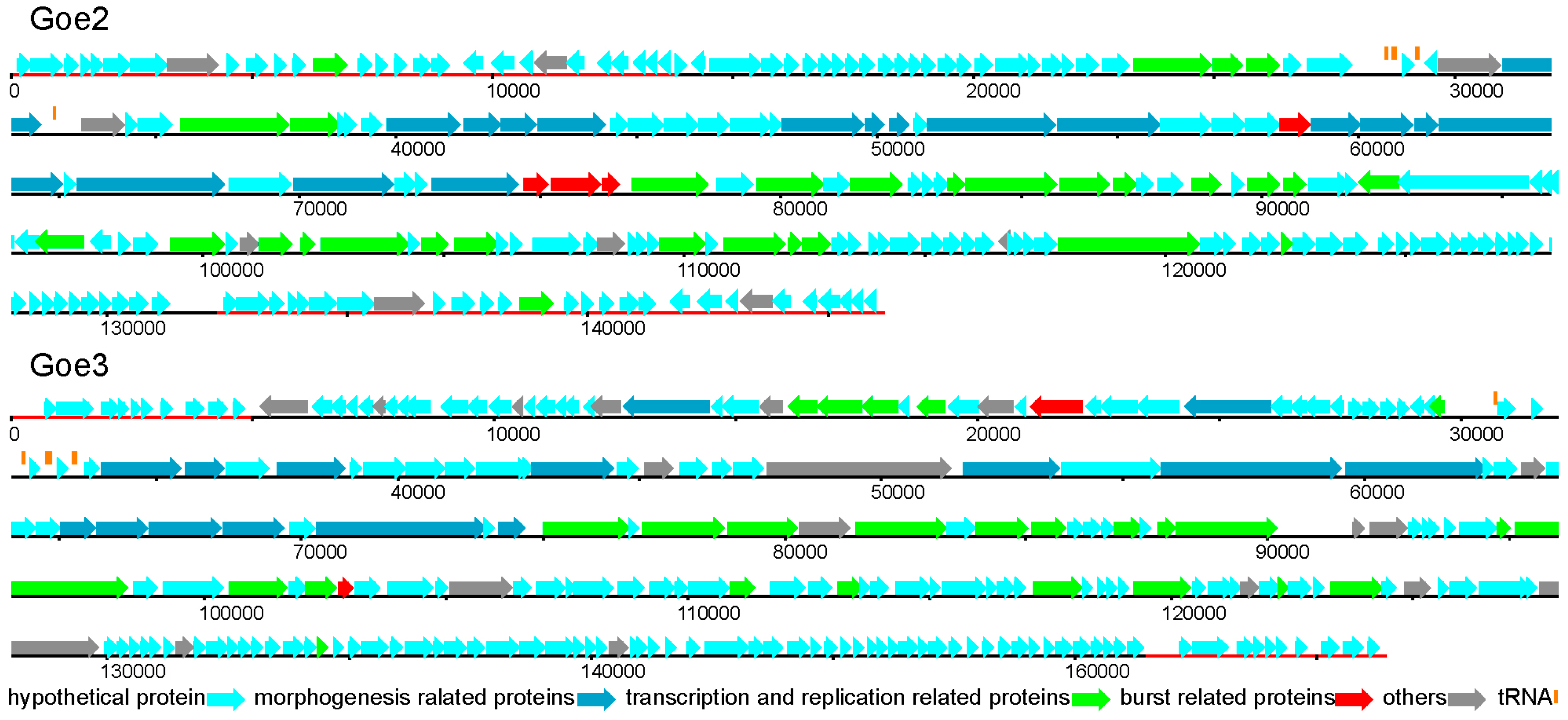


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Species | Goe2 | Goe3 |
|---|---|---|
| B. amyloliquefaciens FZB42 | ++ P | ++ P |
| B. licheniformis 9945A | + | + − |
| B. licheniformis DSM13 | − | + − |
| B. licheniformis 12369 | − | + |
| B. pumilus SAFR-032 | − | + − |
| B. subtilis natto | ++ | + |
| B. subtilis 168 | ++ | + |
| B. subtilis 3610 | ++ | + |
| B. subtilis Δ6 | ++ P | + P |
| Strain | Host | Burst Size in PFU | Latency Period in min | Adsorption in mL·min−1 | Reference |
|---|---|---|---|---|---|
| B4 | B. cereus | >200 | 10–15 min | – | [60] |
| Bc431v3 | B. cereus | 318 | 85 min | – | [61] |
| BCP8-2 | B. cereus | 50 | – | – | [56] |
| BCP1 | B. cereus | 50 | – | – | [56] |
| phiAGATA | B. pumilus | 153 | 35 | 6.44 × 10−9 | [62] |
| SPO1 | B. subtilis | 70 | 80 | – | [57] |
| CP-51 | B. cereus | 90 | 90–100 | – | [63] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willms, I.M.; Hoppert, M.; Hertel, R. Characterization of Bacillus subtilis Viruses vB_BsuM-Goe2 and vB_BsuM-Goe3. Viruses 2017, 9, 146. https://doi.org/10.3390/v9060146
Willms IM, Hoppert M, Hertel R. Characterization of Bacillus subtilis Viruses vB_BsuM-Goe2 and vB_BsuM-Goe3. Viruses. 2017; 9(6):146. https://doi.org/10.3390/v9060146
Chicago/Turabian StyleWillms, Inka M., Michael Hoppert, and Robert Hertel. 2017. "Characterization of Bacillus subtilis Viruses vB_BsuM-Goe2 and vB_BsuM-Goe3" Viruses 9, no. 6: 146. https://doi.org/10.3390/v9060146
APA StyleWillms, I. M., Hoppert, M., & Hertel, R. (2017). Characterization of Bacillus subtilis Viruses vB_BsuM-Goe2 and vB_BsuM-Goe3. Viruses, 9(6), 146. https://doi.org/10.3390/v9060146