
The Human Gut Phage Community and Its Implications for Health and Disease

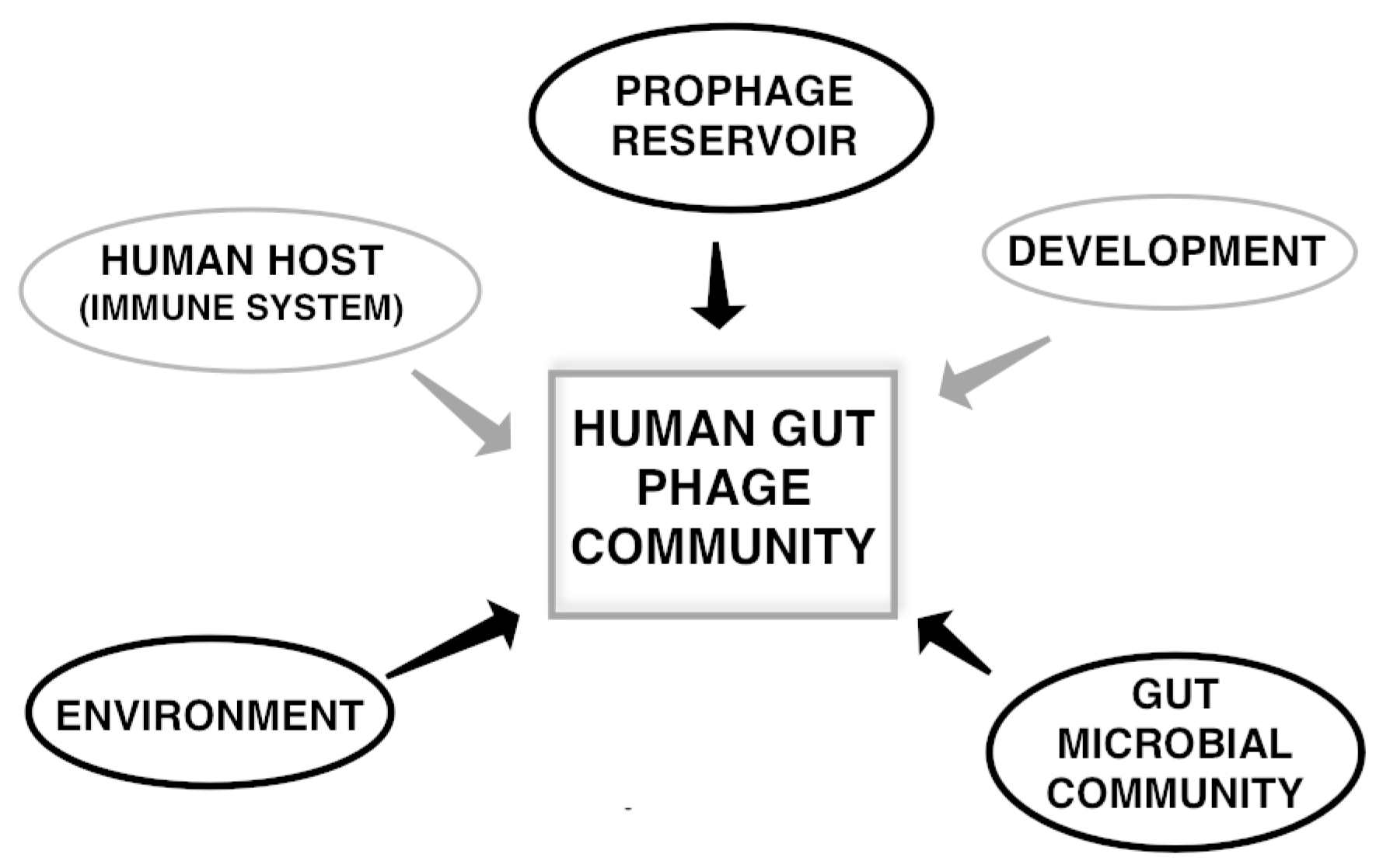{kind=link}
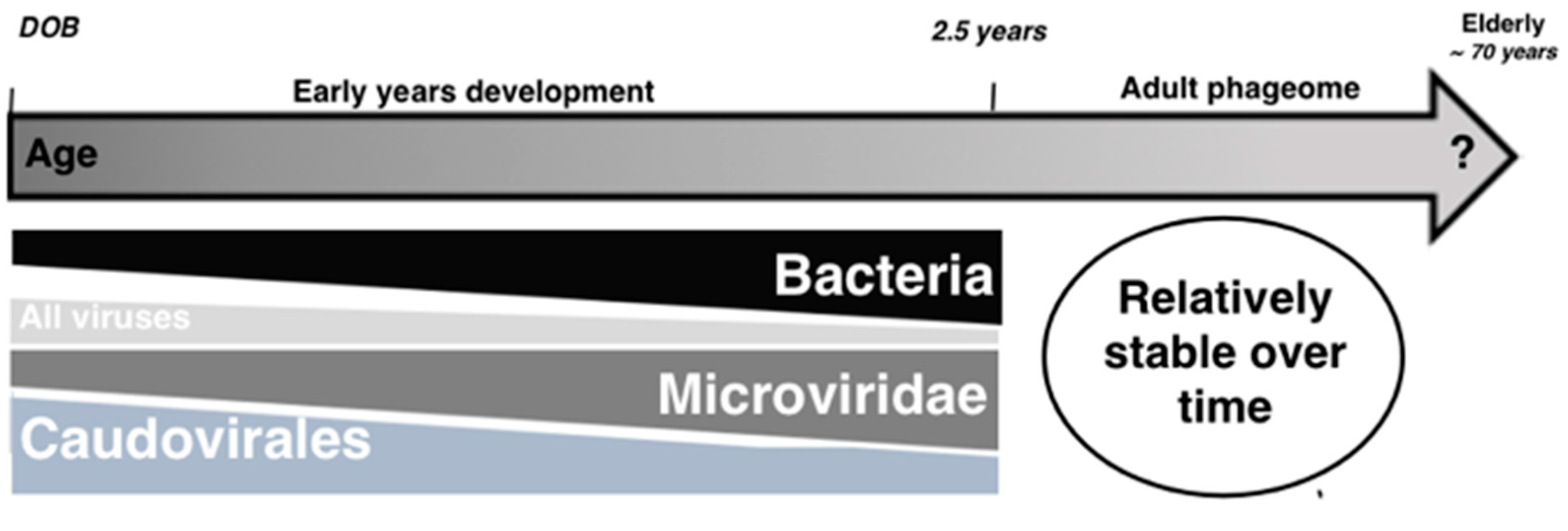{kind=link}
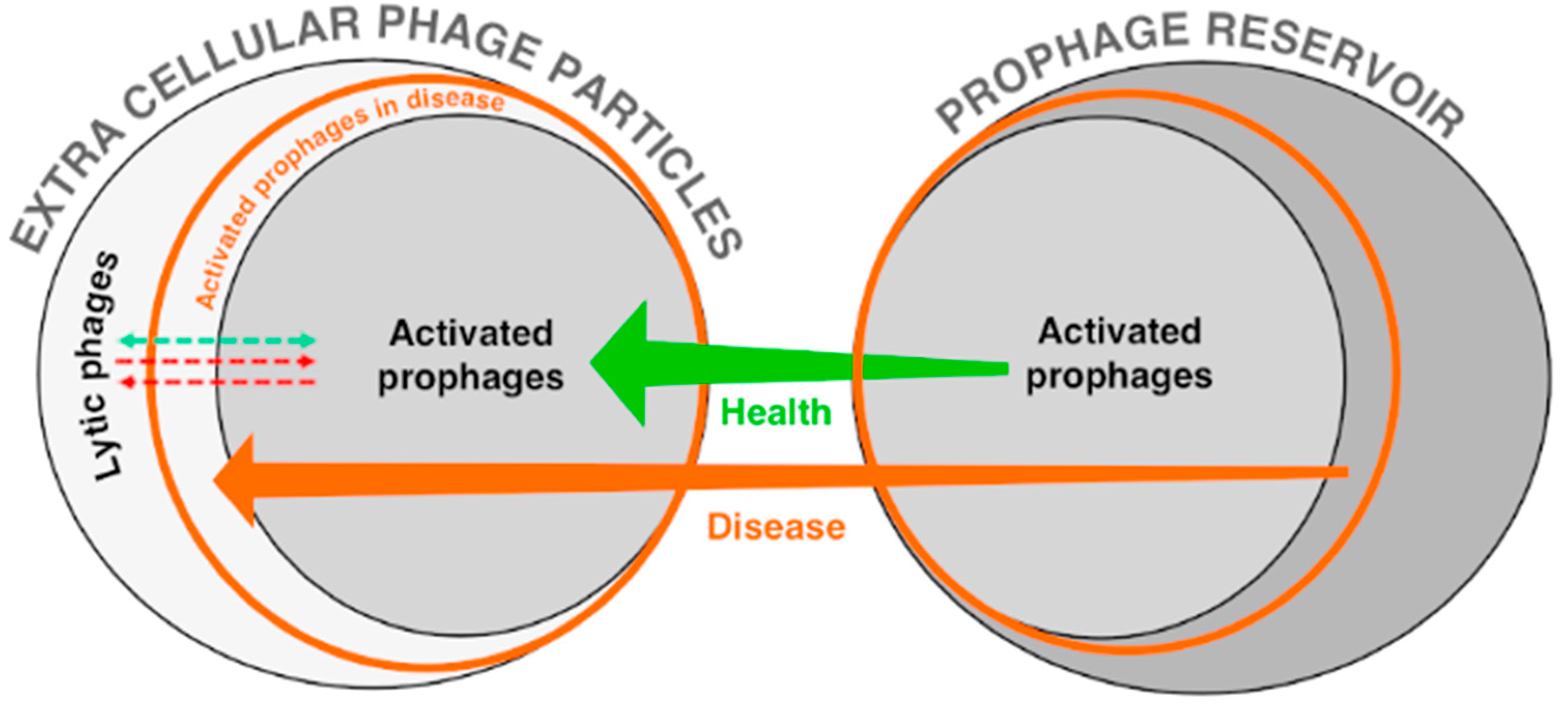{kind=link}
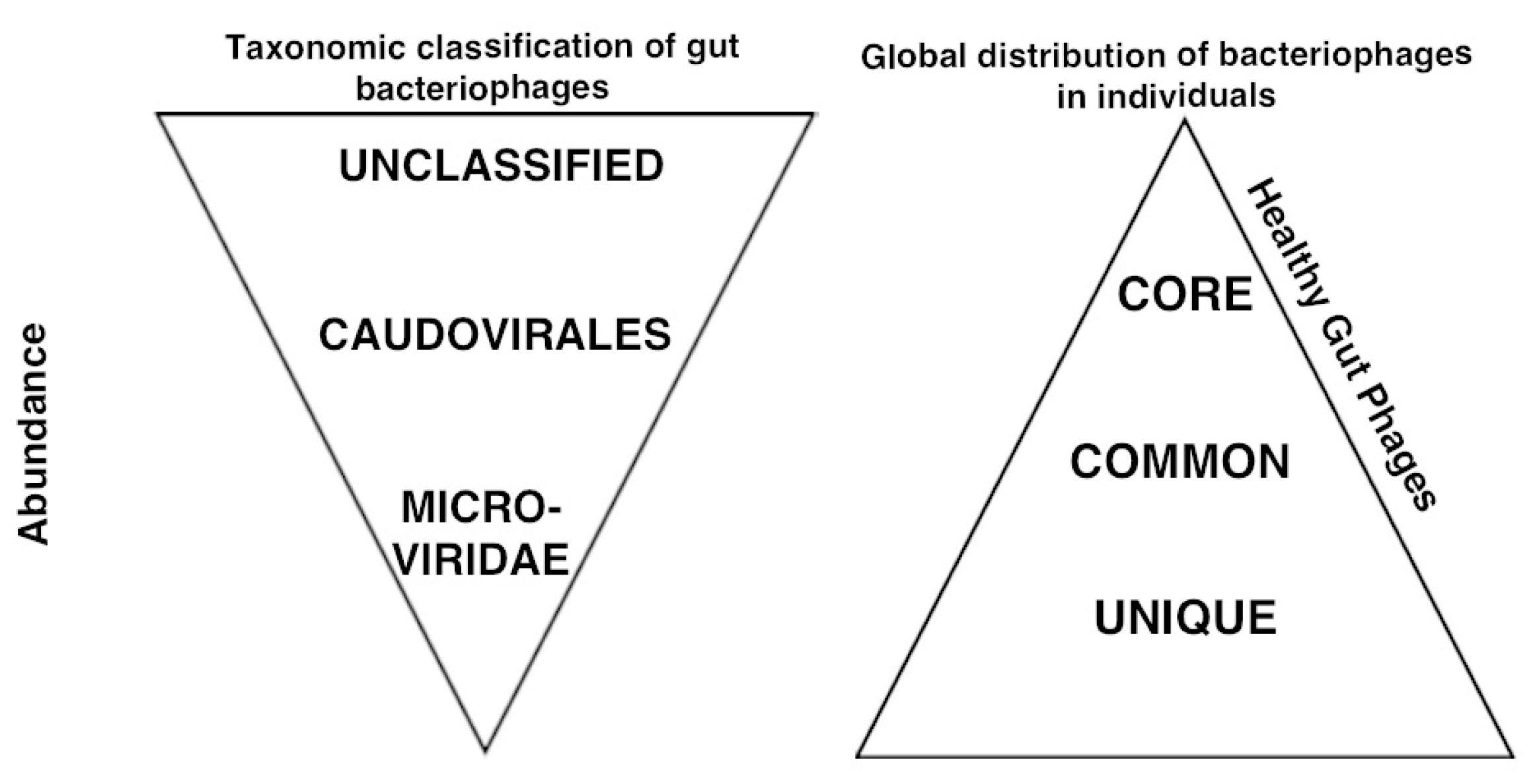{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Outcome of Phage–Bacteria Interactions
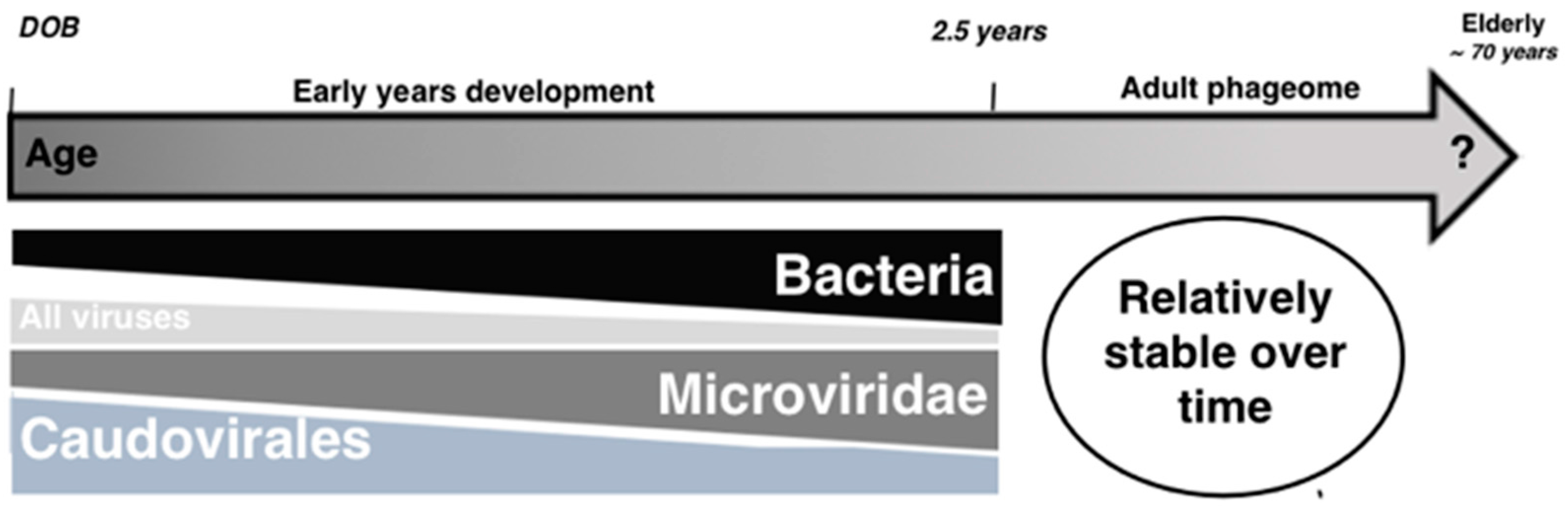
3. Development of the Gut Phage Community
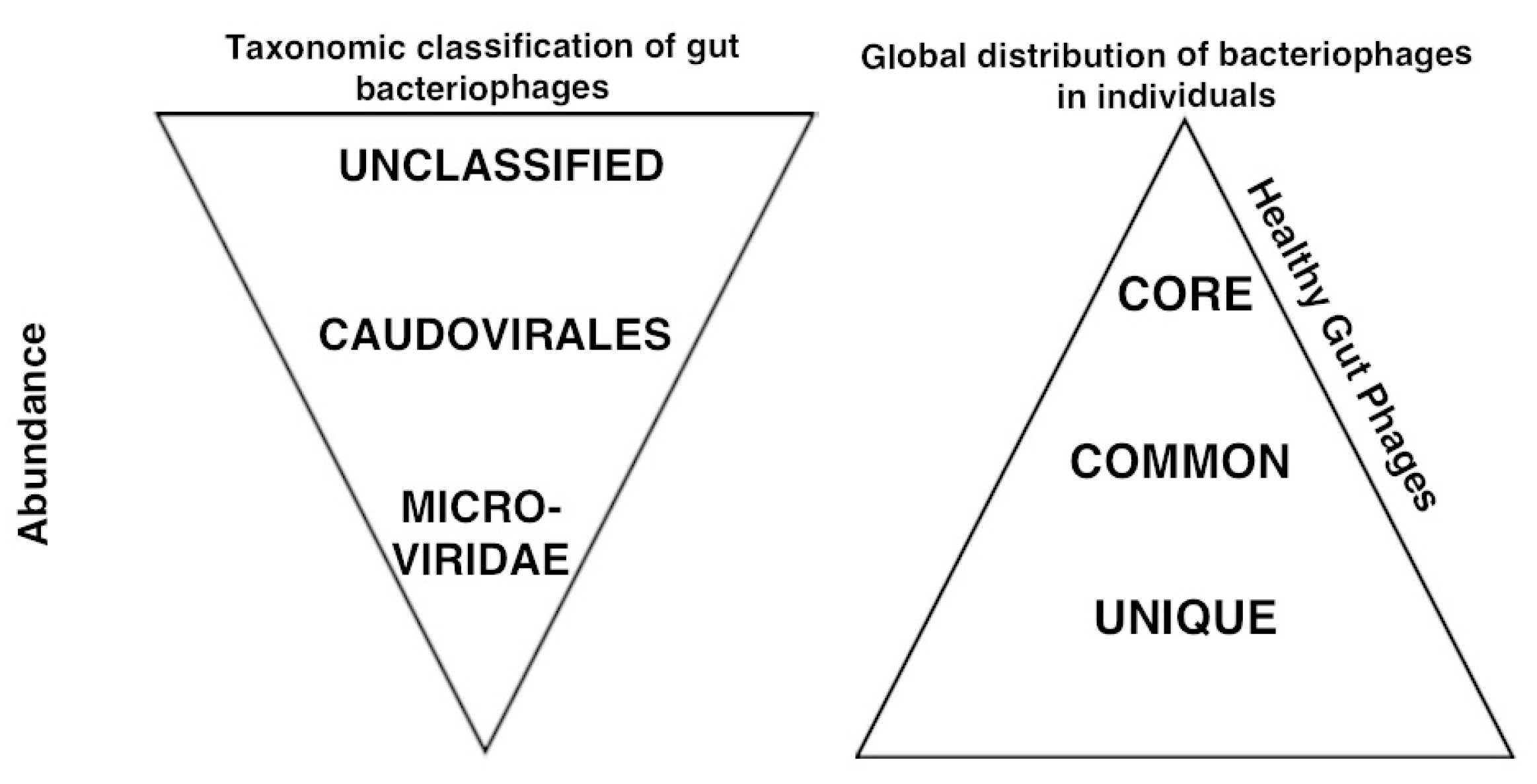
4. Characteristics of the Adult Gut Phage Community
5. Towards an Equilibrium between Lysis and Lysogeny
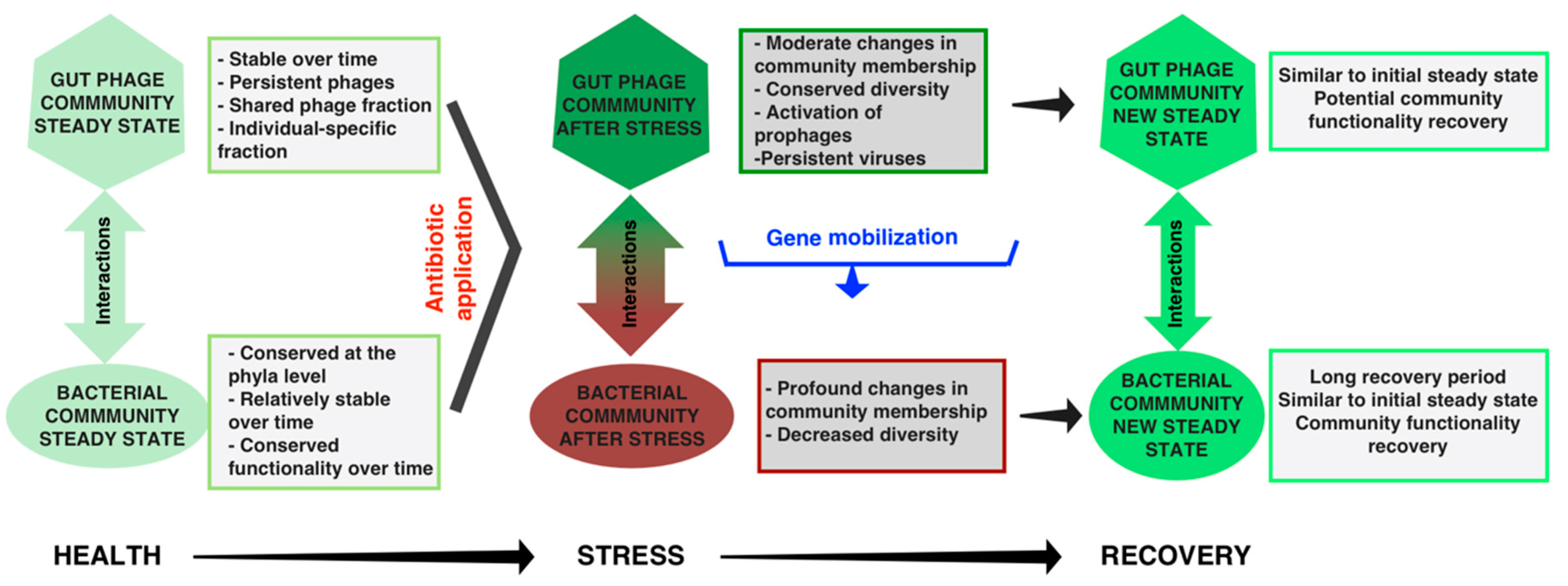
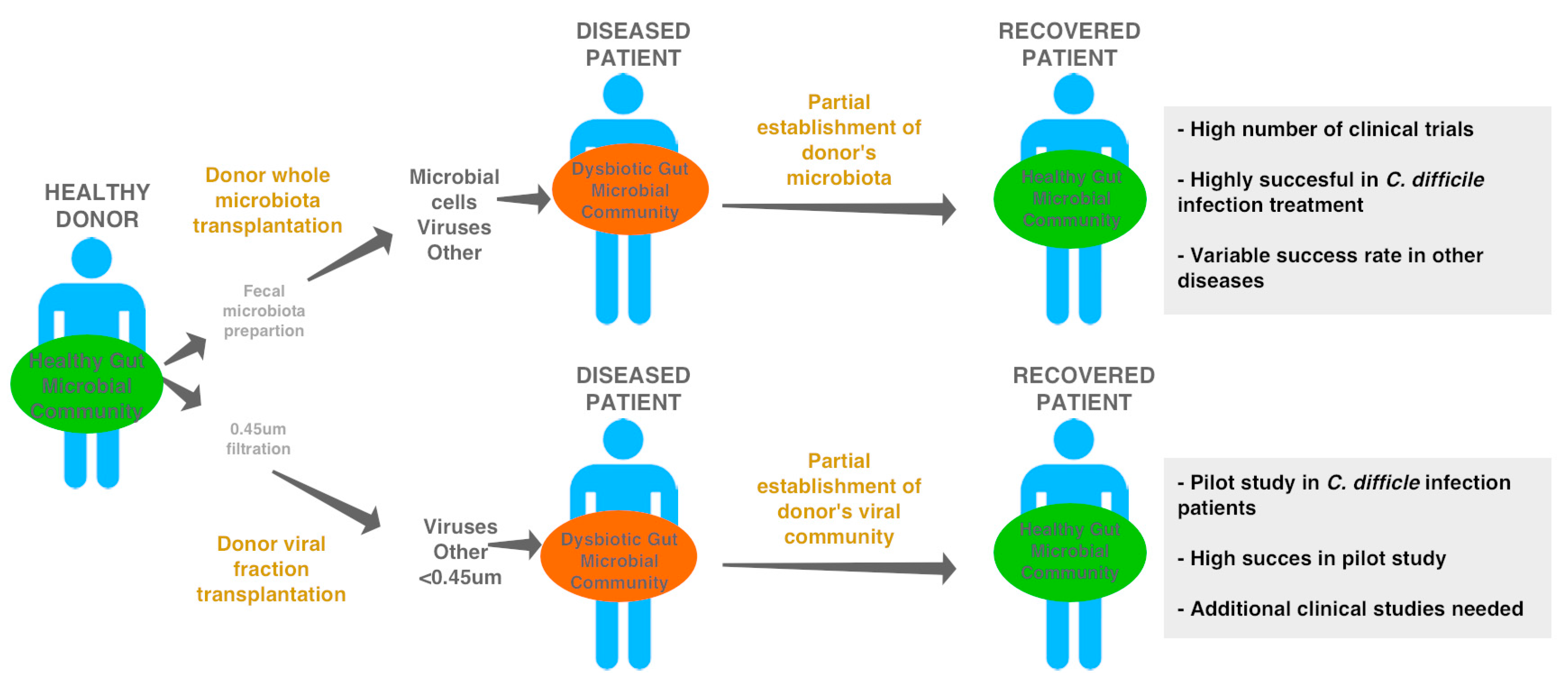
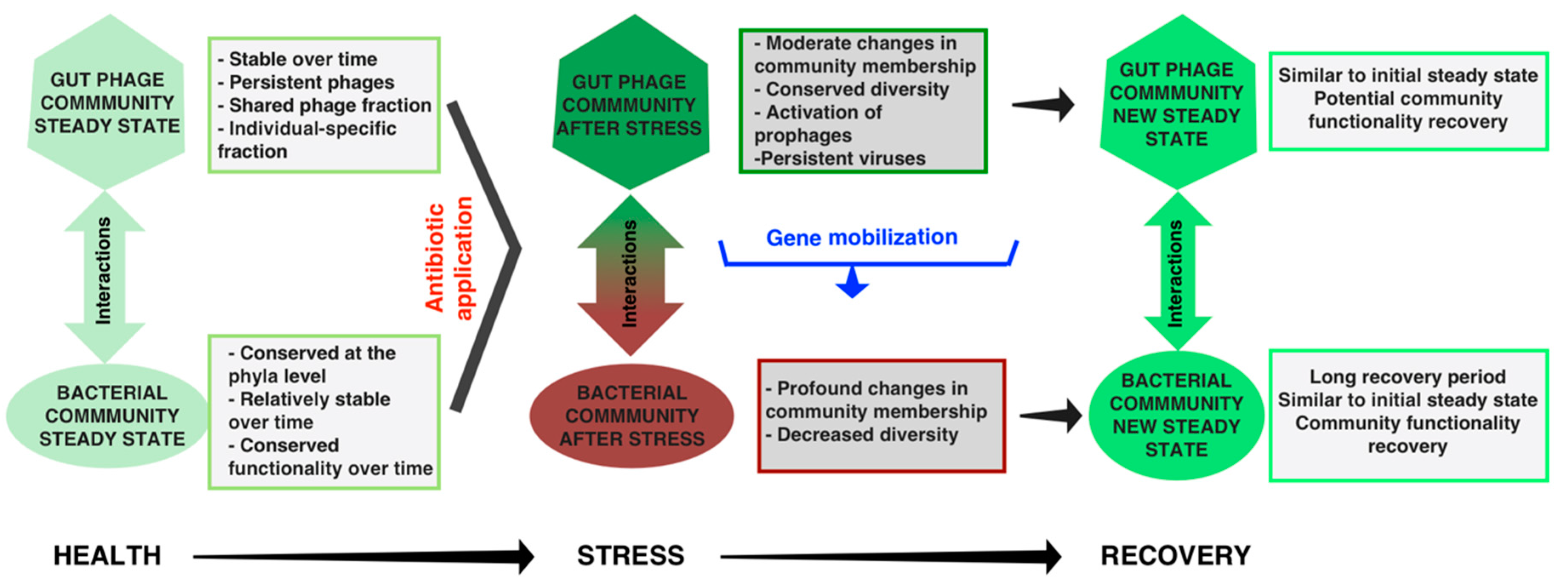
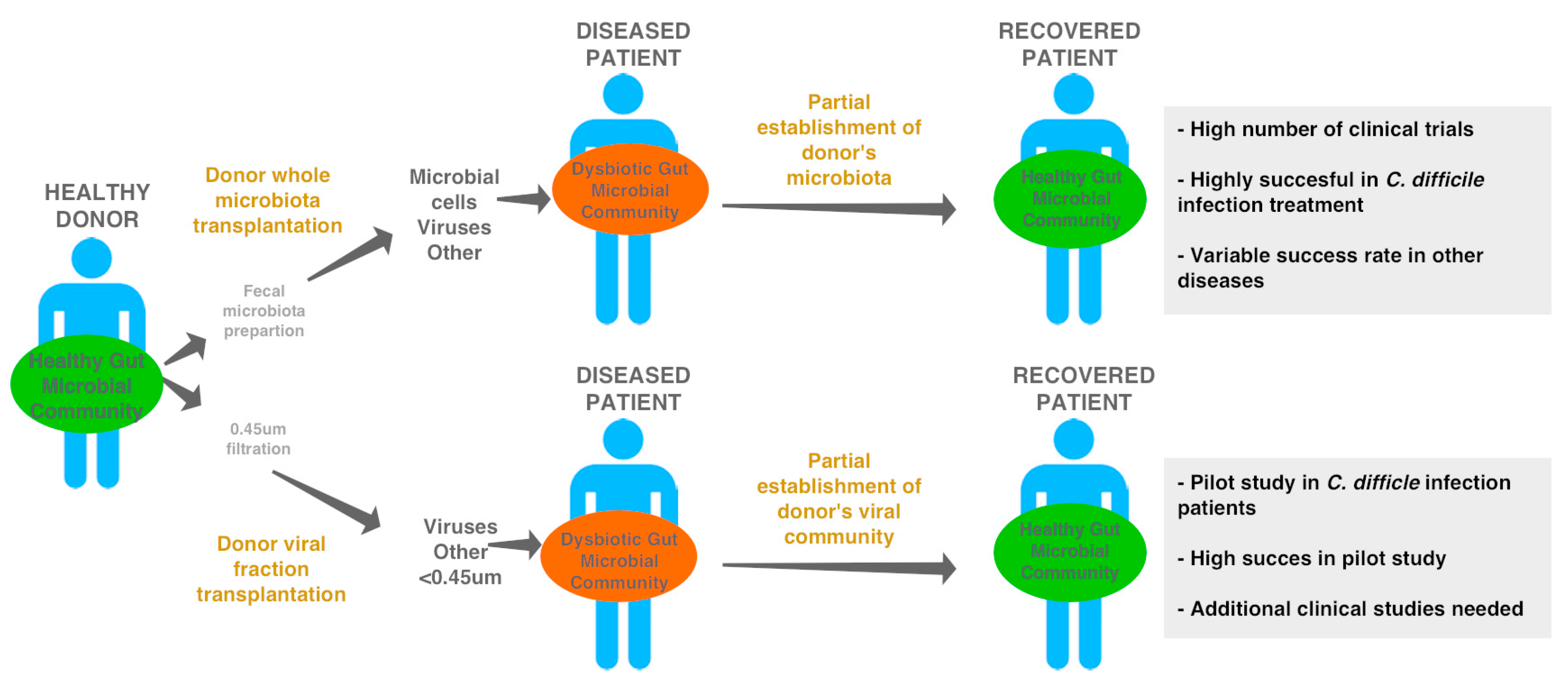
6. Gut Phages, Microbial Resilience, and Health
7. Remarks and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The long-term stability of the human gut microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Fraser, C.M.; Ringel, Y.; Sanders, M.E.; Sartor, R.B.; Sherman, P.M.; Versalovic, J.; Young, V.; Finlay, B.B. Defining a healthy human gut microbiome: Current concepts, future directions, and clinical applications. Cell Host Microbe 2012, 12, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef] [PubMed]
- Relman, D.A. The human microbiome: Ecosystem resilience and health. Nutr. Rev. 2012, 70, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A. Phage therapy: An attractive option for dealing with antibiotic-resistant bacterial infections. Drug Discov. Today 2005, 10, 807–809. [Google Scholar] [CrossRef]
- Brussow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Waldor, M.K. Bacteriophage control of bacterial virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going viral: Next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, M.; Leclerc, M.; Tinsley, C.R.; Petit, M.A. Bacteriophages: An underestimated role in human and animal health? Front. Cell Infect. Microbiol. 2014, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.; Shanahan, F.; Stanton, C.; Hill, C.; Coffey, A.; Ross, R.P. Movers and shakers: Influence of bacteriophages in shaping the mammalian gut microbiota. Gut Microbes 2013, 4, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.A.; Jones, B.V. The human gut virome: A multifaceted majority. Front. Microbiol. 2015, 6, 918. [Google Scholar] [CrossRef] [PubMed]
- Dalmasso, M.; Hill, C.; Ross, R.P. Exploiting gut bacteriophages for human health. Trends Microbiol. 2014, 22, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Skinner, S.O.; Zong, C.; Sippy, J.; Feiss, M.; Golding, I. Decision making at a subcellular level determines the outcome of bacteriophage infection. Cell 2010, 141, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Wegrzyn, G. Pseudolysogeny. Adv. Virus. Res. 2012, 82, 339–349. [Google Scholar] [PubMed]
- Cenens, W.; Makumi, A.; Govers, S.K.; Lavigne, R.; Aertsen, A. Viral Transmission Dynamics at Single-Cell Resolution Reveal Transiently Immune Subpopulations Caused by a Carrier State Association. PLoS Genet. 2015, 11, e1005770. [Google Scholar] [CrossRef] [PubMed]
- Kai, M.; Watanabe, S.; Furuse, K.; Ozawa, A. Bacteroides bacteriophages isolated from human feces. Microbiol. Immunol. 1985, 29, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Maura, D.; Debarbieux, L. On the interactions between virulent bacteriophages and bacteria in the gut. Bacteriophage 2012, 2, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Smeal, S.W.; Schmitt, M.A.; Pereira, R.R.; Prasad, A.; Fisk, J.D. Simulation of the M13 life cycle I: Assembly of a genetically-structured deterministic chemical kinetic simulation. Virology 2017, 500, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.B.; Rodriguez-Brito, B.; Pasic, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009, 7, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015, 13, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Macia, M.D.; Oliver, A.; Schachar, I.; Buckling, A. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 2007, 450, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, P.D.; Hall, A.R.; Blackshields, G.; Friman, V.P.; Davis, M.R., Jr.; Goldberg, J.B.; Buckling, A. Coevolution with bacteriophages drives genome-wide host evolution and constrains the acquisition of abiotic-beneficial mutations. Mol. Biol. Evol. 2015, 32, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, P.D. Bacteria-Bacteriophage Coevolution in the Human Gut: Implications for Microbial Diversity and Functionality. Trends Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lotka, A.J. Fluctuations in the abundance of a species considered mathematically. Nature 1927, 119, 12. [Google Scholar]
- Volterra, V. Fluctuations in the abundance of a species considered mathematically. Nature 1926, 118, 558–560. [Google Scholar] [CrossRef]
- Williams, H.T. Phage-induced diversification improves host evolvability. BMC Evol. Biol. 2013, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.H. Prophages in marine bacteria: Dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M. Marine viruses: Truth or dare. Ann. Rev. Mar. Sci. 2012, 4, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.C.; Paul, J.H. Gene transfer by transduction in the marine environment. Appl. Environ. Microbiol. 1998, 64, 2780–2787. [Google Scholar] [PubMed]
- Rodriguez-Brito, B.; Li, L.; Wegley, L.; Furlan, M.; Angly, F.; Breitbart, M.; Buchanan, J.; Desnues, C.; Dinsdale, E.; Edwards, R.; et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010, 4, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.R.; Lee, H.H.; Spina, C.S.; Collins, J.J. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 2013, 499, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Hurwitz, B.L.; Schofield, O.; Ducklow, H.W.; Sullivan, M.B. Seasonal time bombs: Dominant temperate viruses affect Southern Ocean microbial dynamics. ISME J. 2016, 10, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Knowles, B.; Silveira, C.B.; Bailey, B.A.; Barott, K.; Cantu, V.A.; Cobian-Guemes, A.G.; Coutinho, F.H.; Dinsdale, E.A.; Felts, B.; Furby, K.A.; et al. Lytic to temperate switching of viral communities. Nature 2016, 531, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K. The virome in host health and disease. Immunity 2015, 42, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Cadwell, K. Expanding the role of the virome: Commensalism in the gut. J. Virol. 2015, 89, 1951–1953. [Google Scholar] [CrossRef] [PubMed]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Wylie, K.M.; Mihindukulasuriya, K.A.; Zhou, Y.; Sodergren, E.; Storch, G.A.; Weinstock, G.M. Metagenomic analysis of double-stranded DNA viruses in healthy adults. BMC Biol. 2014, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Pfeiffer, J.K. Viruses and the Microbiota. Annu. Rev. Virol. 2014, 1, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Hooper, L.V. Resident viruses and their interactions with the immune system. Nat. Immunol. 2013, 14, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, P.W.; Jeffery, I.B. Gut microbiota and aging. Science 2015, 350, 1214–1215. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Haynes, M.; Kelley, S.; Angly, F.; Edwards, R.A.; Felts, B.; Mahaffy, J.M.; Mueller, J.; Nulton, J.; Rayhawk, S.; et al. Viral diversity and dynamics in an infant gut. Res. Microbiol. 2008, 159, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Blanton, L.V.; Cao, S.; Zhao, G.; Manary, M.; Trehan, I.; Smith, M.I.; Wang, D.; Virgin, H.W.; Rohwer, F.; et al. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. USA 2015, 112, 11941–11946. [Google Scholar] [CrossRef] [PubMed]
- Morowitz, M.J.; Denef, V.J.; Costello, E.K.; Thomas, B.C.; Poroyko, V.; Relman, D.A.; Banfield, J.F. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc. Natl. Acad. Sci. USA 2011, 108, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Sharon, I.; Morowitz, M.J.; Thomas, B.C.; Costello, E.K.; Relman, D.A.; Banfield, J.F. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome. Res. 2013, 23, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.A.; Milani, C.; Turroni, F.; Tremblay, D.; Ferrario, C.; Mancabelli, L.; Duranti, S.; Ward, D.V.; Ossiprandi, M.C.; Moineau, S.; et al. Prophages of the genus Bifidobacterium as modulating agents of the infant gut microbiota. Environ. Microbiol. 2016, 18, 2196–2213. [Google Scholar] [CrossRef] [PubMed]
- Castro-Mejia, J.L.; Muhammed, M.K.; Kot, W.; Neve, H.; Franz, C.M.; Hansen, L.H.; Vogensen, F.K.; Nielsen, D.S. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 2015, 3, 64. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; McCartney, A.L.; Neve, H.; Gibson, G.R.; Sanderson, J.D.; Heller, K.J.; van Sinderen, D. Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 2014, 165, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. USA 2013, 110, 10771–10776. [Google Scholar] [CrossRef] [PubMed]
- Maura, D.; Galtier, M.; Le Bouguenec, C.; Debarbieux, L. Virulent bacteriophages can target O104:H4 enteroaggregative Escherichia coli in the mouse intestine. Antimicrob. Agents Chemother. 2012, 56, 6235–6242. [Google Scholar] [CrossRef] [PubMed]
- Furuse, K.; Osawa, S.; Kawashiro, J.; Tanaka, R.; Ozawa, A.; Sawamura, S.; Yanagawa, Y.; Nagao, T.; Watanabe, I. Bacteriophage distribution in human faeces: Continuous survey of healthy subjects and patients with internal and leukaemic diseases. J. Gen. Virol. 1983, 64, 2039–2043. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; van der Oost, J.; de Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Breitbart, M.; Lee, W.H.; Run, J.Q.; Wei, C.L.; Soh, S.W.; Hibberd, M.L.; Liu, E.T.; Rohwer, F.; Ruan, Y. RNA viral community in human feces: Prevalence of plant pathogenic viruses. PLoS Biol. 2006, 4, e3. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Janowski, A.B.; Zhao, G.; Barouch, D.; Wang, D. Hyperexpansion of RNA Bacteriophage Diversity. PLoS Biol. 2016, 14, e1002409. [Google Scholar] [CrossRef] [PubMed]
- Havelaar, A.H.; Furuse, K.; Hogeboom, W.M. Bacteriophages and indicator bacteria in human and animal faeces. J. Appl. Bacteriol. 1986, 60, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Wirth, J.F.; Mazurie, A.; Young, M.J. Viral assemblage composition in Yellowstone acidic hot springs assessed by network analysis. ISME J. 2015, 9, 2162–2177. [Google Scholar] [CrossRef] [PubMed]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 2008, 25, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. Elife 2015, 4, e08490. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Ly, M.; Bonilla, N.; Pride, D.T. The human urine virome in association with urinary tract infections. Front. Microbiol. 2015, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.; Rodriguez-Brito, B.; Bangor, D.; McNairnie, P.; Breitbart, M.; Salamon, P.; Felts, B.; Nulton, J.; Mahaffy, J.; Rohwer, F. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics 2005, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Ignacio-Espinoza, J.C.; Solonenko, S.A.; Sullivan, M.B. The global virome: not as big as we thought? Curr. Opin. Virol. 2013, 3, 566–771. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New tools for viral metagenome comparison and assembled virome analysis. BMC Bioinformatics 2014, 15, 76. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A standard operating procedure for analysis of viral metagenome sequences. Stand Genomic Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Gorska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Waller, A.S.; Yamada, T.; Kristensen, D.M.; Kultima, J.R.; Sunagawa, S.; Koonin, E.V.; Bork, P. Classification and quantification of bacteriophage taxa in human gut metagenomes. ISME J. 2014, 8, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Park, E.J.; Roh, S.W.; Bae, J.W. Diversity and abundance of single-stranded DNA viruses in human feces. Appl. Environ. Microbiol. 2011, 77, 8062–8070. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Forterre, P. Microviridae goes temperate: Microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 2011, 6, e19893. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, L.A.; Bowler, L.D.; Caplin, J.; Dedi, C.; Diston, D.; Cheek, E.; Taylor, H.; Ebdon, J.E.; Jones, B.V. Genome signature-based dissection of human gut metagenomes to extract subliminal viral sequences. Nat. Commun. 2013, 4, 2420. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Mick, E.; Tirosh, I.; Sagy, O.; Sorek, R. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Research 2012, 22, 1985–1994. [Google Scholar] [CrossRef] [PubMed]
- Robles-Sikisaka, R.; Ly, M.; Boehm, T.; Naidu, M.; Salzman, J.; Pride, D.T. Association between living environment and human oral viral ecology. ISME J. 2013, 7, 1710–1724. [Google Scholar] [CrossRef] [PubMed]
- Ly, M.; Jones, M.B.; Abeles, S.R.; Santiago-Rodriguez, T.M.; Gao, J.; Chan, I.C.; Ghose, C.; Pride, D.T. Transmission of viruses via our microbiomes. Microbiome 2016, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Colombet, J.; Marteau, P.; Sime-Ngando, T.; Dore, J.; Leclerc, M. Dysbiosis in inflammatory bowel disease: A role for bacteriophages? Gut 2008, 57, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Bae, J.W. Spatial disturbances in altered mucosal and luminal gut viromes of diet-induced obese mice. Environ. Microbiol. 2016, 18, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.B.; Rowler, F.L. Piggy-back-the-Winner in host-associated microbial communities. Biofilms Microbiomes 2016, 18, 1498–1510. [Google Scholar]
- Weitz, J.; Beckett, S.J.; Brum, J.R.; Cael, B.B.; Dushoff, J. Lysis, Lysogeny, and Virus-Microbe Ratios. Biorxiv 2016. [Google Scholar] [CrossRef]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis-lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.K.; Maurice, C.F. Menage a trois in the human gut: Interactions between host, bacteria and phages. Nat. Rev. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, G.W.; Jiang, W.; Bikard, D.; Marraffini, L.A. Conditional tolerance of temperate phages via transcription-dependent CRISPR-Cas targeting. Nature 2014, 514, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Lawrence, C.M.; Wiedenheft, B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 2013, 82, 237–266. [Google Scholar] [CrossRef] [PubMed]
- Mick, E.; Stern, A.; Sorek, R. Holding a grudge: Persisting anti-phage CRISPR immunity in multiple human gut microbiomes. RNA Biol. 2013, 10, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Qimron, U. The Escherichia coli CRISPR system protects from lambda lysogenization, lysogens, and prophage induction. J. Bacteriol. 2010, 192, 6291–6294. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 2013, 493, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Duerkop, B.A.; Clements, C.V.; Rollins, D.; Rodrigues, J.L.; Hooper, L.V. A composite bacteriophage alters colonization by an intestinal commensal bacterium. Proc. Natl. Acad. Sci. USA 2012, 109, 17621–17626. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Wu, M.; McNulty, N.P.; Rohwer, F.L.; Gordon, J.I. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. USA 2013, 110, 20236–20241. [Google Scholar] [CrossRef] [PubMed]
- Maura, D.; Morello, E.; du Merle, L.; Bomme, P.; Le Bouguenec, C.; Debarbieux, L. Intestinal colonization by enteroaggregative Escherichia coli supports long-term bacteriophage replication in mice. Environ. Microbiol. 2012, 14, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Ly, M.; Daigneault, M.C.; Brown, I.H.; McDonald, J.A.; Bonilla, N.; Vercoe, E.A.; Pride, D.T. Chemostat culture systems support diverse bacteriophage communities from human feces. Microbiome 2015, 3, 58. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J.; Auro, R.; Sam-Soon, N.; Kassegne, S.; Peters, G.; Bonilla, N.; Hatay, M.; Mourtada, S.; Bailey, B.; Youle, M.; et al. Subdiffusive motion of bacteriophage in mucosal surfaces increases the frequency of bacterial encounters. Proc. Natl. Acad. Sci. USA 2015, 112, 13675–13680. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Li, H.; Collins, J.J.; Ingber, D.E. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc. Natl. Acad. Sci. USA 2016, 113, E7–E15. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.A.; Fuentes, S.; Schroeter, K.; Heikamp-deJong, I.; Khursigara, C.M.; de Vos, W.M.; Allen-Vercoe, E. Simulating distal gut mucosal and luminal communities using packed-column biofilm reactors and an in vitro chemostat model. J. Microbiol. Methods 2015, 108, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev. 2016, 40, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Marbouty, M.; Baudry, L.; Cournac, A.; Koszul, R. Scaffolding bacterial genomes and probing host-virus interactions in gut microbiome by proximity ligation (chromosome capture) assay. Sci. Adv. 2017, 3, e1602105. [Google Scholar] [CrossRef] [PubMed]
- Kilic, A.O.; Pavlova, S.I.; Alpay, S.; Kilic, S.S.; Tao, L. Comparative study of vaginal Lactobacillus phages isolated from women in the United States and Turkey: Prevalence, morphology, host range, and DNA homology. Clin. Diagn. Lab. Immunol. 2001, 8, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.; Ringus, D.L.; Williams, R.J.; Choo, Z.N.; Greenwald, S.M.; Owens, S.M.; Coleman, M.L.; Meyer, F.; Chang, E.B. Divergent responses of viral and bacterial communities in the gut microbiome to dietary disturbances in mice. ISME J. 2016, 10, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2015, 6, 1543. [Google Scholar] [CrossRef] [PubMed]
- Abeles, S.R.; Ly, M.; Santiago-Rodriguez, T.M.; Pride, D.T. Effects of Long Term Antibiotic Therapy on Human Oral and Fecal Viromes. PLoS ONE 2015, 10, e0134941. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.L.; Relman, D.A. Microbiota’s ‘little helpers’: Bacteriophages and antibiotic-associated responses in the gut microbiome. Genome Biol. 2013, 14, 127. [Google Scholar] [CrossRef] [PubMed]
- Bojanova, D.P.; Bordenstein, S.R. Fecal Transplants: What Is Being Transferred? PLoS Biol. 2016, 14, e1002503. [Google Scholar] [CrossRef] [PubMed]
- Groen, A.K.; Nieuwdorp, M. An evaluation of the therapeutic potential of fecal microbiota transplantation to treat infectious and metabolic diseases. EMBO Mol. Med. 2017, 9, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.; van Nood, E.; Tims, S.; Heikamp-de Jong, I.; ter Braak, C.J.; Keller, J.J.; Zoetendal, E.G.; de Vos, W.M. Reset of a critically disturbed microbial ecosystem: Faecal transplant in recurrent Clostridium difficile infection. ISME J. 2014, 8, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- van Nood, E.; Vrieze, A.; Nieuwdorp, M.; Fuentes, S.; Zoetendal, E.G.; de Vos, W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Moelling, K.; Broecker, F. Fecal microbiota transplantation to fight Clostridium difficile infections and other intestinal diseases. Bacteriophage 2016, 6, e1251380. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Allen-Vercoe, E.; Petrof, E.O. Fecal microbiota transplantation: In perspective. Therap. Adv. Gastroenterol. 2016, 9, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.J.; Borody, T.J. “Putting back the bugs”: Bacterial treatment relieves chronic constipation and symptoms of irritable bowel syndrome. Med. J. Aust. 1993, 159, 633–634. [Google Scholar] [PubMed]
- Li, S.S.; Zhu, A.; Benes, V.; Costea, P.I.; Hercog, R.; Hildebrand, F.; Huerta-Cepas, J.; Nieuwdorp, M.; Salojarvi, J.; Voigt, A.Y.; et al. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 2016, 352, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Broecker, F.; Klumpp, J.; Moelling, K. Long-term microbiota and virome in a Zurich patient after fecal transplantation against Clostridium difficile infection. Ann. N. Y. Acad. Sci. 2016, 1372, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Broecker, F.; Klumpp, J.; Schuppler, M.; Russo, G.; Biedermann, L.; Hombach, M.; Rogler, G.; Moelling, K. Long-term changes of bacterial and viral compositions in the intestine of a recovered Clostridium difficile patient after fecal microbiota transplantation. Cold Spring Harb. Mol. Case Stud. 2016, 2, a000448. [Google Scholar] [CrossRef] [PubMed]
- Broecker, F.; Kube, M.; Klumpp, J.; Schuppler, M.; Biedermann, L.; Hecht, J.; Hombach, M.; Keller, P.M.; Rogler, G.; Moelling, K. Analysis of the intestinal microbiome of a recovered Clostridium difficile patient after fecal transplantation. Digestion 2013, 88, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Chehoud, C.; Dryga, A.; Hwang, Y.; Nagy-Szakal, D.; Hollister, E.B.; Luna, R.A.; Versalovic, J.; Kellermayer, R.; Bushman, F.D. Transfer of Viral Communities between Human Individuals during Fecal Microbiota Transplantation. MBio 2016, 7, e00322. [Google Scholar] [CrossRef] [PubMed]
- Bahra, S.M.; Weidemann, B.J.; Castro, A.N.; Walsh, J.W.; deLeon, O.; Burnett, C.M.; Pearson, N.A.; Murry, D.J.; Grobe, J.L.; Kirby, J.R. Risperidone-induced weight gain is mediated through shifts in the gut microbiome and suppression of energy expenditure. EBioMedicine 2015, 2, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients with Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Parr, N. The evolution of bacterial resistance against bacteriophages in the horse chestnut phyllosphere is general across both space and time. Philos. Trans. R. Soc. Lond. B. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Petrof, E.O.; Gloor, G.B.; Vanner, S.J.; Weese, S.J.; Carter, D.; Daigneault, M.C.; Brown, E.M.; Schroeter, K.; Allen-Vercoe, E. Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manrique, P.; Dills, M.; Young, M.J. The Human Gut Phage Community and Its Implications for Health and Disease. Viruses 2017, 9, 141. https://doi.org/10.3390/v9060141
Manrique P, Dills M, Young MJ. The Human Gut Phage Community and Its Implications for Health and Disease. Viruses. 2017; 9(6):141. https://doi.org/10.3390/v9060141
Chicago/Turabian StyleManrique, Pilar, Michael Dills, and Mark J. Young. 2017. "The Human Gut Phage Community and Its Implications for Health and Disease" Viruses 9, no. 6: 141. https://doi.org/10.3390/v9060141
APA StyleManrique, P., Dills, M., & Young, M. J. (2017). The Human Gut Phage Community and Its Implications for Health and Disease. Viruses, 9(6), 141. https://doi.org/10.3390/v9060141