Protection of Mice from Lethal Vaccinia Virus Infection by Vaccinia Virus Protein Subunits with a CpG Adjuvant


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Viruses
2.3. Recombinant Proteins and Adjuvants
2.4. Mouse Challenges
2.5. ELISA
2.6. Depletion of CD4 and CD8 T cells
2.7. Passive Transfer of Mabs
2.8. Statistical Analysis
3. Results
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nolen, L.D.; Osadebe, L.; Katomba, J.; Likofata, J.; Mukadi, D.; Monroe, B.; Doty, J.; Hughes, C.M.; Kabamba, J.; Malekani, J.; et al. Extended human-to-human transmission during a monkeypox outbreak in the Democratic Republic of the Congo. Emerg. Infect. Dis. 2016, 22, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Rimoin, A.W.; Kisalu, N.; Kebela-Ilunga, B.; Mukaba, T.; Wright, L.L.; Formenty, P.; Wolfe, N.D.; Shongo, R.L.; Tshioko, F.; Okitolonda, E.; et al. Endemic human monkeypox, democratic Republic of Congo, 2001–2004. Emerg. Infect. Dis. 2007, 13, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Fuller, T.; Thomassen, H.A.; Mulembakani, P.M.; Johnston, S.C.; Lloyd-Smith, J.O.; Kisalu, N.K.; Lutete, T.K.; Blumberg, S.; Fair, J.N.; Wolfe, N.D.; et al. Using remote sensing to map the risk of human monkeypox virus in the Congo Basin. Ecohealth 2011, 8, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Leggat, P.A. Human monkeypox: Current state of knowledge and implications for the future. Trop. Med. Infect. Dis. 2016, 1, 8. [Google Scholar] [CrossRef]
- Reed, K.D.; Melski, J.W.; Graham, M.B.; Regnery, R.L.; Sotir, M.J.; Wegner, M.V.; Kazmierczak, J.J.; Stratman, E.J.; Li, Y.; Fairley, J.A.; et al. The detection of monkeypox in humans in the Western Hemisphere. N. Engl. J. Med. 2004, 350, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Fenner, F.; Henderson, D.; Arita, I.; Jezek, Z.; Ladnyi, I. Smallpox and Its Eradication; World Health Organization: Geneva, Switzerland, 1988. [Google Scholar]
- Greenberg, R.N.; Hay, C.M.; Stapleton, J.T.; Marbury, T.C.; Wagner, E.; Kreitmeir, E.; Röesch, S.; von Krempelhuber, A.; Young, P.; Nichols, R.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase II Trial Investigating the Safety and Immunogenicity of Modified Vaccinia Ankara Smallpox Vaccine (MVA-BN®) in 56-80-Year-Old Subjects. PLoS ONE 2016, 11, e0157335. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, R.N.; Hurley, Y.; Dinh, D.V.; Mraz, S.; Vera, J.G.; von Bredow, D.; von Krempelhuber, A.; Roesch, S.; Virgin, G.; Arndtz-Wiedemann, N.; et al. A multicenter, Open-Label, Controlled Phase II Study to Evaluate Safety and Immunogenicity of MVA Smallpox Vaccine (IMVAMUNE) in 18–40 Year Old Subjects with Diagnosed Atopic Dermatitis. PLoS ONE 2015, 10, e0138348. [Google Scholar] [CrossRef]
- Mayr, A.; Stickl, H.; Müller, H.; Danner, K.; Singer, H. The smallpox vaccination strain MVA: Marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism (author’s transl). Zentralbl. Bakteriol. B 1978, 167, 375–390. [Google Scholar] [PubMed]
- Yokote, H.; Shinmura, Y. Reply to “Use of the LC16m8 Smallpox Vaccine in Immunocompromised Individuals Is Still Too Risky”. Clin. Vaccine Immunol. 2015, 22, 605. [Google Scholar] [CrossRef] [PubMed]
- Yokote, H.; Shinmura, Y.; Kanehara, T.; Maruno, S.; Kuranaga, M.; Matsui, H.; Hashizume, S. Safety of attenuated smallpox vaccine LC16m8 in immunodeficient mice. Clin. Vaccine Immunol. 2014, 21, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Kimura, M.; Hirayama, M. Report of the National Smallpox Vaccination Research Committee: Study of side effects, complications and their treatments. Clin. Virol. 1975, 3, 269–278. [Google Scholar]
- Danon, Y.L.; Sutter, G. Use of the LC16m8 smallpox vaccine in immunocompromised individuals is still too risky. Clin. Vaccine Immunol. 2015, 22, 604. [Google Scholar] [CrossRef] [PubMed]
- Eto, A.; Saito, T.; Yokote, H.; Kurane, I.; Kanatani, Y. Recent advances in the study of live attenuated cell-cultured smallpox vaccine LC16m8. Vaccine 2015, 33, 6106–6111. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.; Custer, D.; Thompson, E. Four-gene-combination DNA vaccine protects mice against a lethal vaccinia virus challenge and elicits appropriate antibody responses in nonhuman primates. Virology 2003, 306, 181–195. [Google Scholar] [CrossRef]
- Hooper, J.; Thompson, E.; Wilhelmsen, C.; Zimmerman, M.; Ichou, M.A.; Steffen, S.; Schmaljohn, C.; Schmaljohn, A.; Jahrling, P. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J. Virol. 2004, 78, 4433–4443. [Google Scholar] [CrossRef] [PubMed]
- Pulford, D.; Gates, A.; Bridge, S.; Robinson, J.; Ulaeto, D. Differential efficacy of vaccinia virus envelope proteins administered by DNA immunisation in protection of BALB/c mice from a lethal intranasal poxvirus challenge. Vaccine 2004, 22, 3358–3366. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Smallpox vaccines: Targets of protective immunity. Immunol. Rev. 2011, 239, 8–26. [Google Scholar] [PubMed]
- Sakhatskyy, P.; Wang, S.; Te-hui, W.C.; Lu, S. Immunogenicity and protection efficacy of monovalent and polyvalent poxvirus vaccines that include the D8 antigen. Virology 2006, 355, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, M.C.; Goenaga, J.; Wittek, R.; Rindisbacher, L. Neutralizing and protective antibodies directed against vaccinia virus envelope antigens. Virology 1999, 254, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.H.; McCausland, M.M.; Valdez, C.; Huynh, D.; Hernandez, J.E.; Mu, Y.; Hirst, S.; Villarreal, L.; Felgner, P.L.; Crotty, S. Vaccinia virus H3L envelope protein is a major target of neutralizing antibodies in humans and elicits protection against lethal challenge in mice. J. Virol. 2005, 79, 11724–11733. [Google Scholar] [CrossRef] [PubMed]
- Fogg, C.; Lustig, S.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H.; Moss, B. Protective immunity to vaccinia virus induced by vaccination with multiple recombinant outer membrane proteins of intracellular and extracellular virions. J. Virol. 2004, 78, 10230–10237. [Google Scholar] [CrossRef] [PubMed]
- Fogg, C.N.; Americo, J.L.; Lustig, S.; Huggins, J.W.; Smith, S.K.; Damon, I.; Resch, W.; Earl, P.L.; Klinman, D.M.; Moss, B. Adjuvant-enhanced antibody responses to recombinant proteins correlates with protection of mice and monkeys to orthopoxvirus challenges. Vaccine 2007, 25, 2787–2799. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Gong, S.; Esteban, M. The purified 14-kilodalton envelope protein of vaccinia virus produced in Escherichia coli induces virus immunity in animals. J. Virol. 1991, 65, 5631–5635. [Google Scholar] [PubMed]
- Berhanu, A.; Wilson, R.L.; Kirkwood-Watts, D.L.; King, D.S.; Warren, T.K.; Lund, S.A.; Brown, L.L.; Krupkin, A.K.; VanderMay, E.; Weimers, W. Vaccination of BALB/c mice with Escherichia coli-expressed vaccinia virus proteins A27L, B5R, and D8L protects mice from lethal vaccinia virus challenge. J. Virol. 2008, 82, 3517–3529. [Google Scholar] [CrossRef] [PubMed]
- Buchman, G.W.; Cohen, M.E.; Xiao, Y.; Richardson-Harman, N.; Silvera, P.; DeTolla, L.J.; Davis, H.L.; Eisenberg, R.J.; Cohen, G.H.; Isaacs, S.N. A protein-based smallpox vaccine protects non-human primates from a lethal monkeypox virus challenge. Vaccine 2010, 28, 6627–6636. [Google Scholar] [CrossRef]
- Heraud, J.-M.; Edghill-Smith, Y.; Ayala, V.; Kalisz, I.; Parrino, J.; Kalyanaraman, V.S.; Manischewitz, J.; King, L.R.; Hryniewicz, A.; Trindade, C.J.; et al. Subunit recombinant vaccine protects against monkeypox. J. Immunol. 2006, 177, 2552–2564. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, P. On the mechanism determining the TH1/TH2 phenotype of an immune response, and its pertinence to strategies for the prevention, and treatment, of certain infectious diseases. Scand. J. Immunol. 2014, 79, 361–376. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, J.C.; Gherardi, M.M.; Esteban, M. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: Virus fate and activation of B-and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J. Virol. 2000, 74, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Chu, R.S.; Targoni, O.S.; Krieg, A.M.; Lehmann, P.V.; Harding, C.V. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J. Exp. Med. 1997, 186, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M.; Davis, H.L. CpG ODN as a Th1 immune enhancer for prophylactic and therapeutic vaccines. In Vaccine Adjuvants; Humana Press Inc.: Totowa, NJ, USA, 2006; pp. 87–110. [Google Scholar]
- Davis, H.L.; Weeranta, R.; Waldschmidt, T.J.; Tygrett, L.; Schorr, J.; Krieg, A.M. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J. Immunol. 1998, 160, 870–876. [Google Scholar] [PubMed]
- Cooper, C.; Davis, H.; Morris, M.; Efler, S.; Al Adhami, M.; Krieg, A.; Cameron, D.; Heathcote, J. CpG 7909, an immunostimulatory TLR9 agonist oligodeoxynucleotide, as adjuvant to Engerix-B® HBV vaccine in healthy adults: A double-blind phase I/II study. J. Clin. Immunol. 2004, 24, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.; Davis, H.; Morris, M.; Efler, S.; Krieg, A.; Li, Y.; Laframboise, C.; Al Adhami, M.; Khaliq, Y.; Seguin, I.; et al. Safety and immunogenicity of CpG 7909 injection as an adjuvant to Fluarix influenza vaccine. Vaccine 2004, 22, 3136–3143. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M. CpG still rocks! Update on an accidental drug. Nucleic Acid Ther. 2012, 22, 77–89. [Google Scholar] [PubMed]
- Krieg, A.M. CpG motifs: The active ingredient in bacterial extracts? Nat. Med. 2003, 9, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Phelps, A.; Gates, A.; Hillier, M.; Eastaugh, L.; Ulaeto, D. Comparative efficacy of replicating smallpox vaccine strains in a murine challenge model. Vaccine 2005, 23, 3500–3507. [Google Scholar] [CrossRef] [PubMed]
- Ramı́rez, J.C.; Tapia, E.; Esteban, M. Administration to mice of a monoclonal antibody that neutralizes the intracellular mature virus form of vaccinia virus limits virus replication efficiently under prophylactic and therapeutic conditions. J. Gen. Virol. 2002, 83, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Phelps, A.; Gates, A.; Eastaugh, L.; Hillier, M.; Ulaeto, D. Comparative efficacy of intramuscular and scarification routes of administration of live smallpox vaccine in a murine challenge model. Vaccine 2017, 35, 3889–3896. [Google Scholar] [CrossRef] [PubMed]
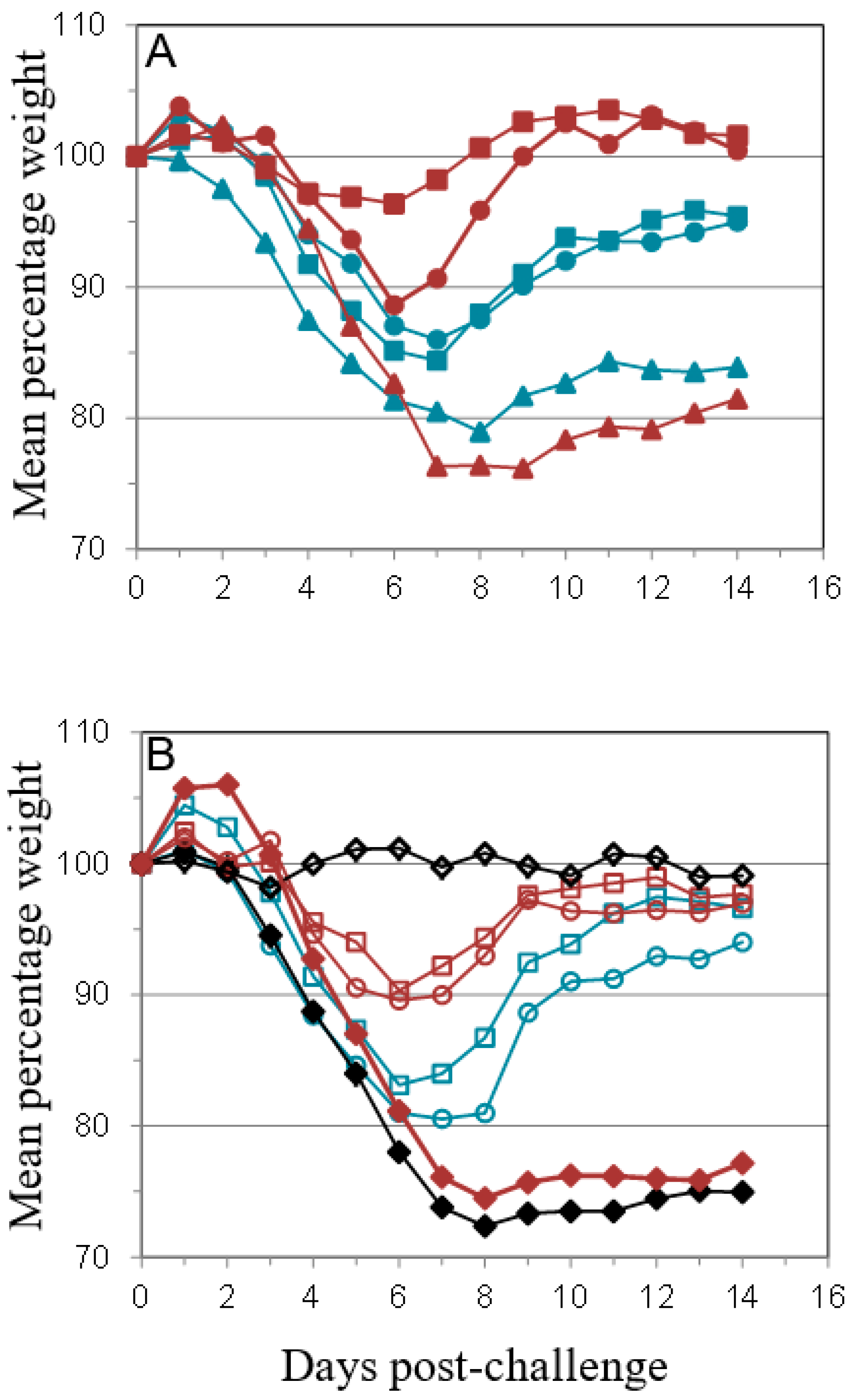
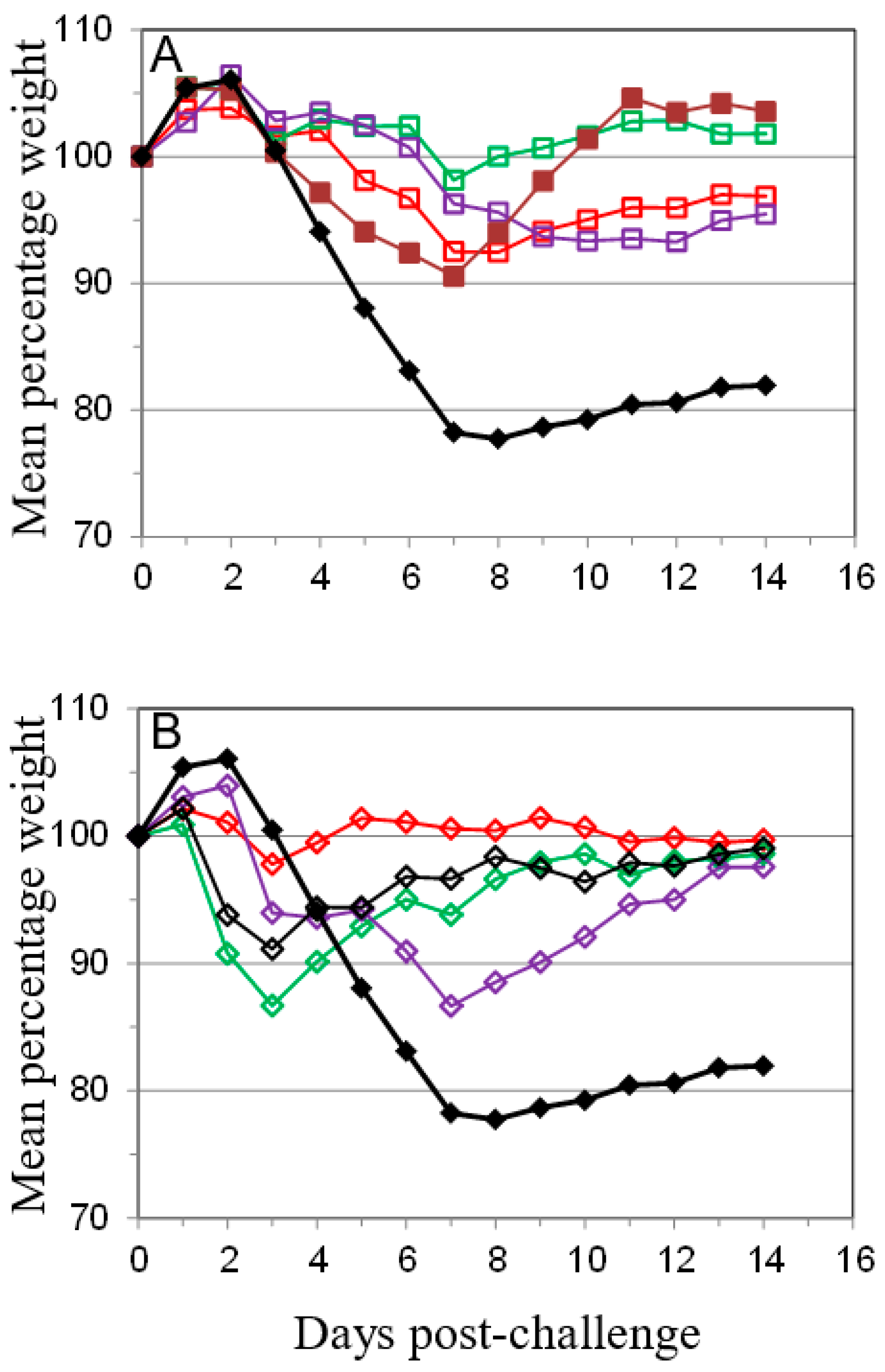
 ); PBS/CpG (
); PBS/CpG (  ); VACV Lister (
); VACV Lister (  ); or protein subunits adjuvanted with alhydrogel or CpG; and challenged with 100 median lethal doses (MLD) of VACV IHD. Protein combinations were A27/B5/alhydrogel (
); or protein subunits adjuvanted with alhydrogel or CpG; and challenged with 100 median lethal doses (MLD) of VACV IHD. Protein combinations were A27/B5/alhydrogel (  );A27/B5/CpG (
);A27/B5/CpG (  ); A33/B5/alhydrogel (
); A33/B5/alhydrogel (  );A33/B5/CpG (
);A33/B5/CpG (  ); A27/A33/alhydrogel (
); A27/A33/alhydrogel (  );A27/A33/CpG (
);A27/A33/CpG (  ); A27/A33/B5/alhydrogel (
); A27/A33/B5/alhydrogel (  ); A27/A33/B5/CpG (
); A27/A33/B5/CpG (  ); A27/A33/B5/L1/alhydrogel (
); A27/A33/B5/L1/alhydrogel (  ); or A27/A33/B5/L1/CpG (
); or A27/A33/B5/L1/CpG (  ). Each animal received 10 µg (divalent, A), 6.7 µg (trivalent, B) or 5 µg (quadrivalent, B) of each protein for each immunization. A total of three immunizations were given at 21-day intervals, with challenge at 21 days after the 3rd immunization. PBS/CpG was administered on the same schedule as protein immunizations. Lister vaccination was given only once, with a dose of 1 × 106 pfu on a shaved flank, as previously described [38]. Lister vaccinated animals were bled once 21 days post vaccination and challenged at 28 days post vaccination, with the other groups. PBS controls were on the same schedule as Lister vaccination. Data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by Analysis of Variance (ANOVA) and 2-way ANOVA.
); PBS/CpG ( ); VACV Lister ( ); or protein subunits adjuvanted with alhydrogel or CpG; and challenged with 100 median lethal doses (MLD) of VACV IHD. Protein combinations were A27/B5/alhydrogel ( );A27/B5/CpG ( ); A33/B5/alhydrogel ( );A33/B5/CpG ( ); A27/A33/alhydrogel ( );A27/A33/CpG ( ); A27/A33/B5/alhydrogel ( ); A27/A33/B5/CpG ( ); A27/A33/B5/L1/alhydrogel ( ); or A27/A33/B5/L1/CpG ( ). Each animal received 10 µg (divalent, A), 6.7 µg (trivalent, B) or 5 µg (quadrivalent, B) of each protein for each immunization. A total of three immunizations were given at 21-day intervals, with challenge at 21 days after the 3rd immunization. PBS/CpG was administered on the same schedule as protein immunizations. Lister vaccination was given only once, with a dose of 1 × 106 pfu on a shaved flank, as previously described [38]. Lister vaccinated animals were bled once 21 days post vaccination and challenged at 28 days post vaccination, with the other groups. PBS controls were on the same schedule as Lister vaccination. Data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by Analysis of Variance (ANOVA) and 2-way ANOVA.
). Each animal received 10 µg (divalent, A), 6.7 µg (trivalent, B) or 5 µg (quadrivalent, B) of each protein for each immunization. A total of three immunizations were given at 21-day intervals, with challenge at 21 days after the 3rd immunization. PBS/CpG was administered on the same schedule as protein immunizations. Lister vaccination was given only once, with a dose of 1 × 106 pfu on a shaved flank, as previously described [38]. Lister vaccinated animals were bled once 21 days post vaccination and challenged at 28 days post vaccination, with the other groups. PBS controls were on the same schedule as Lister vaccination. Data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by Analysis of Variance (ANOVA) and 2-way ANOVA.
); PBS/CpG ( ); VACV Lister ( ); or protein subunits adjuvanted with alhydrogel or CpG; and challenged with 100 median lethal doses (MLD) of VACV IHD. Protein combinations were A27/B5/alhydrogel ( );A27/B5/CpG ( ); A33/B5/alhydrogel ( );A33/B5/CpG ( ); A27/A33/alhydrogel ( );A27/A33/CpG ( ); A27/A33/B5/alhydrogel ( ); A27/A33/B5/CpG ( ); A27/A33/B5/L1/alhydrogel ( ); or A27/A33/B5/L1/CpG ( ). Each animal received 10 µg (divalent, A), 6.7 µg (trivalent, B) or 5 µg (quadrivalent, B) of each protein for each immunization. A total of three immunizations were given at 21-day intervals, with challenge at 21 days after the 3rd immunization. PBS/CpG was administered on the same schedule as protein immunizations. Lister vaccination was given only once, with a dose of 1 × 106 pfu on a shaved flank, as previously described [38]. Lister vaccinated animals were bled once 21 days post vaccination and challenged at 28 days post vaccination, with the other groups. PBS controls were on the same schedule as Lister vaccination. Data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by Analysis of Variance (ANOVA) and 2-way ANOVA.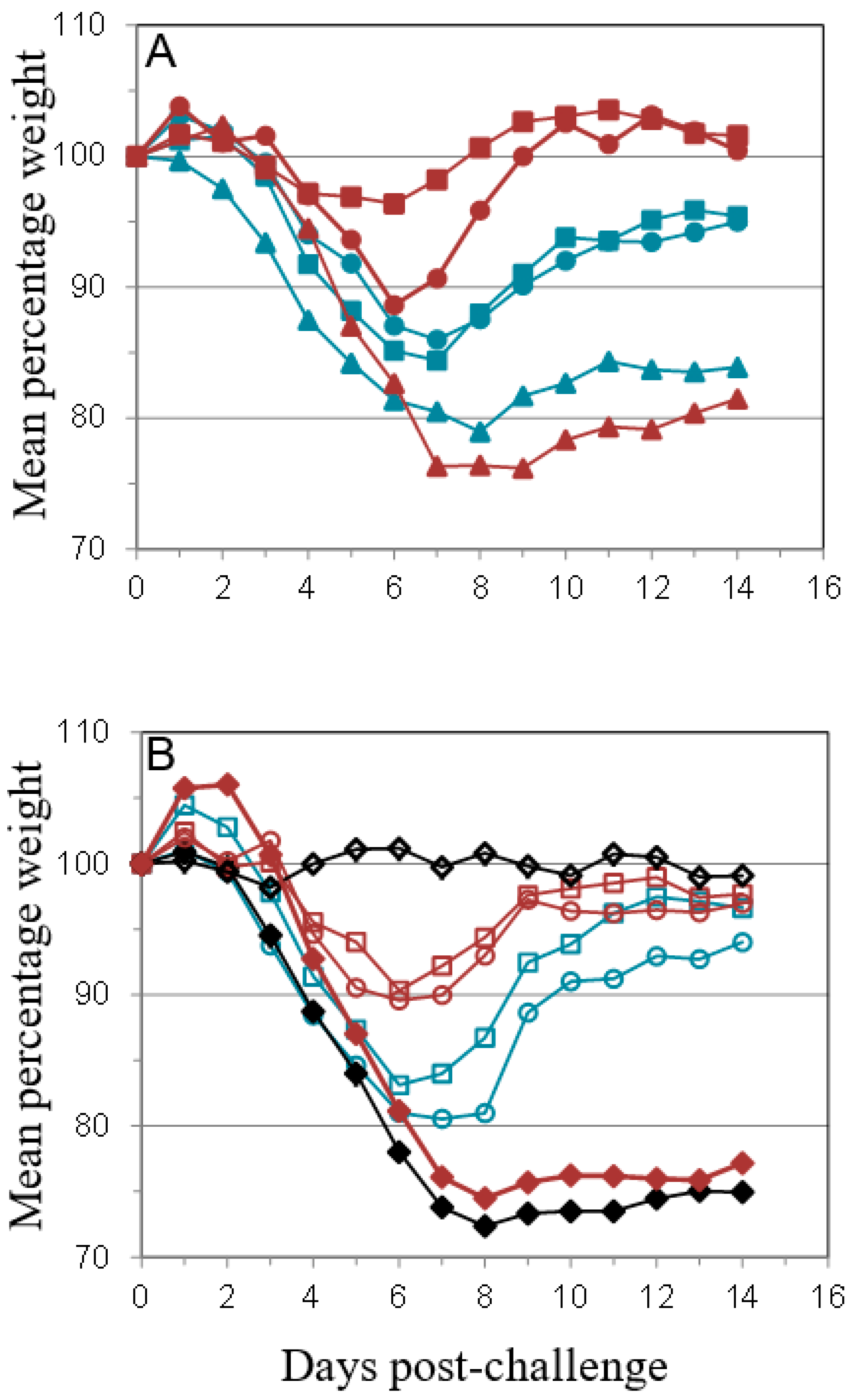
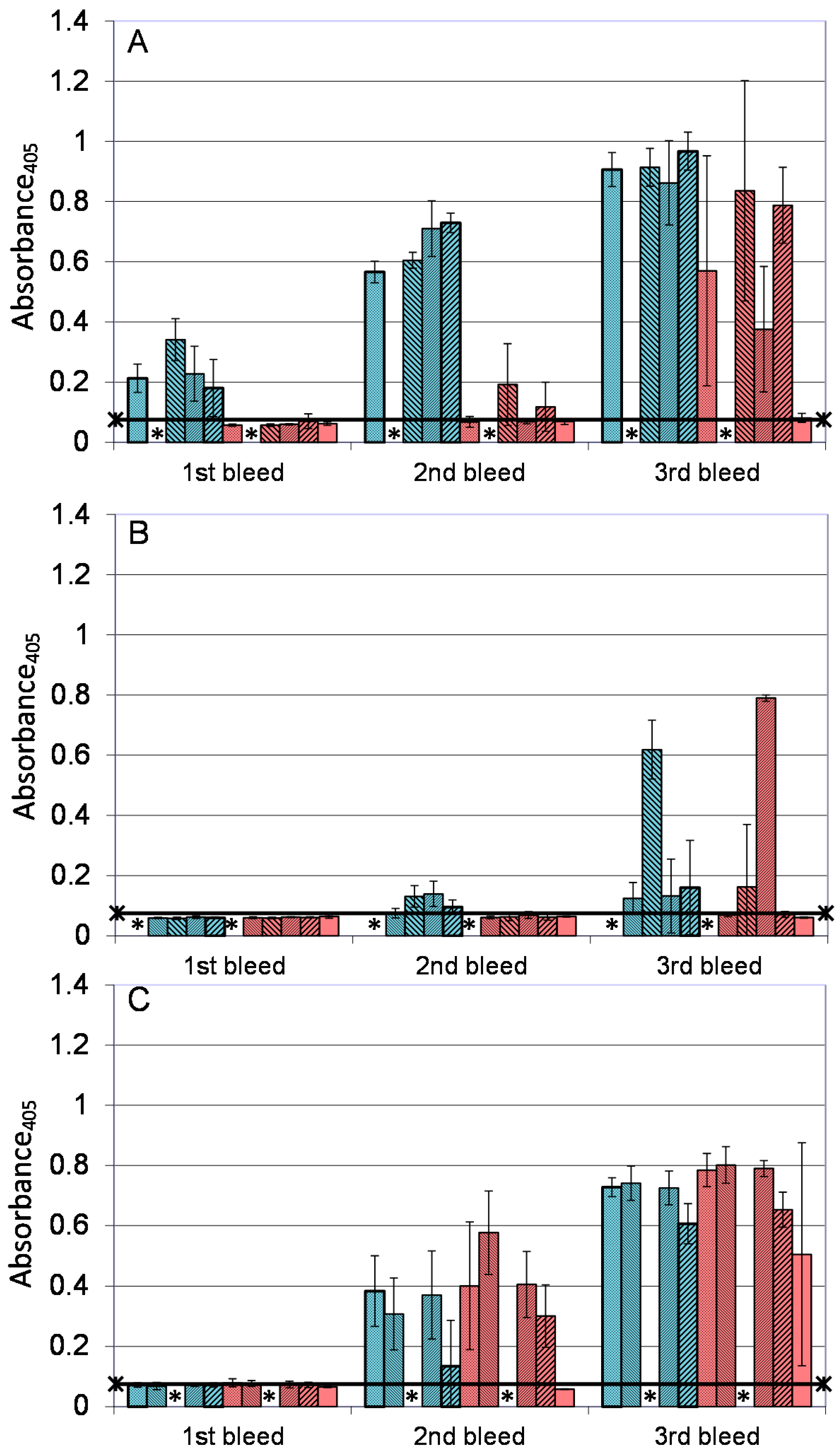
 ); A27/B5/CpG (
); A27/B5/CpG (  ); A33/B5/alhydrogel (
); A33/B5/alhydrogel (  ); A33/B5/CpG (
); A33/B5/CpG (  ); A27/A33/alhydrogel (
); A27/A33/alhydrogel (  ); A27/A33/CpG (
); A27/A33/CpG (  ); A27/A33/B5/alhydrogel (
); A27/A33/B5/alhydrogel (  ); A27/A33/B5/CpG (
); A27/A33/B5/CpG (  ); A27/A33/B5/L1/alhydrogel (
); A27/A33/B5/L1/alhydrogel (  ); A27/A33/B5/L1/CpG (
); A27/A33/B5/L1/CpG (  ); and PBS/CpG (
); and PBS/CpG (  ), and bled 14 days after each immunization. * Not done. ELISA data is presented as means and standard deviations of samples from five animals (1 well/animal). n = five animals/group. Horizontal bar represents the mean of the negative control, plus 2× standard deviations.
); A27/B5/CpG ( ); A33/B5/alhydrogel ( ); A33/B5/CpG ( ); A27/A33/alhydrogel ( ); A27/A33/CpG ( ); A27/A33/B5/alhydrogel ( ); A27/A33/B5/CpG ( ); A27/A33/B5/L1/alhydrogel ( ); A27/A33/B5/L1/CpG ( ); and PBS/CpG ( ), and bled 14 days after each immunization. * Not done. ELISA data is presented as means and standard deviations of samples from five animals (1 well/animal). n = five animals/group. Horizontal bar represents the mean of the negative control, plus 2× standard deviations.
), and bled 14 days after each immunization. * Not done. ELISA data is presented as means and standard deviations of samples from five animals (1 well/animal). n = five animals/group. Horizontal bar represents the mean of the negative control, plus 2× standard deviations.
); A27/B5/CpG ( ); A33/B5/alhydrogel ( ); A33/B5/CpG ( ); A27/A33/alhydrogel ( ); A27/A33/CpG ( ); A27/A33/B5/alhydrogel ( ); A27/A33/B5/CpG ( ); A27/A33/B5/L1/alhydrogel ( ); A27/A33/B5/L1/CpG ( ); and PBS/CpG ( ), and bled 14 days after each immunization. * Not done. ELISA data is presented as means and standard deviations of samples from five animals (1 well/animal). n = five animals/group. Horizontal bar represents the mean of the negative control, plus 2× standard deviations.
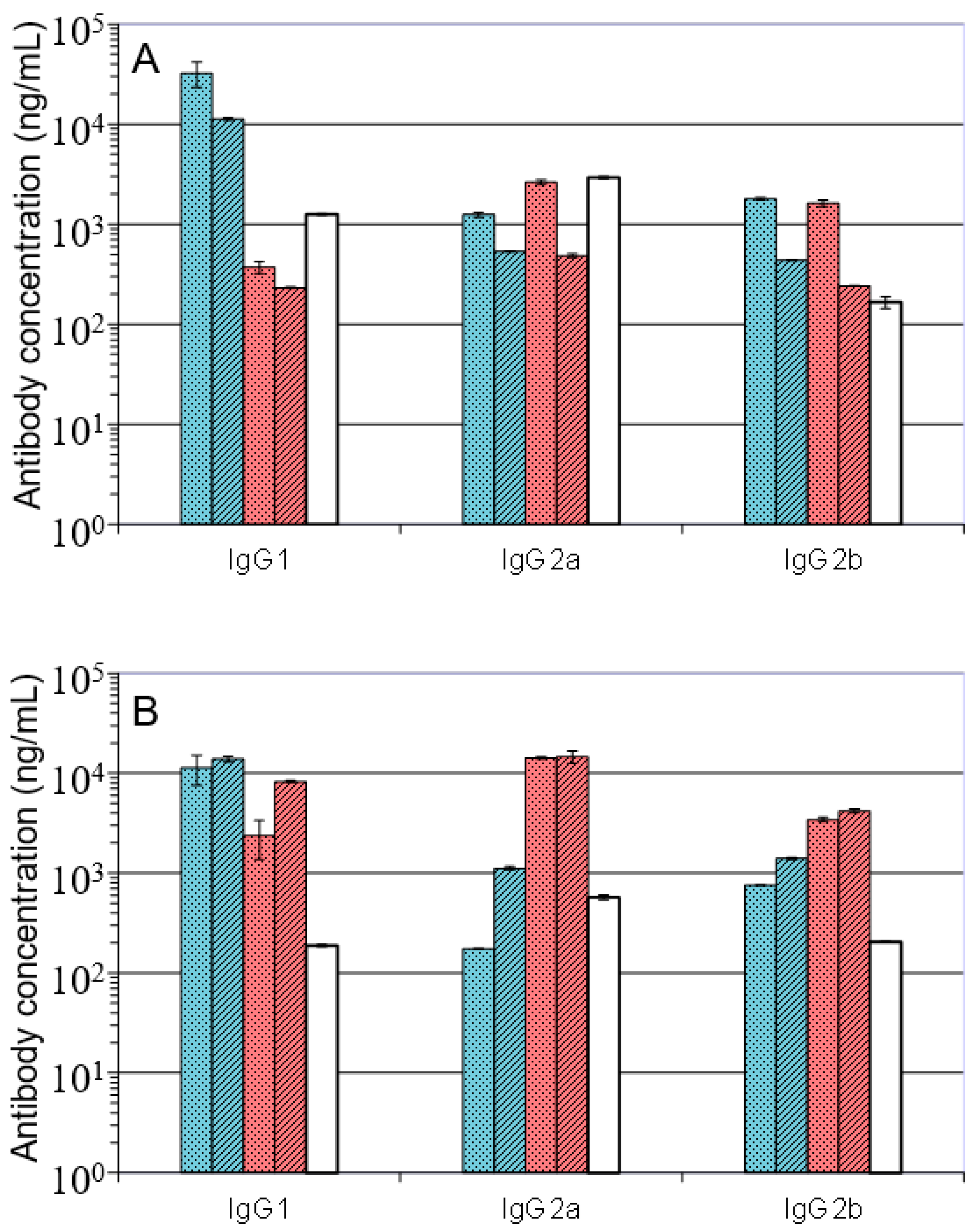
 ); A27/A33/B5/alhydrogel (
); A27/A33/B5/alhydrogel (  ); A27/B5/CpG (
); A27/B5/CpG (  ); A27/A33/B5/CpG (
); A27/A33/B5/CpG (  ); VACV Lister (
); VACV Lister (  ); Data is presented as means and standard deviations of triplicate wells.
); A27/A33/B5/alhydrogel ( ); A27/B5/CpG ( ); A27/A33/B5/CpG ( ); VACV Lister ( ); Data is presented as means and standard deviations of triplicate wells.
); Data is presented as means and standard deviations of triplicate wells.
); A27/A33/B5/alhydrogel ( ); A27/B5/CpG ( ); A27/A33/B5/CpG ( ); VACV Lister ( ); Data is presented as means and standard deviations of triplicate wells.
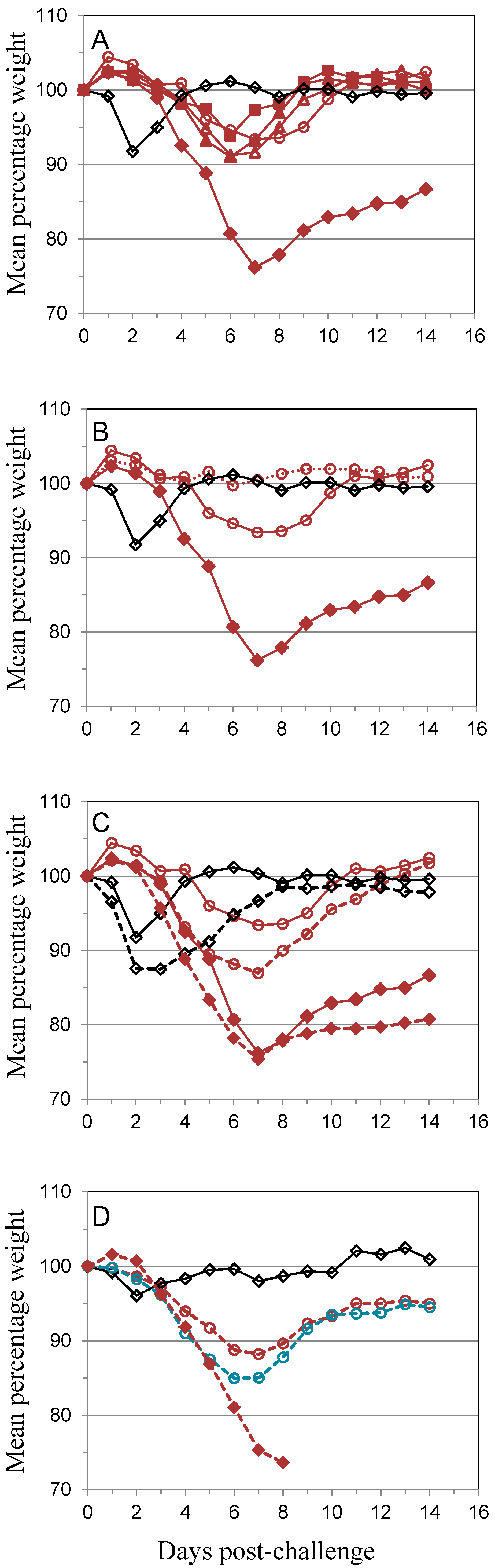
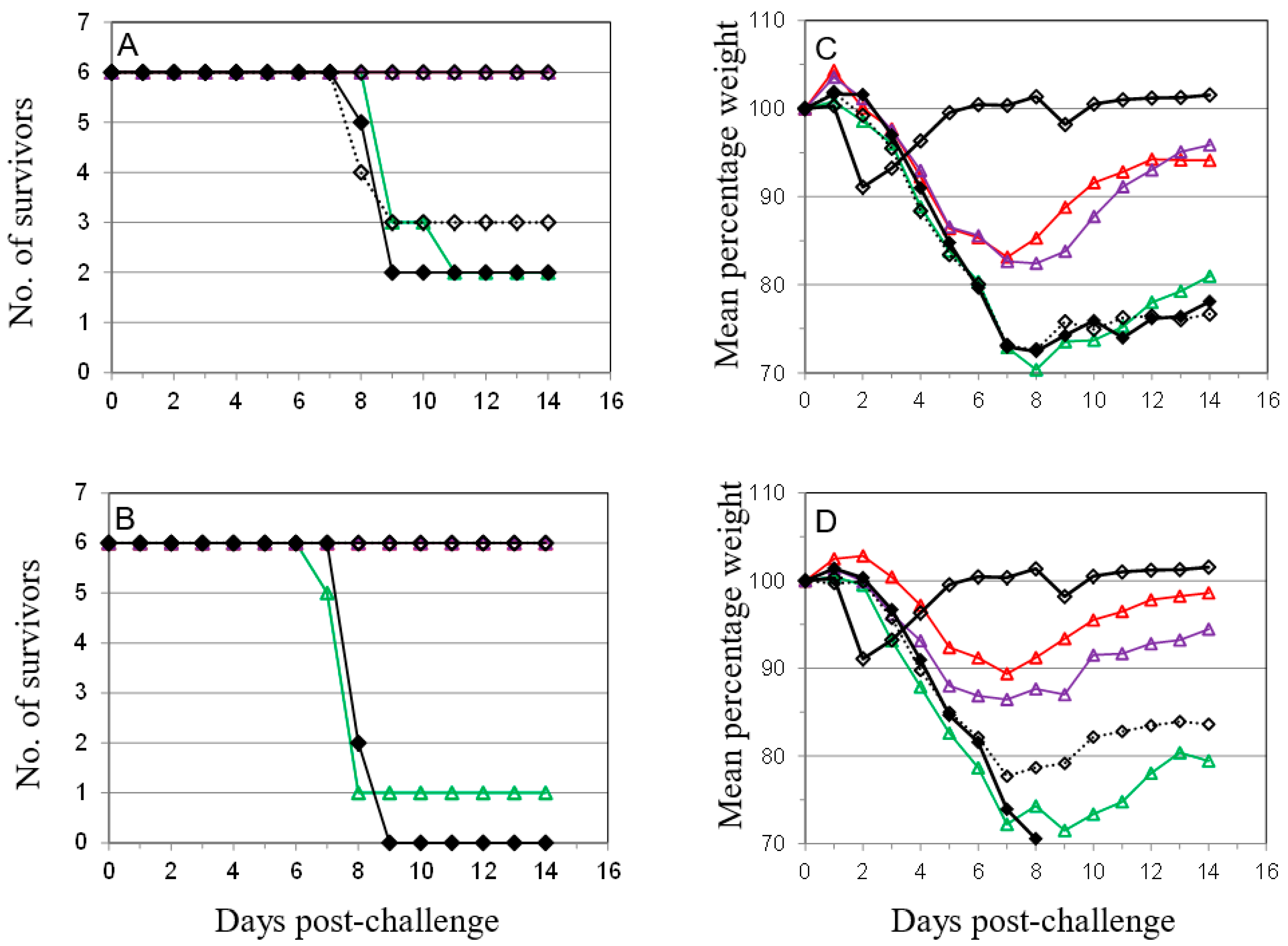
 ); 10 µg A27 and 10 µg B5 (
); 10 µg A27 and 10 µg B5 (  ); 5 µg A27 and 10 µg B5 (
); 5 µg A27 and 10 µg B5 (  ); 10 µg A27 and 5 µg B5 (
); 10 µg A27 and 5 µg B5 (  ); (A). In the same experiment the dosing schedule for animals receiving 5 µg/dose of each protein was varied to reduce the interval between 1st and 2nd immunizations to 14 days (
); (A). In the same experiment the dosing schedule for animals receiving 5 µg/dose of each protein was varied to reduce the interval between 1st and 2nd immunizations to 14 days (  ), with all other aspects unchanged (B). Also in the same experiment the interval between the 3rd immunization and challenge was extended to 56 days for animals receiving 5 µg/dose of each protein (
), with all other aspects unchanged (B). Also in the same experiment the interval between the 3rd immunization and challenge was extended to 56 days for animals receiving 5 µg/dose of each protein (  ); or Lister vaccine (
); or Lister vaccine (  ); or PBS/CpG control (
); or PBS/CpG control (  ); with all other aspects unchanged (C). In a separate experiment the dosing schedule for animals receiving 5 µg/dose of each protein (
); with all other aspects unchanged (C). In a separate experiment the dosing schedule for animals receiving 5 µg/dose of each protein (  ) was varied to reduce all immunization intervals and the 3rd immunization/challenge interval to 14 days for material adjuvanted with CpG7909 (
) was varied to reduce all immunization intervals and the 3rd immunization/challenge interval to 14 days for material adjuvanted with CpG7909 (  ); or alhydrogel (
); or alhydrogel (  ); or PBS/CpG control (
); or PBS/CpG control (  ); with all other aspects unchanged (D). Controls in all panels are Lister vaccine (
); with all other aspects unchanged (D). Controls in all panels are Lister vaccine (  ); and PBS/CpG (
); and PBS/CpG (  ) under the same dosing/challenge schedule as for Figure 1. Weight data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
); 10 µg A27 and 10 µg B5 ( ); 5 µg A27 and 10 µg B5 ( ); 10 µg A27 and 5 µg B5 ( ); (A). In the same experiment the dosing schedule for animals receiving 5 µg/dose of each protein was varied to reduce the interval between 1st and 2nd immunizations to 14 days ( ), with all other aspects unchanged (B). Also in the same experiment the interval between the 3rd immunization and challenge was extended to 56 days for animals receiving 5 µg/dose of each protein ( ); or Lister vaccine ( ); or PBS/CpG control ( ); with all other aspects unchanged (C). In a separate experiment the dosing schedule for animals receiving 5 µg/dose of each protein ( ) was varied to reduce all immunization intervals and the 3rd immunization/challenge interval to 14 days for material adjuvanted with CpG7909 ( ); or alhydrogel ( ); or PBS/CpG control ( ); with all other aspects unchanged (D). Controls in all panels are Lister vaccine ( ); and PBS/CpG ( ) under the same dosing/challenge schedule as for Figure 1. Weight data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
) under the same dosing/challenge schedule as for Figure 1. Weight data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
); 10 µg A27 and 10 µg B5 ( ); 5 µg A27 and 10 µg B5 ( ); 10 µg A27 and 5 µg B5 ( ); (A). In the same experiment the dosing schedule for animals receiving 5 µg/dose of each protein was varied to reduce the interval between 1st and 2nd immunizations to 14 days ( ), with all other aspects unchanged (B). Also in the same experiment the interval between the 3rd immunization and challenge was extended to 56 days for animals receiving 5 µg/dose of each protein ( ); or Lister vaccine ( ); or PBS/CpG control ( ); with all other aspects unchanged (C). In a separate experiment the dosing schedule for animals receiving 5 µg/dose of each protein ( ) was varied to reduce all immunization intervals and the 3rd immunization/challenge interval to 14 days for material adjuvanted with CpG7909 ( ); or alhydrogel ( ); or PBS/CpG control ( ); with all other aspects unchanged (D). Controls in all panels are Lister vaccine ( ); and PBS/CpG ( ) under the same dosing/challenge schedule as for Figure 1. Weight data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
 ), or with divalent A27/B5/CpG (A) or Lister (B) vaccines under the same dosing/challenge schedule as for Figure 1, with protein concentrations (per dose) of 10 µg A27 and 10 µg B5. Animals were further treated with anti-CD4 (
), or with divalent A27/B5/CpG (A) or Lister (B) vaccines under the same dosing/challenge schedule as for Figure 1, with protein concentrations (per dose) of 10 µg A27 and 10 µg B5. Animals were further treated with anti-CD4 (  ,
,  ); anti-CD8 (
); anti-CD8 (  ,
,  ); anti-CD4 and anti-CD8 (
); anti-CD4 and anti-CD8 (  ,
,  ); or non-relevant Mab (
); or non-relevant Mab (  ,
,  ) as described in material methods. Weight data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
), or with divalent A27/B5/CpG (A) or Lister (B) vaccines under the same dosing/challenge schedule as for Figure 1, with protein concentrations (per dose) of 10 µg A27 and 10 µg B5. Animals were further treated with anti-CD4 ( , ); anti-CD8 ( , ); anti-CD4 and anti-CD8 ( , ); or non-relevant Mab ( , ) as described in material methods. Weight data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
) as described in material methods. Weight data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
), or with divalent A27/B5/CpG (A) or Lister (B) vaccines under the same dosing/challenge schedule as for Figure 1, with protein concentrations (per dose) of 10 µg A27 and 10 µg B5. Animals were further treated with anti-CD4 ( , ); anti-CD8 ( , ); anti-CD4 and anti-CD8 ( , ); or non-relevant Mab ( , ) as described in material methods. Weight data is presented as daily means of the weight of each surviving animal as a percentage of its initial weight. Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.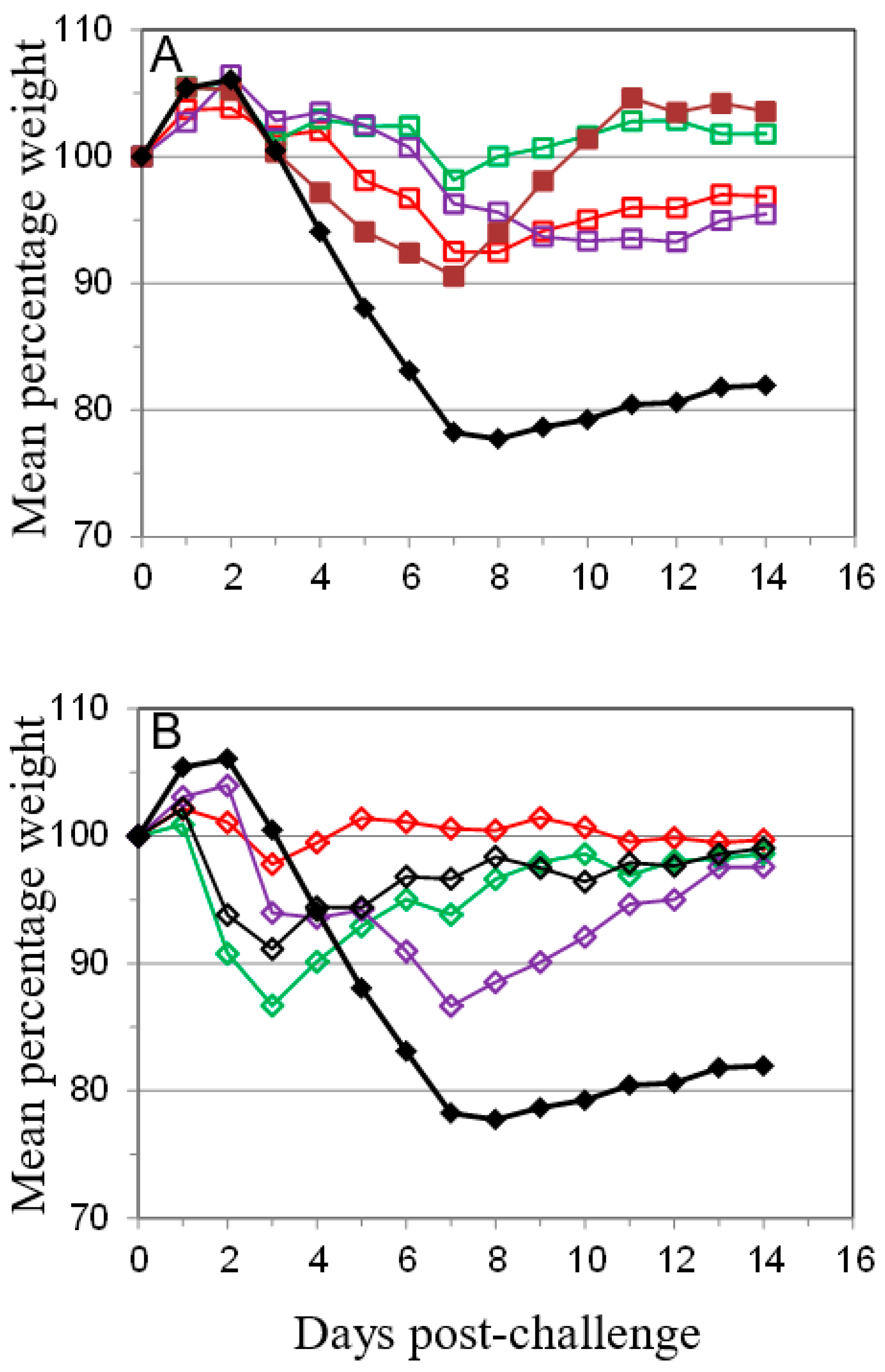
 ), anti-B5 (
), anti-B5 (  ) or both anti-A27 and anti-B5 Mab (
) or both anti-A27 and anti-B5 Mab (  ) was administered as described in Materials and Methods in 50 μg (A,C) or 100 μg (B,D) doses. Control animals were either vaccinated with Lister vaccine on the same schedule as for Figure 1 (
) was administered as described in Materials and Methods in 50 μg (A,C) or 100 μg (B,D) doses. Control animals were either vaccinated with Lister vaccine on the same schedule as for Figure 1 (  ); given purified IgG from animals vaccinated with Lister vaccine (
); given purified IgG from animals vaccinated with Lister vaccine (  ); or given normal rat IgG (
); or given normal rat IgG (  ). Mice were challenged as described above and monitored for 14 days. Data is presented as number of survivors (A,B) and daily means of the weight of each surviving animal as a percentage of its initial weight (C,D). Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
), anti-B5 ( ) or both anti-A27 and anti-B5 Mab ( ) was administered as described in Materials and Methods in 50 μg (A,C) or 100 μg (B,D) doses. Control animals were either vaccinated with Lister vaccine on the same schedule as for Figure 1 ( ); given purified IgG from animals vaccinated with Lister vaccine ( ); or given normal rat IgG ( ). Mice were challenged as described above and monitored for 14 days. Data is presented as number of survivors (A,B) and daily means of the weight of each surviving animal as a percentage of its initial weight (C,D). Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
). Mice were challenged as described above and monitored for 14 days. Data is presented as number of survivors (A,B) and daily means of the weight of each surviving animal as a percentage of its initial weight (C,D). Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.
), anti-B5 ( ) or both anti-A27 and anti-B5 Mab ( ) was administered as described in Materials and Methods in 50 μg (A,C) or 100 μg (B,D) doses. Control animals were either vaccinated with Lister vaccine on the same schedule as for Figure 1 ( ); given purified IgG from animals vaccinated with Lister vaccine ( ); or given normal rat IgG ( ). Mice were challenged as described above and monitored for 14 days. Data is presented as number of survivors (A,B) and daily means of the weight of each surviving animal as a percentage of its initial weight (C,D). Statistical analysis was by ANOVA and 2-way ANOVA. n = 6 animals per group.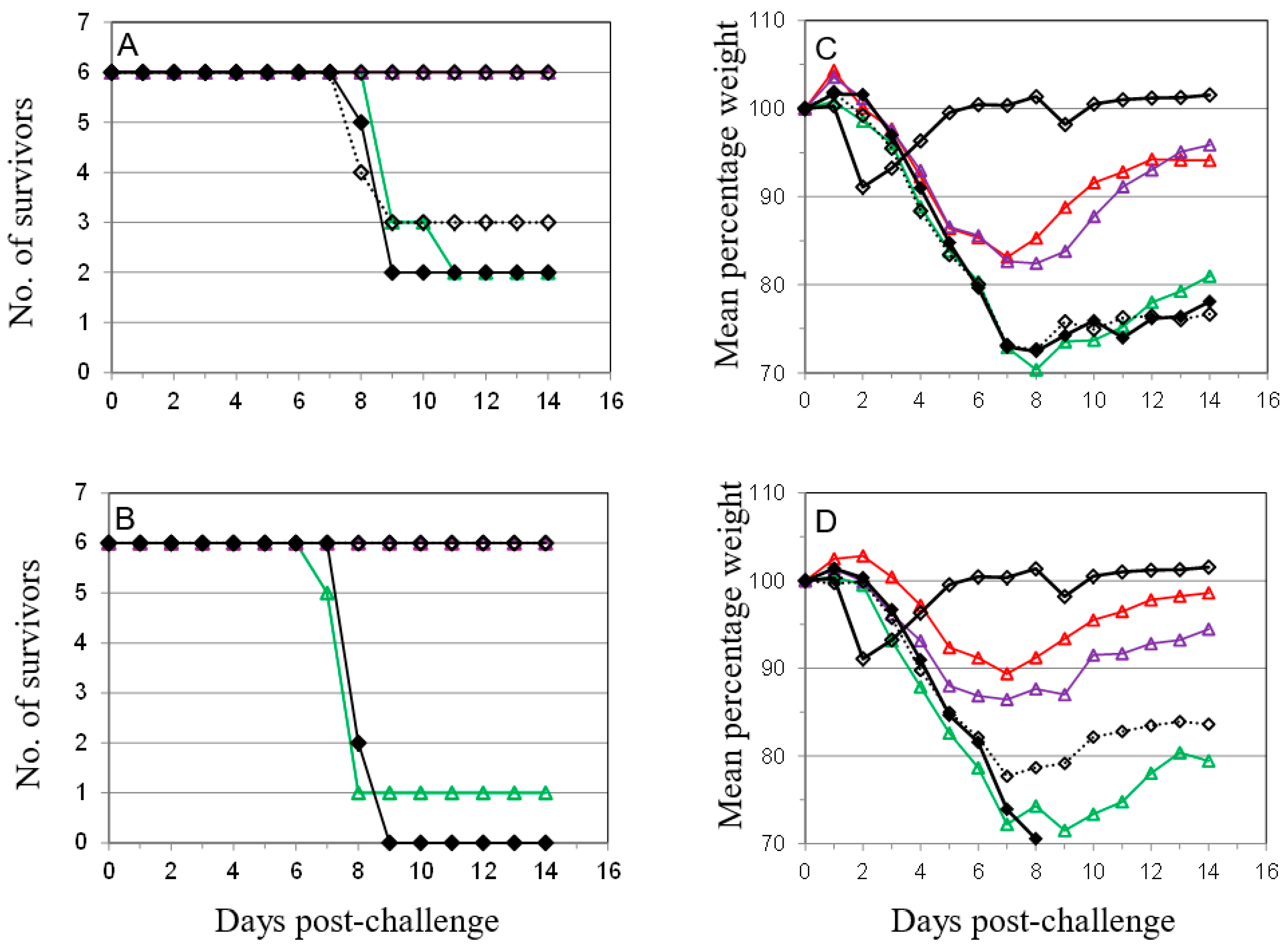
© Crown copyright (2017), Dstl. This material is licensed under the terms of the Open Government Licence except where otherwise stated. To view this licence, visit http://www.nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gsi.gov.uk.
Share and Cite
Reeman, S.; Gates, A.J.; Pulford, D.J.; Krieg, A.; Ulaeto, D.O. Protection of Mice from Lethal Vaccinia Virus Infection by Vaccinia Virus Protein Subunits with a CpG Adjuvant. Viruses 2017, 9, 378. https://doi.org/10.3390/v9120378
Reeman S, Gates AJ, Pulford DJ, Krieg A, Ulaeto DO. Protection of Mice from Lethal Vaccinia Virus Infection by Vaccinia Virus Protein Subunits with a CpG Adjuvant. Viruses. 2017; 9(12):378. https://doi.org/10.3390/v9120378
Chicago/Turabian StyleReeman, Sarah, Amanda J. Gates, David J. Pulford, Art Krieg, and David O. Ulaeto. 2017. "Protection of Mice from Lethal Vaccinia Virus Infection by Vaccinia Virus Protein Subunits with a CpG Adjuvant" Viruses 9, no. 12: 378. https://doi.org/10.3390/v9120378
APA StyleReeman, S., Gates, A. J., Pulford, D. J., Krieg, A., & Ulaeto, D. O. (2017). Protection of Mice from Lethal Vaccinia Virus Infection by Vaccinia Virus Protein Subunits with a CpG Adjuvant. Viruses, 9(12), 378. https://doi.org/10.3390/v9120378