
Coccolithoviruses: A Review of Cross-Kingdom Genomic Thievery and Metabolic Thuggery

 ,
,  ,
,
Abstract
:1. Introduction
2. Methods
2.1. Coccolithoviruses and Their Phylogeny
2.2. Whole Genome Reconstruction, Alignment and Comparison
2.3. “Best BLAST Hit” Analysis of Genomes
3. Results and Discussion
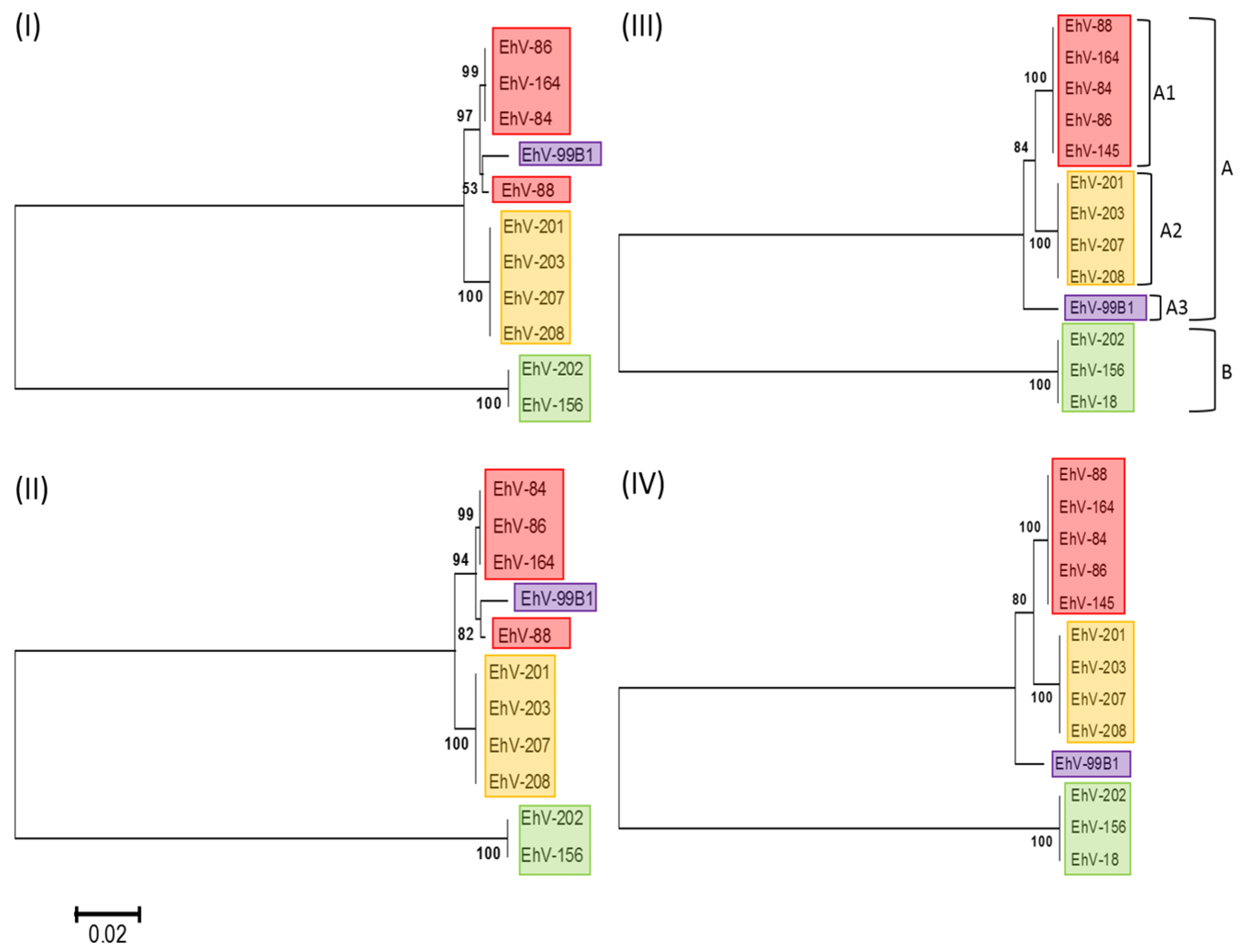
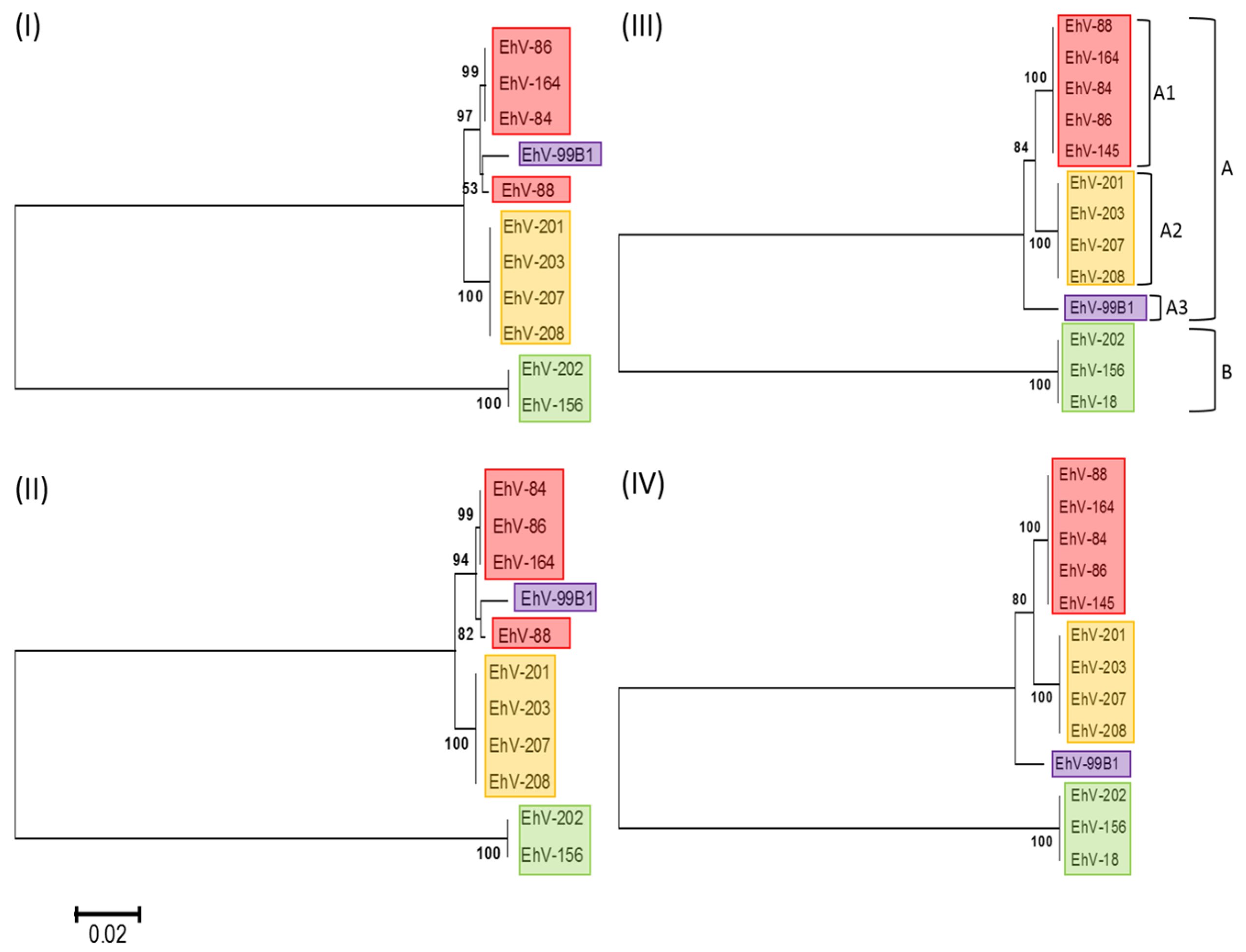
3.1. Phylogenetic Relationships of Coccolithoviruses
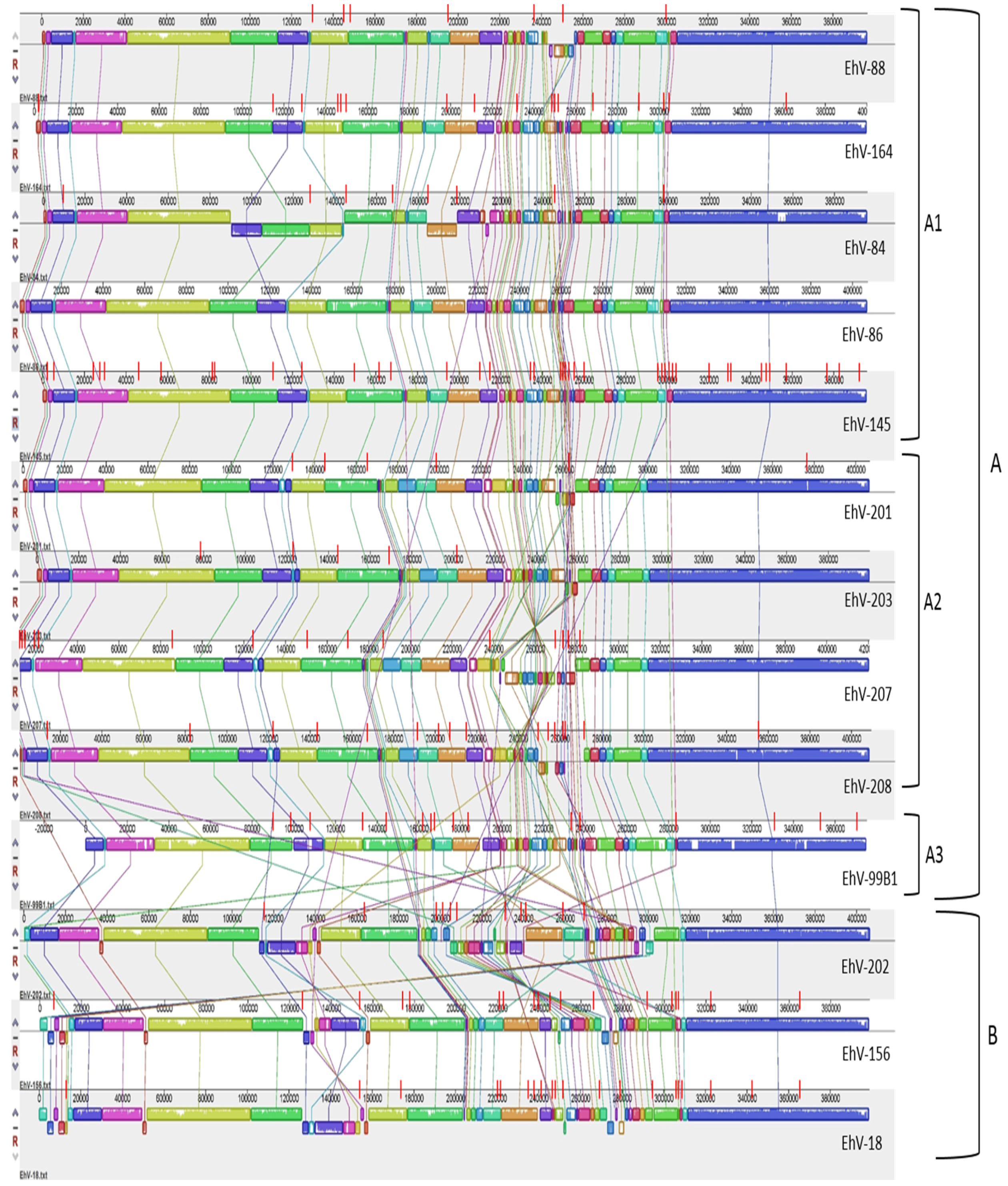
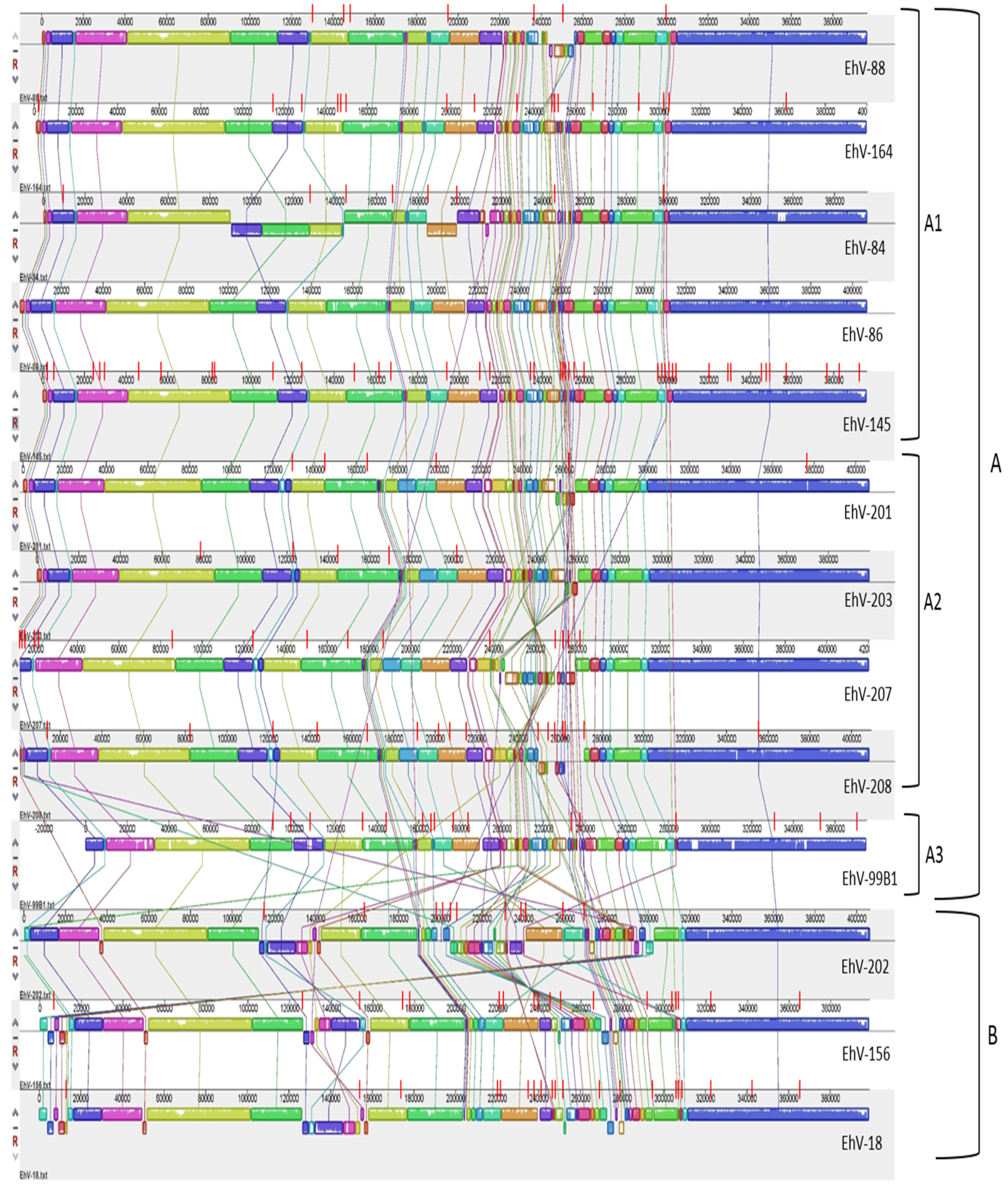
3.2. Homology, Heterology and Genome Structure
3.3. Genome Size and Putative Gene Differences
3.4. Coccolithovirus Gene Similarities to the Emiliania huxleyi Host
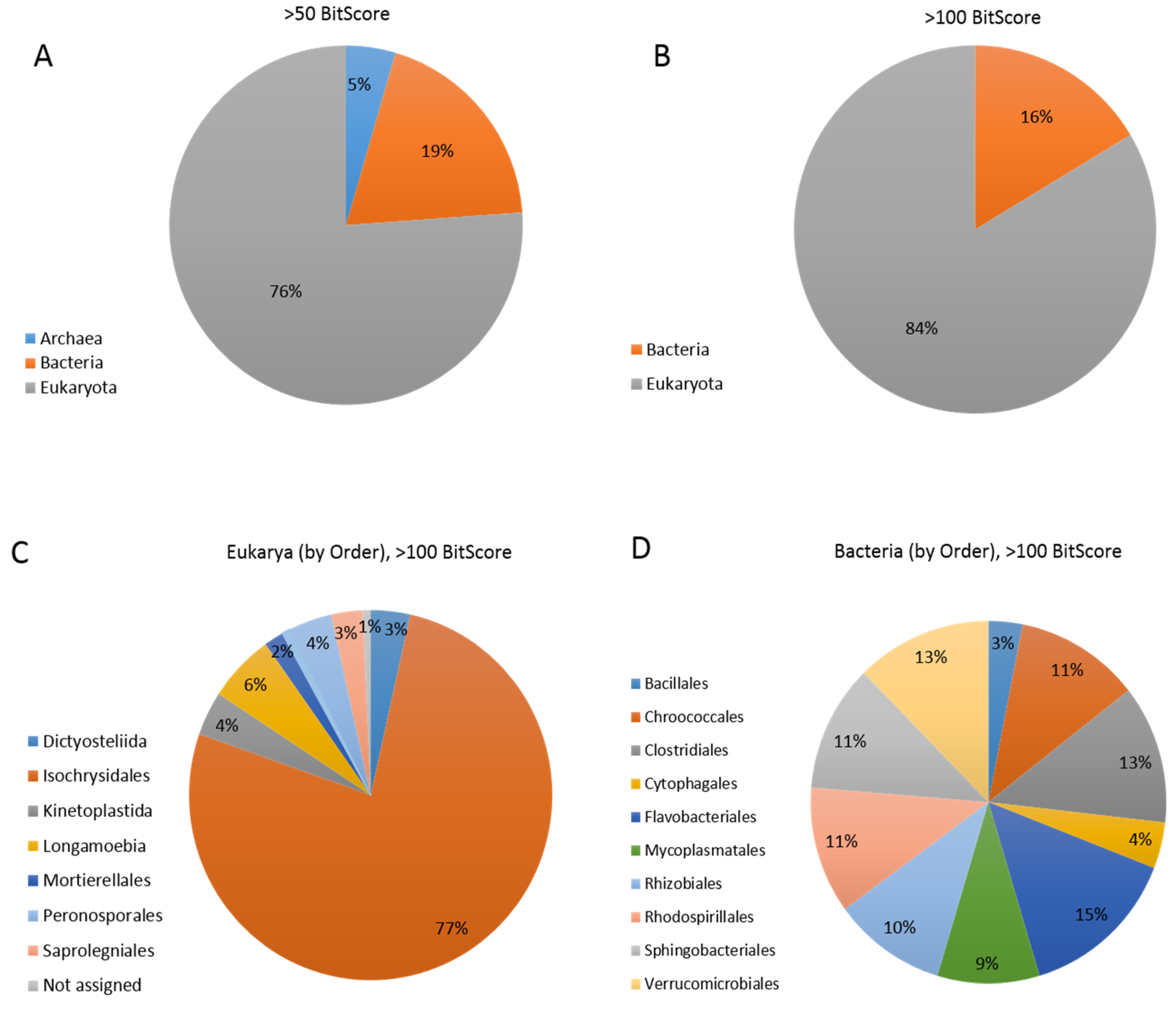
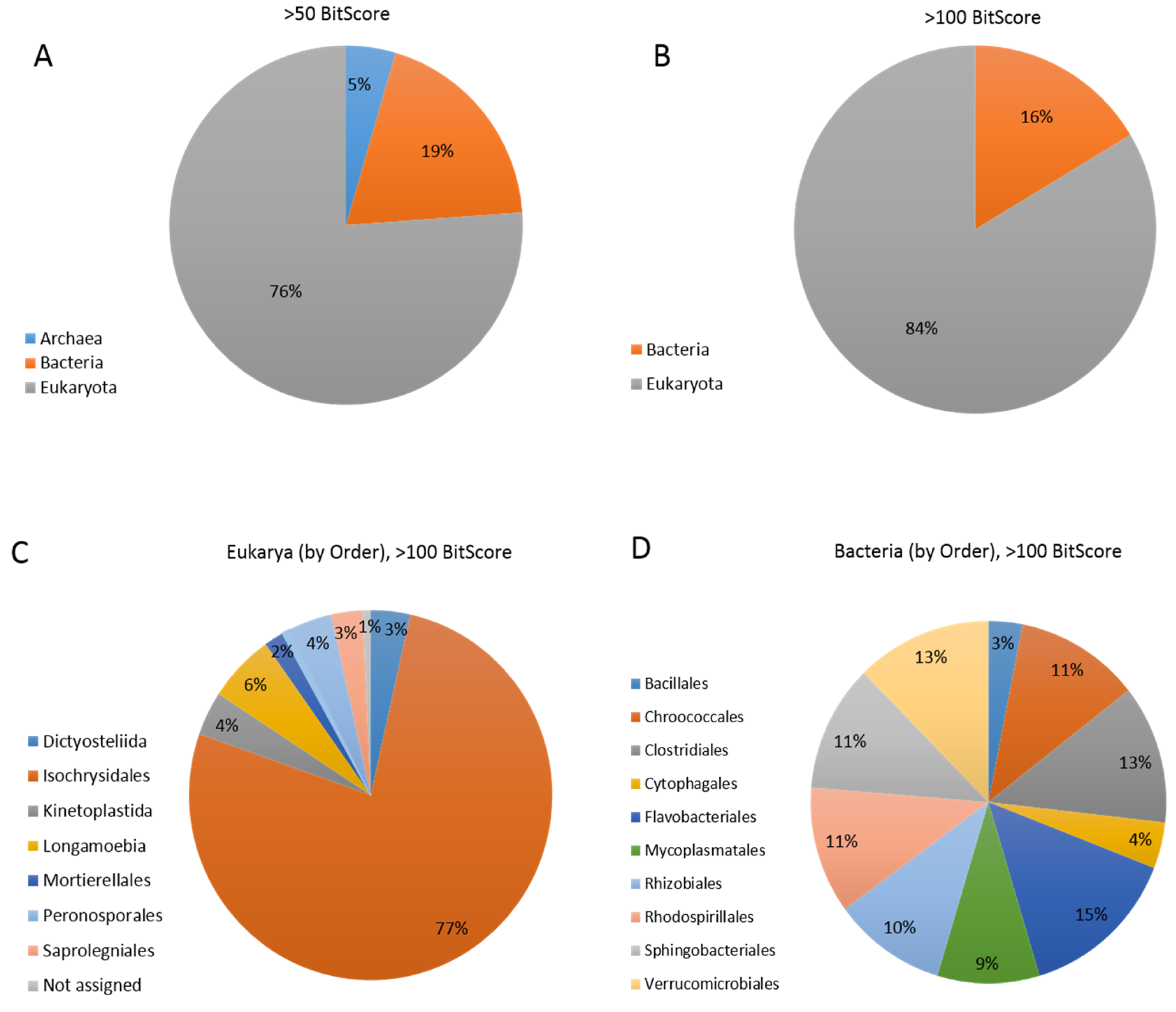
3.5. Coccolithovirus Gene Similarities to Other Eukaryotes
3.6. Coccolithovirus Gene Similarities to Bacteria
3.7. Possible Mode for HGT with Bacteria
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wilson, W.H.; Schroeder, D.C.; Allen, M.J.; Holden, M.T.G.; Parkhill, J.; Barrell, B.G.; Churcher, C.; Hamlin, N.; Mungall, K.; Norbertczak, H.; et al. Complete genome sequence and lytic phase transcription profile of a Coccolithovirus. Science 2005, 309, 1090–1092. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.C.; Oke, J.; Malin, G.; Wilson, W.H. Coccolithovirus (Phycodnaviridae): Characterisation of a new large dsDNA algal virus that infects Emiliana huxleyi. Arch. Virol. 2002, 147, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Tarran, G.A.; Schroeder, D.; Cox, M.J.; Oke, J.; Malin, G. Isolation of viruses responsible for the demise of an Emiliania huxleyi bloom in the English Channel. J. Mar. Biol. Assoc. 2002, 82, 369–377. [Google Scholar] [CrossRef]
- Allen, M.J.; Schroeder, D.C.; Holden, M.T.G.; Wilson, W.H. Evolutionary history of the Coccolithoviridae. Mol. Biol. Evol. 2006, 23, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Dunigan, D.D.; Fitzgerald, L.A.; Van Etten, J.L. Phycodnaviruses: A peek at genetic diversity. Virus Res. 2006, 117, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Van Etten, J.L.; Allen, M.J. The Phycodnaviridae: The story of how tiny giants rule the world. Curr. Top. Microbiol. Immunol. 2009, 328, 1–42. [Google Scholar] [PubMed]
- Martínez, J.M.; Schroeder, D.C.; Larsen, A.; Bratbak, G.; Wilson, W.H. Molecular dynamics of Emiliania huxleyi and cooccurring viruses during two separate mesocosm studies. Appl. Environ. Microbiol. 2007, 73, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.M.; Schroeder, D.C.; Wilson, W.H. Dynamics and genotypic composition of Emiliania huxleyi and their co-occurring viruses during a coccolithophore bloom in the North Sea. FEMS Microbiol. Ecol. 2012, 81, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Pagarete, A.; Allen, M.J.; Wilson, W.H.; Kimmance, S.A.; de Vargas, C. Host-virus shift of the sphingolipid pathway along an Emiliania huxleyi bloom: Survival of the fattest. Environ. Microbiol. 2009, 11, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Vardi, A.; Haramaty, L.; Van Mooy, B.A.S.; Fredricks, H.F.; Kimmance, S.A.; Larsen, A.; Bidle, K.D. Host-virus dynamics and subcellular controls of cell fate in a natural coccolithophore population. Proc. Natl. Acad. Sci. USA 2012, 109, 19327–19332. [Google Scholar] [CrossRef] [PubMed]
- Kimmance, S.A.; Allen, M.J.; Pagarete, A.; Martínez Martínez, J.; Wilson, W.H. Reduction in photosystem II efficiency during a virus-controlled Emiliania huxleyi bloom. Mar. Ecol. Prog. Ser. 2014, 495, 65–76. [Google Scholar] [CrossRef]
- Fulton, J.M.; Fredricks, H.F.; Bidle, K.D.; Vardi, A.; Kendrick, B.J.; DiTullio, G.R.; Van Mooy, B.A.S. Novel molecular determinants of viral susceptibility and resistance in the lipidome of Emiliania huxleyi. Environ. Microbiol. 2014, 16, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I.; Napier, J.A.; Allen, M.J.; Kimmance, S.A. Intragenus competition between coccolithoviruses: An insight on how a select few can come to dominate many. Environ. Microbiol. 2015, 18, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Frada, M.J.; Fredricks, H.F.; Vardi, A.; Van Mooy, B.A.S. Targeted and untargeted lipidomics of Emiliania huxleyi viral infection and life cycle phases highlights molecular biomarkers of infection, susceptibility, and ploidy. Front. Mar. Sci. 2015, 2. [Google Scholar] [CrossRef]
- Schroeder, D.C.; Oke, J.; Hall, M.; Malin, G.; Wilson, W.H. Virus succession observed during an Emiliania huxleyi bloom. Appl. Environ. Microbiol. 2003, 69, 2484–2490. [Google Scholar] [CrossRef] [PubMed]
- Lehahn, Y.; Koren, I.; Schatz, D.; Frada, M.; Sheyn, U.; Boss, E.; Efrati, S.; Rudich, Y.; Trainic, M.; Sharoni, S.; et al. Decoupling Physical from Biological Processes to Assess the Impact of Viruses on a Mesoscale Algal Bloom. Curr. Biol. 2014, 24, 2041–2046. [Google Scholar] [CrossRef] [PubMed]
- Bidle, K.D. The Molecular Ecophysiology of Programmed Cell Death in Marine Phytoplankton. Ann. Rev. Mar. Sci. 2015, 7, 341–375. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Lanzén, A.; Bratbak, G. Characterisation of the coccolithovirus intein. Mar. Genomics 2011, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Wilson, W.H. The coccolithovirus microarray: An array of uses. Brief. Funct. Genomic. Proteom. 2006, 5, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Martinez-Martinez, J.; Schroeder, D.C.; Somerfield, P.J.; Wilson, W.H. Use of microarrays to assess viral diversity: from genotype to phenotype. Environ. Microbiol. 2007, 9, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Kegel, J.U.; Blaxter, M.; Allen, M.J.; Metfies, K.; Wilson, W.H.; Valentin, K. Transcriptional host–virus interaction of Emiliania huxleyi (Haptophyceae) and EhV-86 deduced from combined analysis of expressed sequence tags and microarrays. Eur. J. Phycol. 2010, 45, 1–12. [Google Scholar] [CrossRef]
- Mackinder, L.C.M.; Worthy, C.A.; Biggi, G.; Hall, M.; Ryan, K.P.; Varsani, A.; Harper, G.M.; Wilson, W.H.; Brownlee, C.; Schroeder, D.C. A unicellular algal virus, Emiliania huxleyi virus 86, exploits an animal-like infection strategy. J. Gen. Virol. 2009, 90, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.L.; Fulton, J.M.; Brown, C.M.; Natale, F.; Van Mooy, B.A.S.; Bidle, K.D. Isolation and characterization of lipid rafts in Emiliania huxleyi: A role for membrane microdomains in host-virus interactions. Environ. Microbiol. 2014, 16, 1150–1166. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.; Malin, G.; Mills, G.P.; Wilson, W.H. Viral Infection of Emiliania huxleyi (Prymnesiophyceae) Leads to Elevated Production of Reactive Oxygen Species. J. Phycol. 2006, 42, 1040–1047. [Google Scholar] [CrossRef]
- Bidle, K.D.; Haramaty, L.; Barcelos, E.; Ramos, J.; Falkowski, P. Viral activation and recruitment of metacaspases in the unicellular coccolithophore, Emiliania huxleyi. Proc. Natl. Acad. Sci. USA 2007, 104, 6049–6054. [Google Scholar] [CrossRef] [PubMed]
- Bidle, K.D.; Kwityn, C.J. Assessing the Role of Caspase Activity and Metacaspase Expression on Viral Susceptibility of the Coccolithophore, Emiliania Huxleyi (Haptophyta). J. Phycol. 2012, 48, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Schatz, D.; Shemi, A.; Rosenwasser, S.; Sabanay, H.; Wolf, S.G.; Ben-Dor, S.; Vardi, A. Hijacking of an autophagy-like process is critical for the life cycle of a DNA virus infecting oceanic algal blooms. New Phytol. 2014, 204, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Vardi, A.; Van Mooy, B.A.S.; Fredricks, H.F.; Popendorf, K.J.; Ossolinski, J.E.; Haramaty, L.; Bidle, K.D. Viral glycosphingolipids induce lytic infection and cell death in marine phytoplankton. Science 2009, 326, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Bidle, K.D.; Vardi, A. A chemical arms race at sea mediates algal host-virus interactions. Curr. Opin. Microbiol. 2011, 14, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Rosenwasser, S.; Mausz, M.A.; Schatz, D.; Sheyn, U.; Malitsky, S.; Aharoni, A.; Weinstock, E.; Tzfadia, O.; Ben-Dor, S.; Feldmesser, E.; et al. Rewiring Host Lipid Metabolism by Large Viruses Determines the Fate of Emiliania huxleyi, a Bloom-Forming Alga in the Ocean. Plant Cell 2014, 26, 2689–2707. [Google Scholar] [CrossRef] [PubMed]
- Ziv, C.; Malitsky, S.; Othman, A.; Ben-Dor, S.; Wei, Y.; Zheng, S.; Aharoni, A.; Hornemann, T.; Vardi, A. Viral serine palmitoyltransferase induces metabolic switch in sphingolipid biosynthesis and is required for infection of a marine alga. Proc. Natl. Acad. Sci. USA 2016, 113, E1907–E1916. [Google Scholar] [CrossRef] [PubMed]
- Frada, M.J.; Schatz, D.; Farstey, V.; Ossolinski, J.E.; Sabanay, H.; Ben-Dor, S.; Koren, I.; Vardi, A. Zooplankton May Serve as Transmission Vectors for Viruses Infecting Algal Blooms in the Ocean. Curr. Biol. 2014, 24, 2592–2597. [Google Scholar] [CrossRef] [PubMed]
- Sharoni, S.; Trainic, M.; Schatz, D.; Lehahn, Y.; Flores, M.J.; Bidle, K.D.; Ben-Dor, S.; Rudich, Y.; Koren, I.; Vardi, A. Infection of phytoplankton by aerosolized marine viruses. Proc. Natl. Acad. Sci. USA 2015, 112, 6643–6647. [Google Scholar] [CrossRef] [PubMed]
- Coolen, M.J.L. 7000 years of Emiliania huxleyi viruses in the Black Sea. Science 2011, 333, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.M.; Fabre, M.-F.; Gobena, D.; Wilson, W.H.; Wilhelm, S.W. Application of the major capsid protein as a marker of the phylogenetic diversity of Emiliania huxleyi viruses. FEMS Microbiol. Ecol. 2011, 76, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I.; Jones, M.; Napier, J.A.; Munn, C.B.; Kimmance, S.A.; Allen, M.J. Functional inferences of environmental coccolithovirus biodiversity. Virol. Sin. 2013, 28, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Pagarete, A.; Kusonmano, K.; Petersen, K.; Kimmance, S.A.; Martínez Martínez, J.; Wilson, W.H.; Hehemann, J.H.; Allen, M.J.; Sandaa, R.A. Dip in the gene pool: Metagenomic survey of natural coccolithovirus communities. Virology 2014, 466–467, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Highfield, A.; Evans, C.; Walne, A.; Miller, P.I.; Schroeder, D.C. How many Coccolithovirus genotypes does it take to terminate an Emiliania huxleyi bloom? Virology 2014, 466-467, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Pagarete, A.; de Vargas, C.; Allen, M.J.; Read, B.; Claverie, J.; Ogata, H. Horizontal gene transfer of an entire metabolic pathway between a eukaryotic alga and its DNA virus. Genome Res. 2009, 19, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Pagarete, A.; Le Corguillé, G.; Tiwari, B.; Ogata, H.; de Vargas, C.; Wilson, W.H.; Allen, M.J. Unveiling the transcriptional features associated with coccolithovirus infection of natural Emiliania huxleyi blooms. FEMS Microbiol. Ecol. 2011, 78, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Lindell, D.; Sullivan, M.B.; Johnson, Z.I.; Tolonen, A.C.; Rohwer, F.; Chisholm, S.W. Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc. Natl. Acad. Sci. USA 2004, 101, 11013–11018. [Google Scholar] [CrossRef] [PubMed]
- Monier, A.; Welsh, R.M.; Gentemann, C.; Weinstock, G.; Sodergren, E.; Armbrust, E.V.; Eisen, J.A.; Worden, A.Z. Phosphate transporters in marine phytoplankton and their viruses: Cross-domain commonalities in viral-host gene exchanges. Environ. Microbiol. 2011, 14, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Schroeder, D.C.; Donkin, A.; Crawfurd, K.J.; Wilson, W.H. Genome comparison of two Coccolithoviruses. Virol. J. 2006, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Pagarete, A.; Lanzén, A.; Puntervoll, P.; Sandaa, R.A.; Larsen, A.; Larsen, J.B.; Allen, M.J.; Bratbak, G. Genomic sequence and analysis of EhV-99B1, a new coccolithovirus from the Norwegian fjords. Intervirology 2013, 56, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I.; Worthy, C.A.; Rooks, P.; Napier, J.A.; Kimmance, S.A.; Henn, M.R.; Ogata, H.; Allen, M.J. Draft genome sequence of the coccolithovirus EhV-84. Stand. Genomic Sci. 2011, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I.; Worthy, C.A.; Rooks, P.; Napier, J.A.; Kimmance, S.A.; Henn, M.R.; Ogata, H.; Allen, M.J. Draft Genome Sequence of Four Coccolithoviruses: Emiliania huxleyi Virus EhV-88, EhV-201, EhV-207, and EhV-208. J. Virol. 2012, 86, 2896–2897. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I.; Worthy, C.A.; Rooks, P.; Napier, J.A.; Kimmance, S.A.; Henn, M.R.; Ogata, H.; Allen, M.J. Draft Genome Sequence of the Coccolithovirus Emiliania huxleyi Virus 202. J. Virol. 2012, 86, 2380–2381. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I.; Worthy, C.A.; Rooks, P.; Napier, J.A.; Kimmance, S.A.; Henn, M.R.; Ogata, H.; Allen, M.J. Draft genome sequence of the Coccolithovirus Emiliania huxleyi virus 203. J. Virol. 2011, 85, 13468–13469. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I.; Napier, J.A.; Kimmance, S.A.; Allen, M.J. Permanent draft genomes of four new coccolithoviruses: EhV-18, EhV-145, EhV-156 and EhV-164. Mar. Genomics 2014, 15, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Fenchel, T.; Field, J. The ecological role of water-column microbes in the sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar] [CrossRef]
- Azam, F.; Fandino, L.; Grossart, H.; Long, R. Microbial loop: Its significance in oceanic productivity and global change. Rapp. Comrn. inl. Mer Médit. 1998, 35, 1–3. [Google Scholar]
- Mayers, T.J.; Bramucci, A.R.; Yakimovich, K.M.; Case, R.J. A Bacterial Pathogen Displaying Temperature-Enhanced Virulence of the Microalga Emiliania huxleyi. Front. Microbiol. 2016, 7, 892. [Google Scholar] [CrossRef] [PubMed]
- Egan, S.; Fernandes, N.D.; Kumar, V.; Gardiner, M.; Thomas, T. Bacterial pathogens, virulence mechanism and host defence in marine macroalgae. Environ. Microbiol. 2014, 16, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Lachnit, T.; Thomas, T.; Steinberg, P. Expanding our understanding of the seaweed holobiont: RNA Viruses of the Red Alga Delisea pulchra. Front. Microbiol. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Filée, J.; Siguier, P.; Chandler, M. I am what I eat and I eat what I am: acquisition of bacterial genes by giant viruses. Trends Genet. 2007, 23, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Filée, J.; Pouget, N.; Chandler, M. Phylogenetic evidence for extensive lateral acquisition of cellular genes by Nucleocytoplasmic large DNA viruses. BMC Evol. Biol. 2008, 8, 320. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The Neighbor-joining Method : A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.-A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis Comparison Tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, V.M.; Mavromatis, K.; Ivanova, N.N.; Chen, I.-M.A.; Chu, K.; Kyrpides, N.C. IMG ER: A system for microbial genome annotation expert review and curation. Bioinformatics 2009, 25, 2271–2278. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Klappenbach, J.A.; Goris, J.; Vandamme, P.; Coenye, T.; Konstantinidis, K.T.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Louie, B.; Higdon, R.; Kolker, E. A statistical model of protein sequence similarity and function similarity reveals overly-specific function predictions. PLoS ONE 2009, 4, e7546. [Google Scholar] [CrossRef] [PubMed]
- Read, B.A.; Kegel, J.; Klute, M.J.; Kuo, A.; Lefebvre, S.C.; Maumus, F.; Mayer, C.; Miller, J.; Monier, A.; Salamov, A.; et al. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 2013, 499, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I. Competitive ecology of coccolithoviruses as revealed through biochemical diversity of serine palmitoyltransferase. 2017; unpublished, in review. [Google Scholar]
- Allen, M.J.; Schroeder, D.C.; Wilson, W.H. Preliminary characterisation of repeat families in the genome of EhV-86, a giant algal virus that infects the marine microalga Emiliania huxleyi. Arch. Virol. 2006, 151, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, L.P.; DeFilippis, V.R. A hypothesis for DNA viruses as the origin of eukaryotic replication proteins. J. Virol. 2000, 74, 7079–7084. [Google Scholar] [CrossRef] [PubMed]
- Maat, D.S.; Blok, R.; De Brussaard, C.P.D. Combined Phosphorus Limitation and Light Stress Prevent Viral Proliferation in the Phytoplankton Species Phaeocystis globosa, but Not in Micromonas pusilla. Front. Micr. 2016, 3. [Google Scholar] [CrossRef]
- Wilson, W.H.; Carr, N.G.; Mann, N.H. The effect of phosphate status on the kinetics of cyanophage infection in the oceanic cyanobacterium Synechococcus sp. wh7803. J. Phycol. 1996, 32, 506–516. [Google Scholar] [CrossRef]
- Bratbak, G.; Jacobsen, A.; Heldal, M.; Nagasaki, K.; Thingstad, F. Virus production in Phaeocystis pouchetii and its relation to host cell growth and nutrition. Aquat. Microb. Ecol. 1998, 16, 1–9. [Google Scholar] [CrossRef]
- Coleman, M.L.; Chisholm, S.W. Ecosystem-specific selection pressures revealed through comparative population genomics. Proc. Natl. Acad. Sci. USA 2010, 107, 18634–18639. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.M. Re-evaluation of the physiological ecology of coccolithophores. In Coccolithophores; Springer: Berlin, Germany, 2004; pp. 165–190. [Google Scholar]
- Johannessen, T.V.; Bratbak, G.; Larsen, A.; Ogata, H.; Egge, E.S.; Edvardsen, B.; Eikrem, W.; Sandaa, R.-A. Characterisation of three novel giant viruses reveals huge diversity among viruses infecting Prymnesiales (Haptophyta). Virology 2015, 476, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Gonza´lez, J.M.; Simo, R.; Massana, R.; Covert, J.S.; Casamayor, E.O.; Pedro, C.; Moran, M.A. Bacterial Community Structure Associated with a North Atlantic Algal Bloom. Appl. Environ. Microb. 2000, 66, 4237–4246. [Google Scholar] [CrossRef]
- Green, D.H.; Echavarri-Bravo, V.; Brennan, D.; Hart, M.C.; Green, D.H.; Echavarri-Bravo, V.; Brennan, D.; Hart, M.C. Bacterial Diversity Associated with the Coccolithophorid Algae Emiliania huxleyi and Coccolithus pelagicus f. braarudii. Biomed Res. Int. 2015, 1–15. [Google Scholar]
- Carrias, J.-F.; Serre, J.-P.; Sime-Ngando, T.; Amblard, C. Distribution, size, and bacterial colonization of pico- and nano-detrital organic particles (DOP) in two lakes of different trophic status. Limnol. Oceanogr. 2002, 47, 1202–1209. [Google Scholar] [CrossRef]
- Yabuuchi, E.; Kaneko, T.; Yano, I.; Moss, C.W.; Miyoshi, N. Sphingobacterium gen. nov., Sphingobacterium spiritivorum comb. nov., Sphingobacterium multivorum comb. nov., Sphingobacterium mizutae sp. nov., and Flavobacterium indologenes sp. nov.: Glucose-Nonfermenting Gram-Negative Rods in CDC Groups IIK-2 and IIb. Int. J. Syst. Bacteriol. 1983, 33, 580–598. [Google Scholar] [CrossRef]
- Ikushiro, H.; Islam, M.M.; Tojo, H.; Hayashi, H. Molecular characterization of membrane-associated soluble serine palmitoyltransferases from Sphingobacterium multivorum and Bdellovibrio stolpii. J. Bacteriol. 2007, 189, 5749–5761. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Gable, K.; Yan, L.; Allen, M.J.; Wilson, W.H.; Moitra, P.; Harmon, J.M.; Dunn, T.M. Expression of a novel marine viral single-chain serine palmitoyltransferase and construction of yeast and mammalian single-chain chimera. J. Biol. Chem. 2006, 281, 39935–39942. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, K.; Kuroyanagi, H.; Inoue, S.; Ye, J.; Troy, F.A.; Inoue, Y. Discovery of a new type of sialidase, “KDNase,” which specifically hydrolyzes deaminoneuraminyl (3-deoxy-D-glycero-D-galacto-2-nonulosonic acid) but not N-acylneuraminyl linkages. J. Biol. Chem. 1994, 269, 21415–21419. [Google Scholar] [PubMed]
- Nishino, S.; Kuroyanagi, H.; Terada, T.; Inoue, S.; Inoue, Y.; Troy, F.A.; Kitajima, K. Induction, localization, and purification of a novel sialidase, deaminoneuraminidase (KDNase), from Sphingobacterium multivorum. J. Biol. Chem. 1996, 271, 2909–2913. [Google Scholar] [CrossRef] [PubMed]
- Stray, S.J.; Cummings, R.D.; Air, G.M. Influenza virus infection of desialylated cells. Glycobiology 2000, 10, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Neu, U.; Bauer, J.; Stehle, T. Viruses and sialic acids: rules of engagement. Curr. Opin. Struct. Biol. 2011, 21, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.T.; Matrosovich, M.; Rodgers, M.E.; McGregor, M.; Kawaoka, Y. Influenza A viruses lacking sialidase activity can undergo multiple cycles of replication in cell culture, eggs, or mice. J. Virol. 2000, 74, 5206–5212. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.; Pond, D.; Wilson, W. Changes in Emiliania huxleyi fatty acid profiles during infection with E. huxleyi virus 86: physiological and ecological implications. Aquat. Microb. Ecol. 2009, 55, 219–228. [Google Scholar] [CrossRef]
- Stoecker, D.K.; Hansen, P.J.; Caron, D.A.; Mitra, A. Mixotrophy in the Marine Plankton. Ann. Rev. Mar. Sci. 2016, 9, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [PubMed]
- Rokitta, S.D.; de Nooijer, L.J.; Trimborn, S.; de Vargas, C.; Rost, B.; John, U. Transcriptome analyses reveal differential gene expression patterns between the life-cycle stages of Emiliania huxleyi (haptophyta) and reflect specialization to different ecological niches. J. Phycol. 2011, 47, 829–838. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Strain # | Isolate | Isolation | Lat/Long | Depth (m) | Date | NCBI | Reference * |
|---|---|---|---|---|---|---|---|
| Location | Date | Sequenced | Accession # | ||||
| EhV-84 | E.C. | 1999 | 50°15′ N, 04°13′ W | 15 | 2011 | JF974290 | Schroeder et al., 2002 [2]; Nissimov et al., 2011 [45] |
| EhV-86 | E.C. | 1999 | 50°30′ N, 04°20′ W | surface | 2005 | AJ890364 | Schroeder et al., 2002 [2]; Wilson et al., 2005 [1] |
| EhV-88 | E.C. | 1999 | 50°15′ N, 04°13′ W | 5 | 2011 | JF974310 | Schroeder et al., 2002 [2]; Nissimov et al., 2012 [46] |
| EhV-201 | E.C. | 2001 | 49°56′ N, 04°19′ W | 2 | 2011 | JF974311 | Schroeder et al., 2002 [2]; Nissimov et al., 2012 [46] |
| EhV-202 | E.C. | 2001 | 50°00′ N, 04°18′ W | 15 | 2011 | HQ634145 | Schroeder et al., 2002 [2]; Nissimov et al., 2012 [47] |
| EhV-203 | E.C. | 2001 | 50°00′ N, 04°18′ W | 15 | 2011 | JF974291 | Schroeder et al., 2002 [2]; Nissimov et al., 2011 [48] |
| EhV-207 | E.C. | 2001 | 50°15′ N, 04°13′ W | 5 | 2011 | JF974317 | Schroeder et al., 2002 [2]; Nissimov et al., 2012 [46] |
| EhV-208 | E.C. | 2001 | 50°15′ N, 04°13′ W | 5 | 2011 | JF974318 | Schroeder et al., 2002 [2]; Nissimov et al., 2012 [46] |
| EhV-99B1 | N.F. | 1999 | 60°20′ N, 05°20′ E | surface | 2013 | FN429076 | Pagarete et al., 2013 [44] |
| EhV-18 | E.C. | 2008 | 50°15′ N, 04°13′ W | surface | 2013 | KF481685 | Nissimov et al., 2014 [49] |
| EhV-145 | Loss. | 2008 | 57°72′ N, 03°29′W | surface | 2013 | KF481686 | Nissimov et al., 2014 [49] |
| EhV-156 | E.C. | 2009 | 50°15′ N, 04°13′ W | surface | 2013 | KF481687 | Nissimov et al., 2014 [49] |
| EhV-164 | SSF. | 2009 | 56°26′ N, 02°63′ W | surface | 2013 | KF481688 | Nissimov et al., 2014 [49] |
| Reference Genome | Draft Genome | ANI Score | Total BBH * | Clade |
|---|---|---|---|---|
| EhV-86 | EhV-164 | 99.95 | 443 | A1 |
| EhV-145 | 99.93 | 456 | A1 | |
| EhV-84 | 99.07 | 434 | A1 | |
| EhV-88 | 98.96 | 442 | A1 | |
| EhV-99B1 | 98.23 | 421 | A3 | |
| EhV-208 | 96.78 | 399 | A2 | |
| EhV-207 | 96.67 | 411 | A2 | |
| EhV-201 | 96.6 | 399 | A2 | |
| EhV-203 | 96.6 | 402 | A2 | |
| EhV-18 | 79.52 | 307 | B | |
| EhV-202 | 79.42 | 312 | B | |
| EhV-156 | 79.4 | 308 | B |
| Genome Name | Genes | Total Bases | CDS | Coding Bases | Genes with Function Prediction | tRNAs | GC (%) | Number of Gaps in Genome |
|---|---|---|---|---|---|---|---|---|
| E. huxleyi virus 84 | 486 | 396620 | 482 | 334463 | 85 | 4 | 40.17 | 8 |
| E. huxleyi virus 86 | 478 | 407339 | 472 | 369157 | 90 | 5 | 40.18 | 0 |
| E. huxleyi virus 88 | 480 | 397298 | 475 | 357803 | 90 | 5 | 40.18 | 7 |
| E. huxleyi virus 201 | 457 | 407301 | 451 | 363714 | 89 | 6 | 40.46 | 6 |
| E. huxleyi virus 202 | 488 | 407516 | 485 | 352215 | 93 | 3 | 40.3 | 11 |
| E. huxleyi virus 203 | 470 | 400520 | 464 | 364178 | 91 | 6 | 40.12 | 5 |
| E. huxleyi virus 207 | 479 | 421891 | 473 | 371313 | 93 | 6 | 40.49 | 15 |
| E. huxleyi virus 208 | 461 | 411003 | 455 | 348386 | 90 | 6 | 40.42 | 16 |
| E. huxleyi virus 99B1 | 451 | 376759 | 444 | 333400 | 90 | 6 | 40.04 | 16 |
| E. huxleyi virus 18 | 508 | 399651 | 503 | 346161 | 91 | 5 | 40.49 | 21 |
| E. huxleyi virus 145 | 552 | 397508 | 548 | 350414 | 103 | 4 | 39.94 | 41 |
| E. huxleyi virus 156 | 498 | 399344 | 493 | 351083 | 88 | 5 | 40.47 | 19 |
| E. huxleyi virus 164 | 514 | 400675 | 510 | 354290 | 95 | 4 | 40.11 | 17 |
| Phylogenetic Group | A | B | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | A2 | A3 | ||||||||||||
| tRNA | Genome | EhV-84 | EhV-86 | EhV-88 | EhV-164 | EhV-145 | EhV-201 | EhV-203 | EhV-207 | EhV-208 | EhV-99B1 | EhV-18 | EhV-156 | EhV-202 |
| Arg | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Asn | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Gln | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Glu | + | + | + | + | ||||||||||
| Ile | + | + | + | + | + | + | ||||||||
| Leu | + | + | + | + | + | + | + | + | ||||||
| Lys | + | + | + | + | + | + | + | + | ||||||
| Phylogenetic Group | A | B | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | A2 | A3 | |||||||||||
| Predicted CDS | EhV-84 | EhV-86 | EhV-88 | EhV-145 | EhV-164 | EhV-201 | EhV-203 | EhV-207 | EhV-208 | EhV-99B1 | EhV-202 | EhV-18 | EhV-156 |
| hypothetical protein | 27 | 3 | 4 | 7 | 8 | 4 | 4 | 9 | 9 | 6 | 18 | 9 | 6 |
| putative endonuclease | 2 | ||||||||||||
| putative membrane protein | 2 | 1 | 1 | 4 | |||||||||
| putative transposase | 1 | ||||||||||||
| putative DUF814 domain containing protein | 1 | ||||||||||||
| zinc finger protein | 1 | ||||||||||||
| putative ribonuclease | 1 | 1 | |||||||||||
| glycosyltransferase family 29 (sialyltransferase) | 1 | 1 | |||||||||||
| Total | 27 | 5 | 4 | 8 | 9 | 4 | 5 | 9 | 9 | 15 | 18 | 11 | 8 |
| Major Hits against Eukarya | ||||||||
| Ehv Predicted Gene | COG ID | Identity (%) | E-Value | Bit-Score | Phylum | Class | Order | Genus |
| DNA-directed RNA polymerase subunit B | K | 40.39 | 0 | 825 | NA | NA | Dictyosteliida | Dictyostelium |
| DNA ligase | L | 51.29 | 0 | 612 | Chlorophyta | Mamiellophyceae | Mamiellales | Micromonas |
| DNA topoisomerase | L | 33 | 0 | 586 | Microsporidia | NA | NA | Nematocida |
| DNA-dependent RNA polymerase II largest subunit | K | 34.09 | 7 × 10−171 | 555 | Ascomycota | Sordariomycetes | Xylariales | Eutypa |
| thymidylate synthase | F | 49.7 | 1 × 10−168 | 497 | NA | NA | Peronosporales | Phytophthora |
| DNA polymerase delta catalytic subunit | L | 35.11 | 2 × 10−132 | 436 | Arthropoda | Malacostraca | Decapoda | Procambarus |
| DNA helicase | L | 49.3 | 3 × 10−137 | 419 | Bacillariophyta | Coscinodiscophyceae | Thalassiosirales | Thalassiosira |
| Major Hits against Bacteria | ||||||||
| deoxycytidylate deaminase | F | 62.96 | 8 × 10−64 | 206 | Firmicutes | Clostridia | Clostridiales | Clostridium |
| Sialidase | G | 30.03 | 2 × 10−37 | 148 | Bacteroidetes | Sphingobacteriia | Sphingobacteriales | Sphingobacterium |
| DNA-binding protein | R | 34.36 | 7 × 10−32 | 129 | Verrucomicrobia | NA | NA | NA |
| endonuclease | L | 45.67 | 4 × 10−32 | 121 | Proteobacteria | Alphaproteobacteria | Rhizobiales | Pseudochrobactrum |
| fatty acid desaturase | I | 34.76 | 2 × 10−28 | 121 | Proteobacteria | Alphaproteobacteria | Rhodospirillales | Niveispirillum |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nissimov, J.I.; Pagarete, A.; Ma, F.; Cody, S.; Dunigan, D.D.; Kimmance, S.A.; Allen, M.J. Coccolithoviruses: A Review of Cross-Kingdom Genomic Thievery and Metabolic Thuggery. Viruses 2017, 9, 52. https://doi.org/10.3390/v9030052
Nissimov JI, Pagarete A, Ma F, Cody S, Dunigan DD, Kimmance SA, Allen MJ. Coccolithoviruses: A Review of Cross-Kingdom Genomic Thievery and Metabolic Thuggery. Viruses. 2017; 9(3):52. https://doi.org/10.3390/v9030052
Chicago/Turabian StyleNissimov, Jozef I., António Pagarete, Fangrui Ma, Sean Cody, David D. Dunigan, Susan A. Kimmance, and Michael J. Allen. 2017. "Coccolithoviruses: A Review of Cross-Kingdom Genomic Thievery and Metabolic Thuggery" Viruses 9, no. 3: 52. https://doi.org/10.3390/v9030052
APA StyleNissimov, J. I., Pagarete, A., Ma, F., Cody, S., Dunigan, D. D., Kimmance, S. A., & Allen, M. J. (2017). Coccolithoviruses: A Review of Cross-Kingdom Genomic Thievery and Metabolic Thuggery. Viruses, 9(3), 52. https://doi.org/10.3390/v9030052