How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery


 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The DDR Machinery
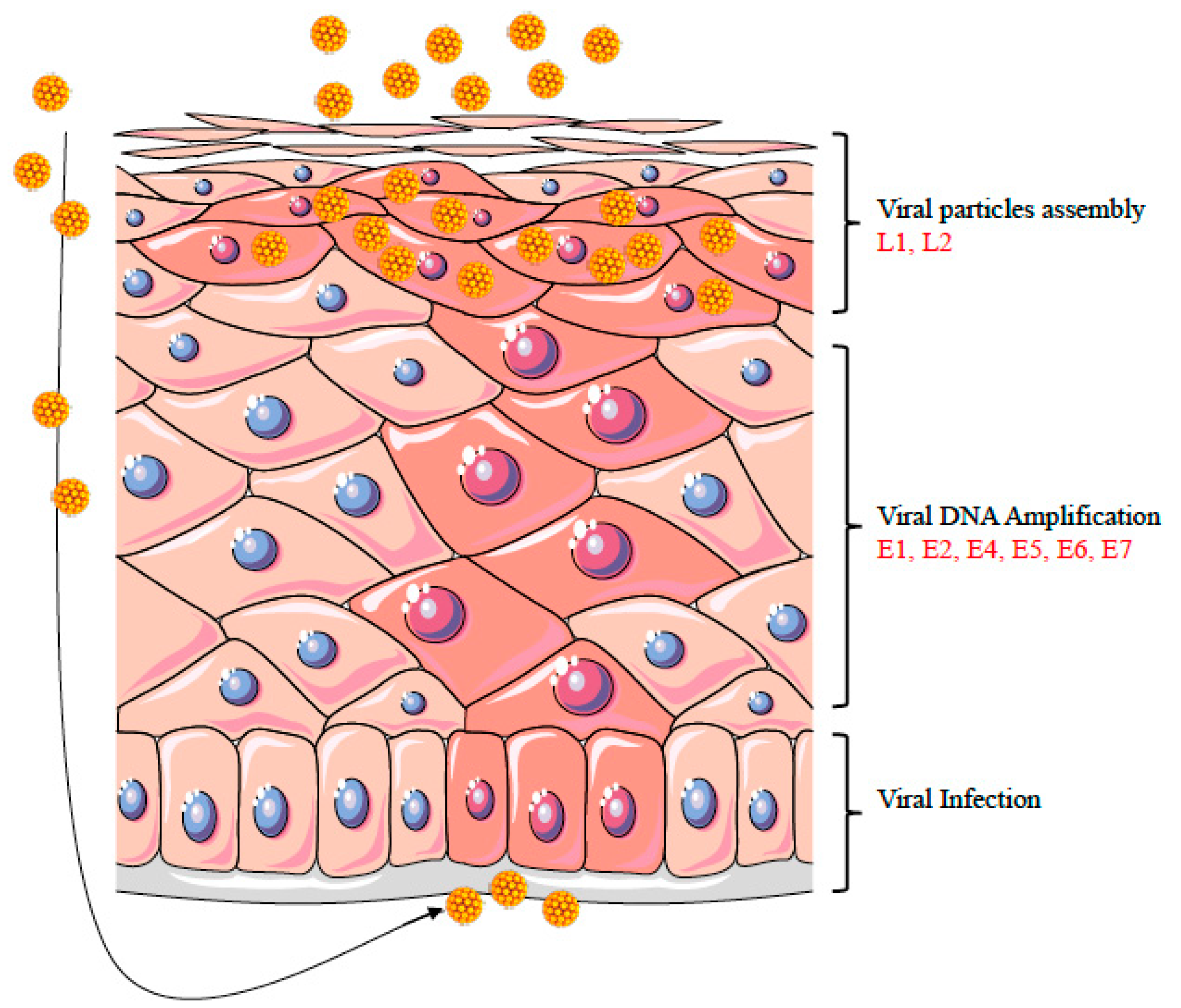
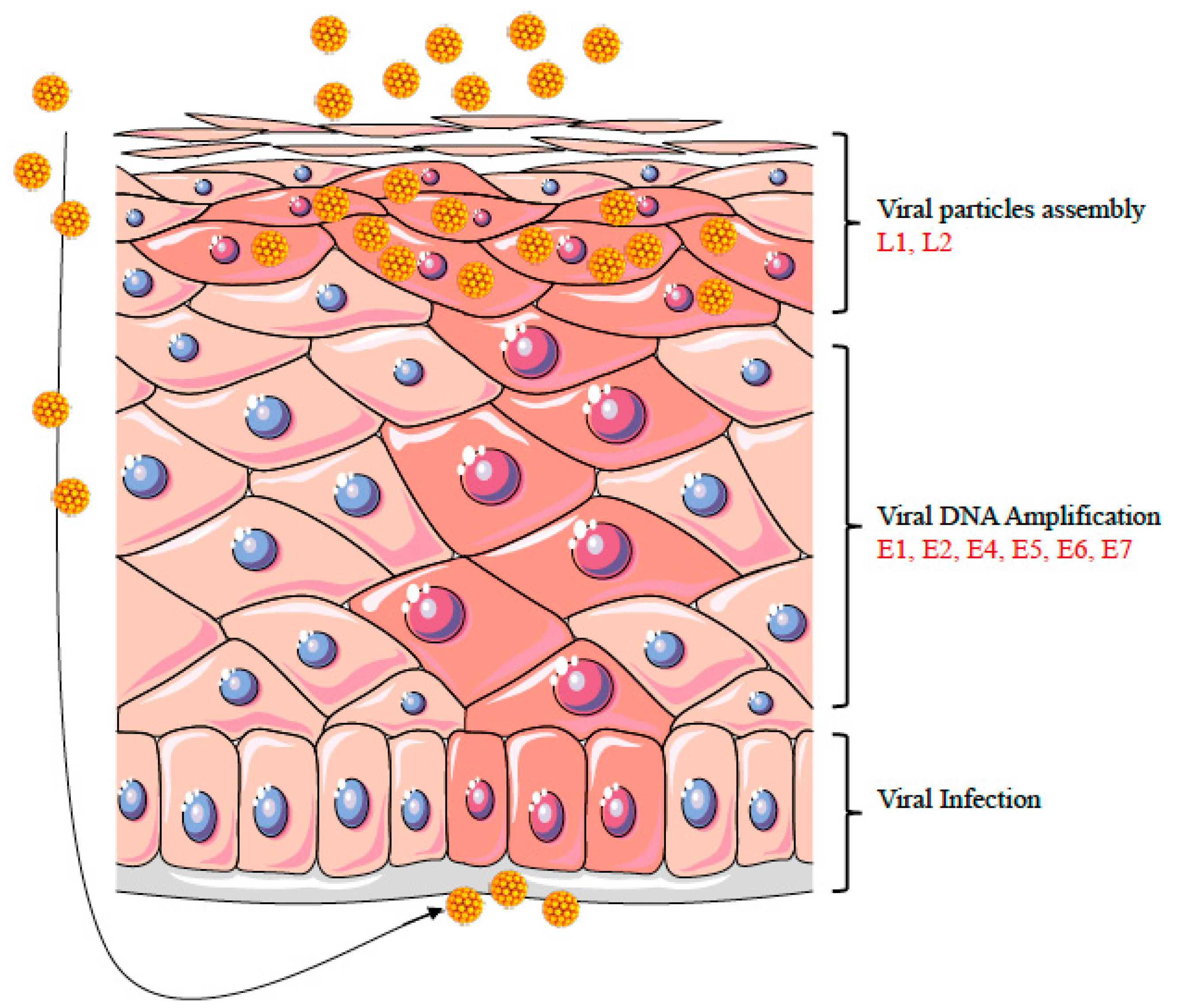
3. HPV Life Cycle
4. Normal Immune Response in the Uterine Cervix
5. HPV and DDR: Strategies to Escape Immune Surveillance
6. Deregulation of Interferon Synthesis
7. STAT Signalling is Part of the IFN Pathway
8. STAT-5 Activation Promotes Both ATM and ATR Signalling
9. HPV and Genomic Instability: Modulation of Apoptosis
10. HPV E6 and p53
11. HPV E7 and pRB
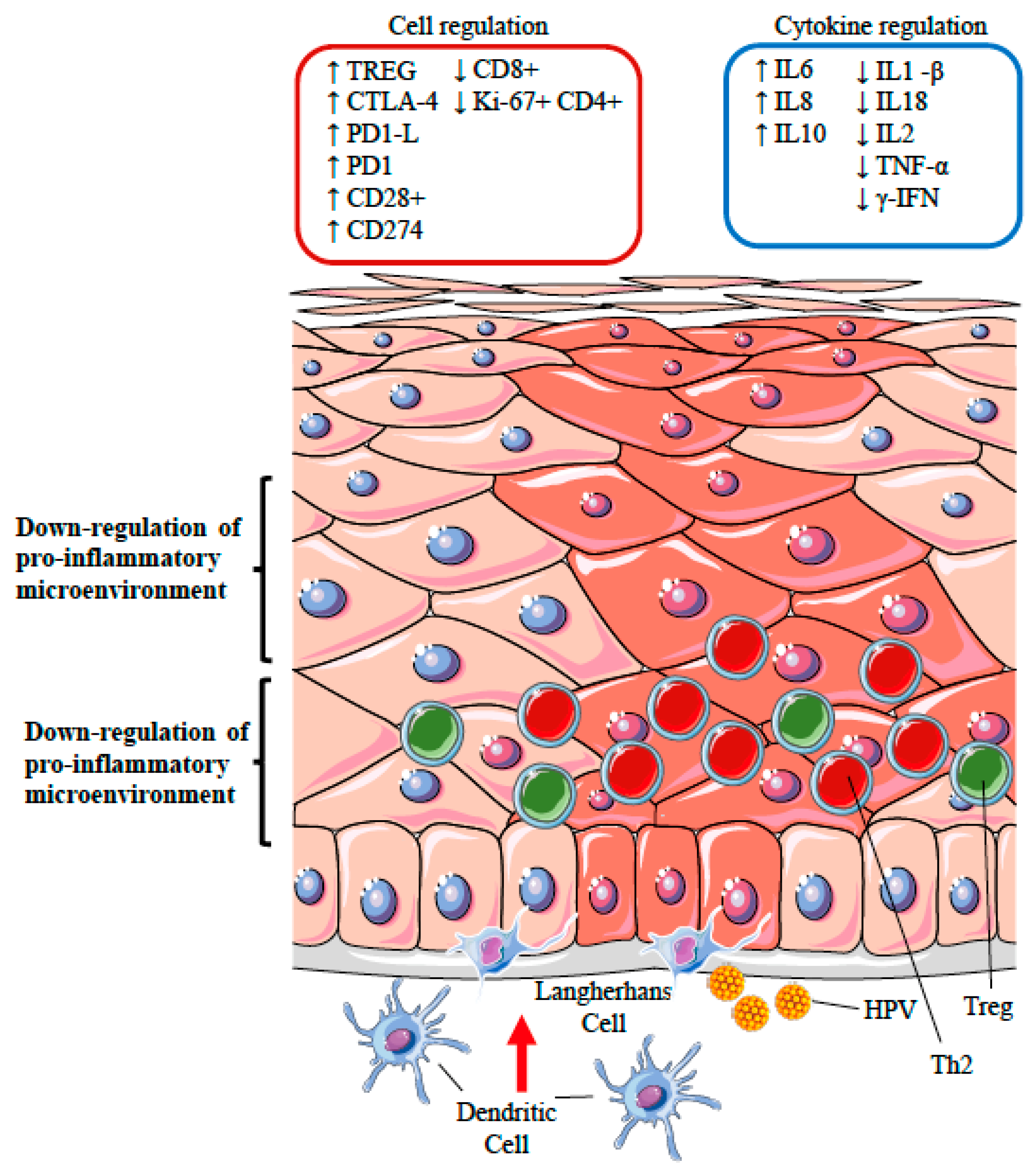
12. HPV and the Immune System: Subversion and Piracy
12.1. Perturbation of Antigen Processing and Presentation
12.2. Polarization of T Cell Phenotypes
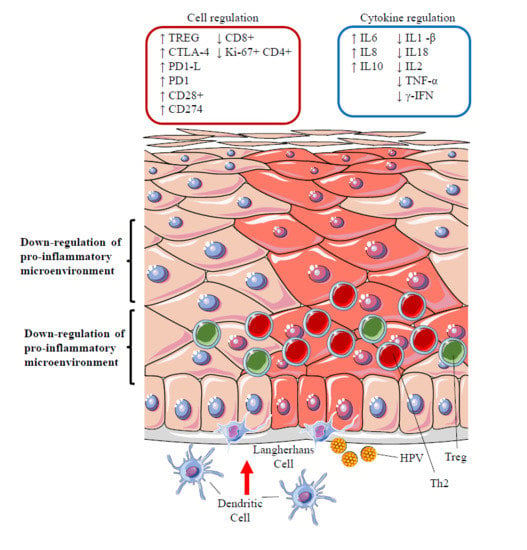
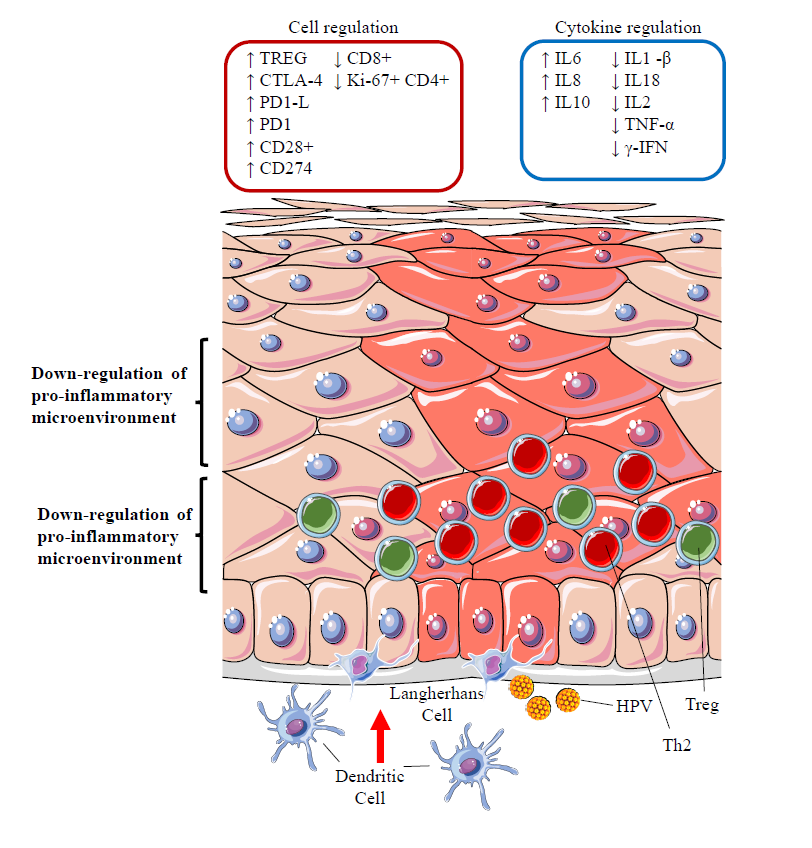
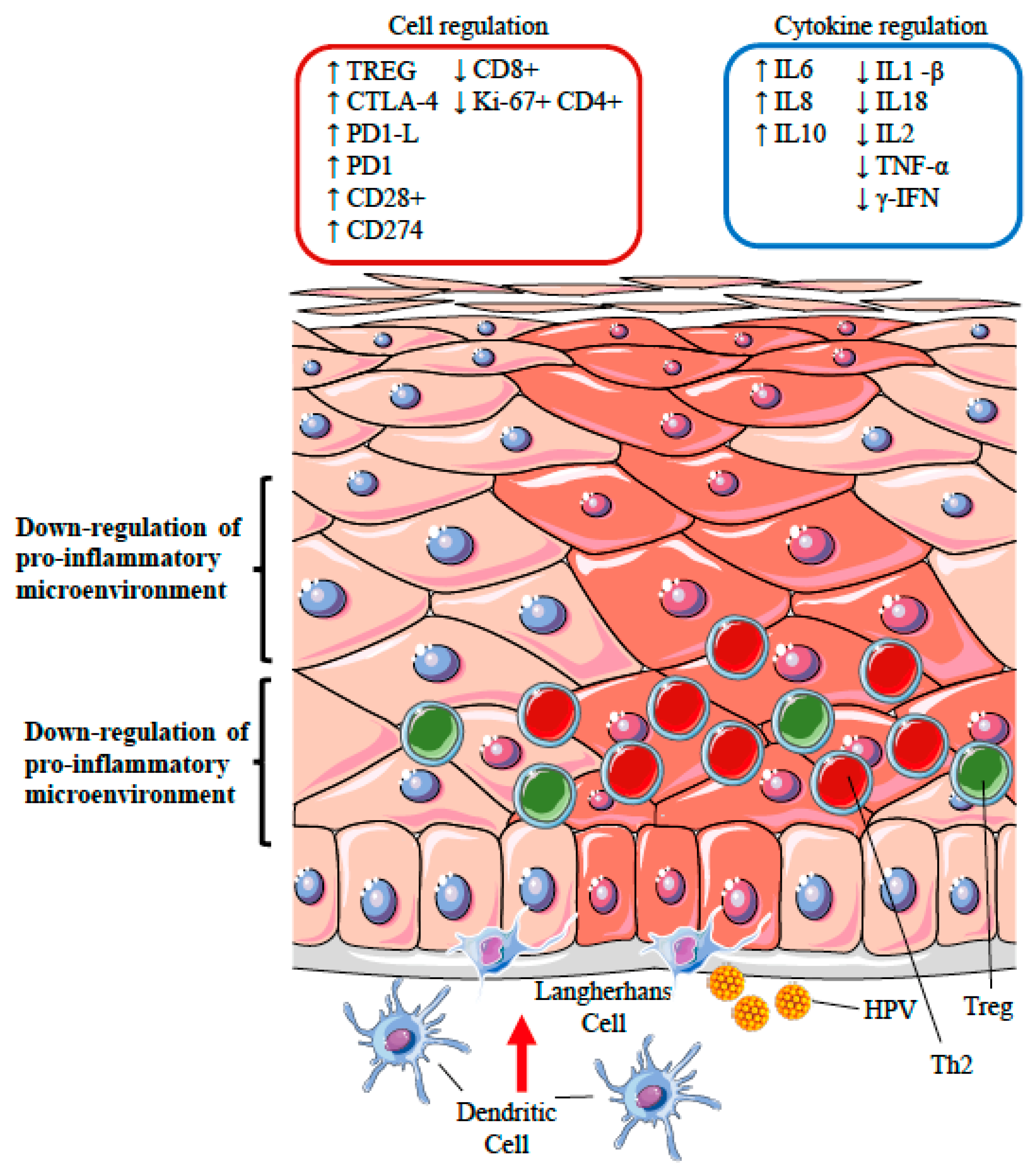
12.3. Silencing of the Inflammatory Response: the Role of Regulatory T Cells
13. Methods
14. Conclusions
Author Contributions
Conflicts of Interest
References
- Frazer, I.H. Interaction of human papillomaviruses with the host immune system: A well evolved relationship. Virology 2009, 384, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, A.K.; Riemer, A.B. The invisible enemy—How human papillomaviruses avoid recognition and clearance by the host immune system. Open Virol. J. 2012, 6, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.I. Mission impossible: How HPV-associated head and neck cancers escape a primed immune response. Oral Oncol. 2013, 49, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Senba, M.; Mori, N. Mechanisms of virus immune evasion lead to development from chronic inflammation to cancer formation associated with human papillomavirus infection. Oncol. Rev. 2012, 5, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Luftig, M.A. Viruses and the DNA damage response: Activation andantagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Moody, C.A. Impact of the DNA damage response on human papillomavirus chromatin. PLoS Pathog. 2016, 12, e1005613. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Moody, C.A. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017, 231, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Vidmar, T.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. DNA damage repair genes controlling human papillomavirus (HPV) episome levels under conditions of stability and extreme instability. PLoS ONE 2013, 8, e75406. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Helmus, M.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J. Virol. 2013, 87, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play withknives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Hübscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Alt, F.W.; Zhang, Y.; Meng, F.L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.; Hearing, P. Relocalization of the Mre11-Rad50-Nbs1 complex bythe adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 2005, 79, 6207–6215. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, G.; Stacey, S.N. Adeno-associated virus Rep78 protein interacts with protein kinase A and its homolog PRKX and inhibits CREB-dependent transcriptional activation. J. Virol. 1998, 72, 7916–7925. [Google Scholar] [PubMed]
- Haig, D.M. Subversion and piracy: DNA viruses and immune evasion. Res. Vet. Sci. 2001, 70, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C. Recent insights into the control of human papillomavirus (HPV) genome stability, loss, and degradation. J. Clin. Med. 2015, 4, 204–230. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin. Cancer Biol. 2014, 26, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.E.; Weller, S.K. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 2006, 119, 2695–2703. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Nakamura, H.; Liang, X.; Feng, P.; Chang, H.; Kowalik, T.F.; Jung, J.U. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006, 80, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.; Luftig, M.A. Interplay between DNA tumor viruses and the host DNA damage response. Curr. Top. Microbiol. Immunol. 2013, 371, 229–257. [Google Scholar] [PubMed]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dodson, G.E.; Shaikh, S.; Rundell, K.; Tibbetts, R.S. Ataxia-telangiectasia-mutated (ATM) is a T-antigen kinase that controls SV40 viral replication invivo. J. Biol. Chem. 2005, 280, 40195–40200. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.; You, J.; Benjamin, T.L. Induction and utilization of an ATM signaling pathway by polyomavirus. J. Virol. 2005, 79, 13007–13017. [Google Scholar] [CrossRef] [PubMed]
- Shirata, N.; Kudoh, A.; Daikoku, T.; Tatsumi, Y.; Fujita, M.; Kiyono, T.; Sugaya, Y.; Isomura, H.; Ishizaki, K.; Tsurumi, T. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J. Biol. Chem. 2005, 280, 30336–30341. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.E.; Weller, S.K. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 2004, 78, 4783–4796. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer. 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.C.; Hussmann, K.L.; McBride, A.A. The role of the DNA damage response throughout the papillomavirus life cycle. Viruses 2015, 7, 2450–2469. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 2013, 9, e1003321. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Wang, H.K.; Broker, T.R.; Chow, L.T. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J. Biol. Chem. 2011, 286, 15473–15482. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Lambert, P.F. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997, 71, 7167–7179. [Google Scholar] [PubMed]
- Medzhitov, R.; Janeway, J.C. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Cella, M.; Scheidegger, D.; Palmer-Lehmann, K.; Lane, P.; Lanzavecchia, A.; Alber, G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J. Exp. Med. 1996, 184, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Broere, F.; Apasov, S.G.; Sitkovsky, M.V. Cell subsets and T cell-mediated immunity. In Principles of Immunopharmacology, 3rd Revised and Extended ed.; Nijkamp, F.P., Parnham, M.J., Eds.; Springer: Basel, Switzerland, 2011; pp. 15–27. [Google Scholar]
- Zhang, X.; Dawson, C.W.; He, Z.; Huang, P. Immune evasion strategies of the human gamma-herpesviruses: Implications for viral tumorigenesis. J. Med. Virol. 2012, 84, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of innate immunity against human papillomavirus (HPV) infections and effect of adjuvants in promoting specific immune response. Viruses 2013, 5, 2624–2642. [Google Scholar] [CrossRef] [PubMed]
- Beglin, M.; Melar-New, M.; Laimins, L. Human papillomaviruses and the interferon response. J. Interferon Cytokine Res. 2009, 29, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef]
- Arany, I.; Goel, A.; Tyring, S.K. Interferon response depends on viral transcription in human papillomavirus-containing lesions. Anticancer Res. 1995, 15, 2865–2869. [Google Scholar] [PubMed]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7- mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef] [PubMed]
- Um, S.J.; Rhyu, J.W.; Kim, E.J.; Jeon, K.C.; Hwang, E.S.; Park, J.S. Abrogation of IRF-1 response by high-risk HPV E7 protein in vivo. Cancer Lett. 2002, 179, 205–212. [Google Scholar] [CrossRef]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Kanodia, S.; Fahey, L.M.; Kast, W.M. Mechanisms used by human papillomaviruses to escape the host immune response. Curr. Cancer Drug Targets 2007, 7, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAKSTAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef] [PubMed]
- Reich, N.C.; Liu, L. Tracking STAT nuclear traffic. Nat. Rev. Immunol. 2006, 6, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Van Boxel-Dezaire, A.H.; Rani, M.R.; Stark, G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006, 25, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dutta, A.; Laimins, L.A. The acetyltransferase Tip60 is a critical regulator of the differentiationdependent amplification of human papillomaviruses. J. Virol. 2015, 89, 4668–4675. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.J.; Heo, D.S. PPARgamma ligands induce growth inhibition and apoptosis through p63 and p73 in human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2011, 406, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Olivo-Marston, S.E.; Zhu, Y.; Lee, R.Y.; Cabanes, A.; Khan, G.; Zwart, A.; Wang, Y.; Clarke, R.; Hilakivi-Clarke, L. Gene signaling pathways mediating the opposite effects of prepubertal low-fat and high-fat n-3 polyunsaturated fatty acid diets on mammary cancer risk. Cancer Prev. Res. 2008, 1, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Garnett, T.O.; Duerksen-Hughes, P.J. Modulation of apoptosis by human papillomavirus (HPV) oncoproteins. Arch. Virol. 2006, 151, 2321–2335. [Google Scholar] [CrossRef] [PubMed]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Potapova, T.A.; Li, S.; Rankin, S.; Gorbsky, G.J.; Angeletti, P.C.; Ceresa, B.P. Expression of HPV16 E5 produces enlarged nuclei and polyploidy through endoreplication. Virology 2010, 405, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, K.; Alonso, A. The human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J. Virol. 2002, 76, 12162–12172. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, Q.; Xu, K.; Gao, C.; Chen, C.; Li, X.L.; Wang, G.R.; Tian, C.; Han, J.; Dong, X.P. Familial CJD associated PrP mutants within transmembrane region induced Ctm-PrP retention in ER and triggered apoptosis by ER stress in SH-SY5Y cells. PLoS ONE 2011, 6, e14602. [Google Scholar] [CrossRef] [PubMed]
- Sudarshan, S.R.; Schlegel, R.; Liu, X.F. The HPV-16 E5 protein represses expression of stress pathway genes XBP-1 and COX-2 in genital keratinocytes. Biochem. Biophys. Res. Commun. 2010, 399, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Castle, P.E. The promise of global cervical-cancer prevention. N. Engl. J. Med. 2005, 353, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Muench, P.; Probst, S.; Schuetz, J.; Leiprecht, N.; Busch, M.; Wesselborg, S.; Stubenrauch, F.; Iftner, T. Cutaneous papillomavirus E6 proteins must interact with p300 and blockp53-mediated apoptosis for cellular immortalization and tumorigenesis. Cancer Res. 2010, 70, 6913–6924. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.M.; Campo, M.S. Papillomaviruses: A correlation between immune evasion and oncogenicity? Trends Microbiol. 2003, 11, 300–305. [Google Scholar] [CrossRef]
- Murray-Zmijewski, F.; Slee, E.A.; Lu, X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008, 9, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Ristriani, T.; Nominé, Y.; Masson, M.; Weiss, E.; Travé, G. Specific recognition of four-way DNA junctions by the C-terminal zinc-binding domain of HPV oncoprotein E6. J. Mol. Biol. 2001, 305, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus type 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Gu, Z.; Pim, D.; Labrecque, S.; Banks, L.; Matlashewski, G. DNA damage inducedp53 mediated transcription is inhibited by human papillomavirus type 18 E6. Oncogene 1994, 9, 629–633. [Google Scholar] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acet-ylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Patel, D.; Huang, S.M.; Baglia, L.A.; Mc Cance, D.J. The E6 protein of human papillo-mavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. p53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Roberts, J.; Dakic, A.; Zhang, Y.; Schlegel, R. HPV E7 contributes to the telomerase activity of immortalized and tumorigenic cells and augments E6-induced hTERT promoter function. Virology 2008, 375, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Thompson, D.A.; Münger, K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 1997, 239, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papillomavirus-16 induces degradation of retinoblastoma protein through the ubiquitin–proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- White, E.A.; Sowa, M.E.; Tan, M.J.; Jeudy, S.; Hayes, S.D.; Santha, S.; Münger, K.; Harper, J.W. Howley PM. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. USA 2012, 109, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, E.G.; Romano, E.; del Porto, P.; Ascenzioni, F. Multifunctional role of ATM/Tel1 kinase in genome stability: From the DNA damage response to telomere maintenance. BioMed. Res. Int. 2014, 2014, 78740–78744. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Tindle, R.W. Immune evasion in human papillomavirus-associated cervical cancer. Nat. Rev. Cancer 2002, 2, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.; Giannini, T.; Lucchese, A.; Kanduc, D. Computer-assisted analysis ofmolecular mimicry between human papillomavirus 16 E7 oncoprotein and human protein sequences. Immunol. Cell Biol. 2000, 78, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Caberg, J.H.; Hubert, P.M.; Begon, D.Y.; Herfs, M.F.; Roncarati, P.J.; Boniver, J.J. Delvenne PO. Silencing of E7 oncogene restores functional E-cadherin expression in human papillomavirus 16-transformed keratinocytes. Carcinogenesis 2008, 29, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Suprynowicz, F.A.; Disbrow, G.L.; Krawczyk, E.; Simic, V.; Lantzky, K.; Schlegel, R. HPV-16 E5 oncoprotein upregulates lipid raft components caveolin-1 and ganglioside GM1 at the plasma membrane of cervical cells. Oncogene 2008, 27, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Bates, E.; Takeshita, F.; Biliato, A.; Accardi, R.; Bouvard, V.; Mansour, M.; Vincent, I.; Gissmann, L.; Iftner, T.; et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J. Immunol. 2007, 178, 3186–3197. [Google Scholar] [CrossRef] [PubMed]
- Monnier-Benoit, S.; Mauny, F.; Riethmuller, D. Immunohistochemical analysis of CD4+ and CD8+ T-cell subsets in high risk human papillomavirus-associated pre-malignant and malignant lesions of the uterine cervix. Gynecol. Oncol. 2006, 102, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.L.; van den Hende, M.; Sterling, J.C. A prospective study on the natural course of low-grade squamous intraepithelial lesions and the presence of HPV16 E2-, E6- and E7-specific T-cell responses. Int. J. Cancer 2010, 126, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Kawana, K.; Schust, D.J. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: A possible mechanism for immune evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.Y.; Song, S.H.; You, S.; Lee, J.; Kim, J.; Racanelli, V.; Son, H.; Shin, E.C. Programmed death-1 (PD-1)-dependent functional impairment of CD4 (+) T cells in recurrent genital papilloma. Clin. Exp. Med. 2014, 14, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Adurthi, S.; Krishna, S.; Mukherjee, G.; Bafna, U.D.; Devi, U.; Jayshree, R.S. Regulatory T cells in a spectrum of HPV-induced cervical lesions: Cervicitis, cervical intraepithelial neoplasia and squamous cell carcinoma. Am. J. Reprod. Immunol. 2008, 60, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Weinberg, V.; Darragh, T.; Smith-McCune, K. Evolving immunosuppressive microenvironment during human cervical carcinogenesis. Mucosal Immunol. 2008, 1, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Piersma, S.J.; Welters, M.J.; van der Burg, S.H. Tumor-specific regulatory T cells in cancer patients. Hum. Immunol. 2008, 69, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Arribillaga, L.; Sarobe, P. CD4+/CD25+ regulatory cells inhibit activation of tumor-primed CD4+T cells with IFN-γ-dependent antiangiogenic activity, as well as long-lasting tumor immunity elicited by peptide vaccination. J. Immunol. 2003, 171, 5931–5939. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bordignon, V.; Di Domenico, E.G.; Trento, E.; D’Agosto, G.; Cavallo, I.; Pontone, M.; Pimpinelli, F.; Mariani, L.; Ensoli, F. How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery. Viruses 2017, 9, 390. https://doi.org/10.3390/v9120390
Bordignon V, Di Domenico EG, Trento E, D’Agosto G, Cavallo I, Pontone M, Pimpinelli F, Mariani L, Ensoli F. How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery. Viruses. 2017; 9(12):390. https://doi.org/10.3390/v9120390
Chicago/Turabian StyleBordignon, Valentina, Enea Gino Di Domenico, Elisabetta Trento, Giovanna D’Agosto, Ilaria Cavallo, Martina Pontone, Fulvia Pimpinelli, Luciano Mariani, and Fabrizio Ensoli. 2017. "How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery" Viruses 9, no. 12: 390. https://doi.org/10.3390/v9120390
APA StyleBordignon, V., Di Domenico, E. G., Trento, E., D’Agosto, G., Cavallo, I., Pontone, M., Pimpinelli, F., Mariani, L., & Ensoli, F. (2017). How Human Papillomavirus Replication and Immune Evasion Strategies Take Advantage of the Host DNA Damage Repair Machinery. Viruses, 9(12), 390. https://doi.org/10.3390/v9120390