Genomic Characterisation of Vinegar Hill Virus, An Australian Nairovirus Isolated in 1983 from Argas Robertsi Ticks Collected from Cattle Egrets

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus Culture and Genomic Sequencing
2.2. Bioinformatic Analysis
2.3. Phylogenetic Analysis
3. Results and Discussion
3.1. VINHV Genome and Terminal Sequences
3.2. L Protein
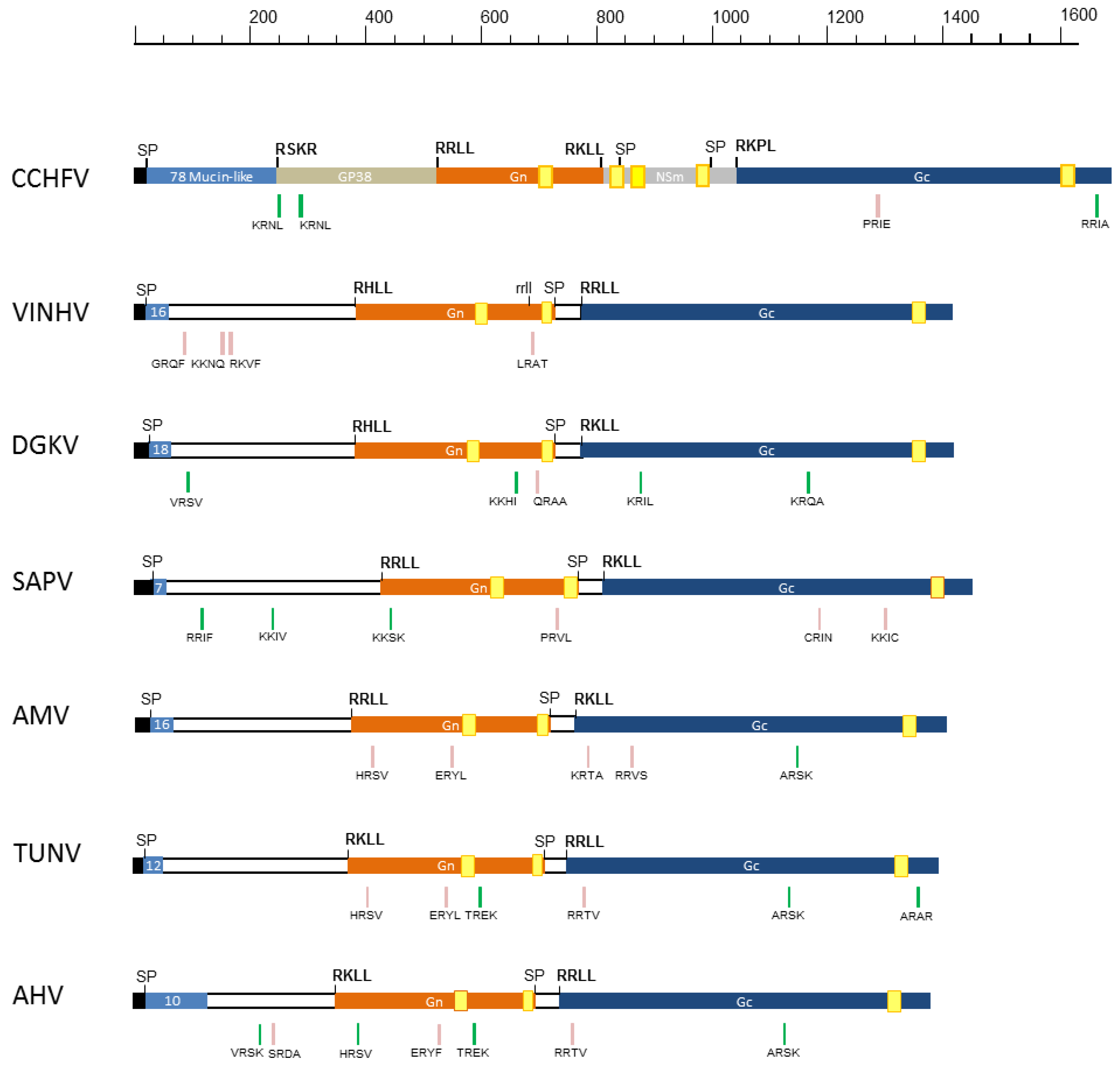
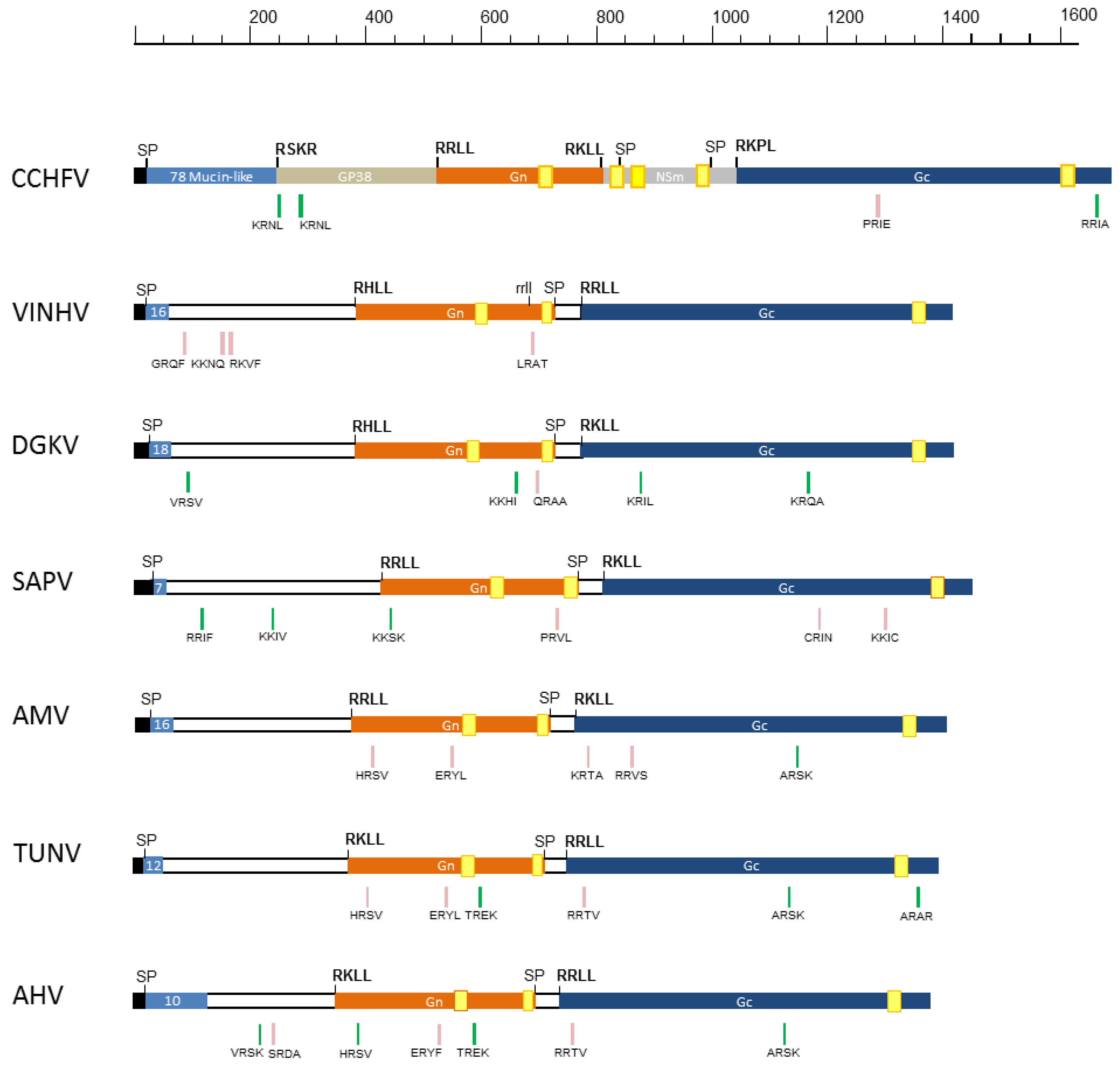
3.3. GPC
3.4. N Protein
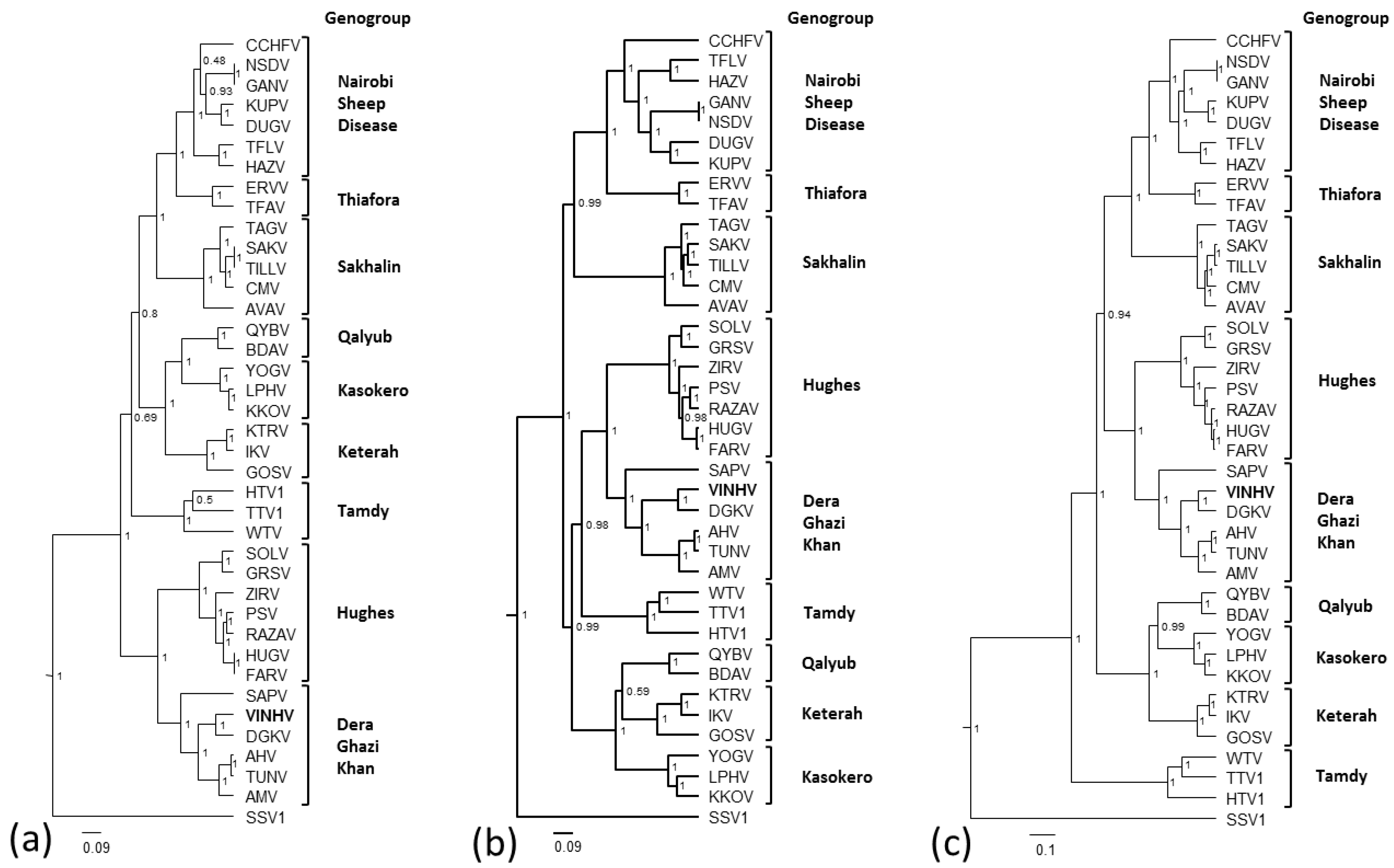
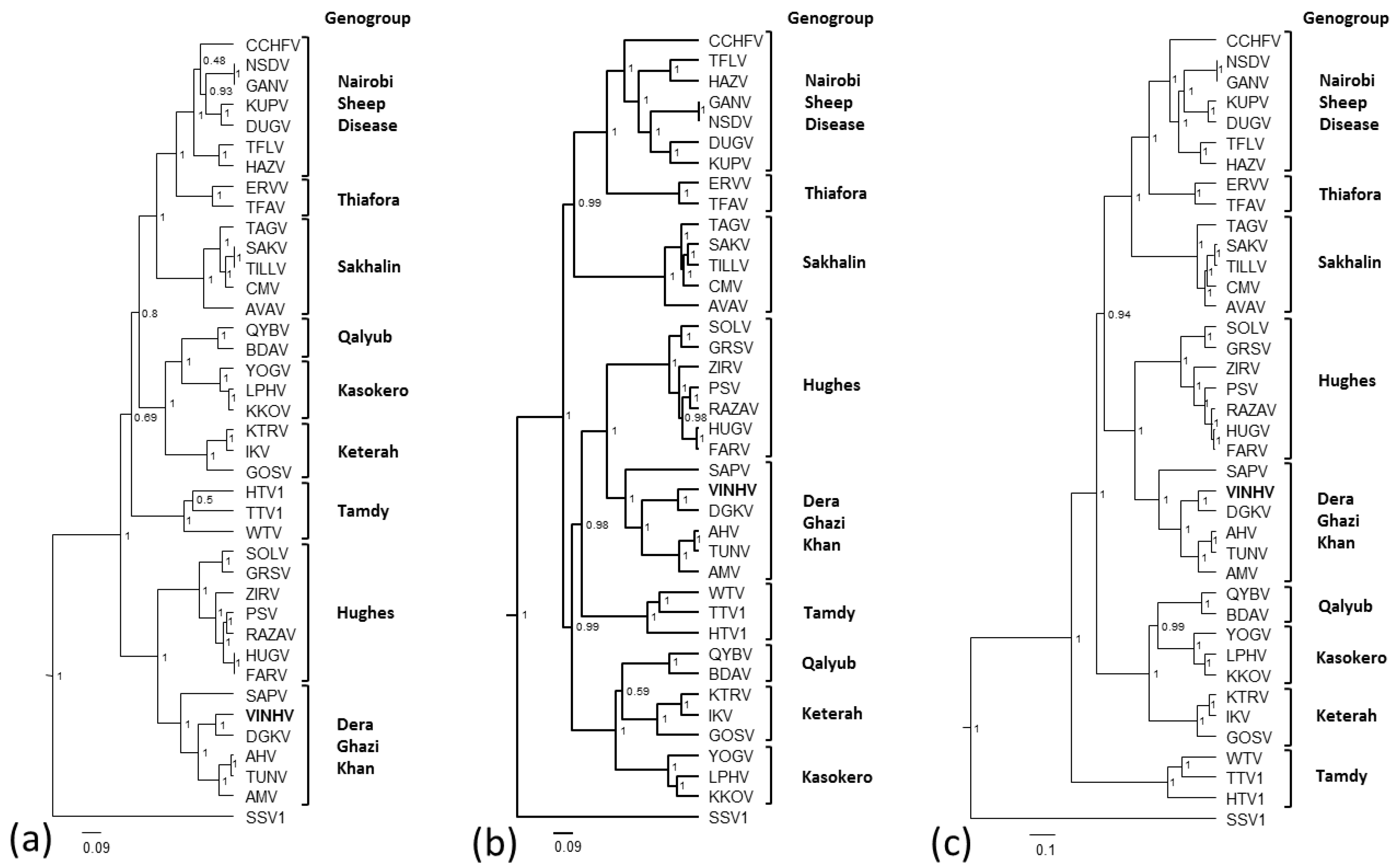
3.5. Phylogenetic Analysis
3.6. Ticks and Emerging Viruses in Australia
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Plyusnin, A.; Beaty, B.J.; Elliott, R.M.; Goldbach, R.; Kormelink, R.; Lundkvist, A.; Schmaljohn, C.S.; Tesh, R.B. Bunyaviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2012; pp. 725–741. [Google Scholar]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Wiley, M.R.; Rodriguez, S.E.; Yīmíng, B.; Prieto, K.; Travassos da Rosa, A.P.A.; Guzman, H.; Savji, N.; Ladner, J.T.; Tesh, R.B.; et al. Genomic Characterization of the Genus Nairovirus (Family Bunyaviridae). Viruses 2016, 8, 164. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Widen, S.G.; Firth, C.; Blasdell, K.R.; Wood, T.G.; Travassos Da Rosa, A.P.A.; Guzman, H.; Tesh, R.B.; Vasilakis, N. Genomic characterization of yogue, kasokero, issyk-kul, keterah, gossas, and thiafora viruses: Nairoviruses naturally infecting bats, shrews, and ticks. Am. J. Trop. Med. Hyg. 2015, 93, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Widen, S.G.; Wood, T.G.; Guzman, H.; Tesh, R.B.; Vasilakis, N. A Global genomic characterization of nairoviruses identifies nine discrete genogroups with distinctive structural characteristics and host-vector associations. Am. J. Trop. Med. Hyg. 2016, 94, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Doherty, R.L.; Carley, J.G.; Murray, M.D.; Main, A.J., Jr.; Kay, B.H.; Domrow, R. Isolation of arboviruses (Kemerovo group, Sakhalin group) from Ixodes uriae collected at Macquarie Island, Southern ocean. Am. J. Trop. Med. Hyg. 1975, 24, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Major, L.; La Linn, M.; Slade, R.W.; Schroder, W.A.; Hyatt, A.D.; Gardner, J.; Cowley, J.; Suhrbier, A. Ticks Associated with macquarie island penguins carry arboviruses from four genera. PLoS ONE 2009, 4, e4375. [Google Scholar] [CrossRef] [PubMed]
- Doherty, R.L.; Kay, B.H.; Carley, J.G.; Filippich, C. Isolation of virus strains related to kao shuan virus from argas robertsi in northern territory, Australia. Search 1976, 7, 484. [Google Scholar]
- St George, T.D. Research on Milk Fever, Bluetongue, Akabane, Ephemeral Fever and Other Arboviruses at CSIRO Long Pocket Laboratories, Brisbane, Australia. Ph.D. Thesis, University of Queensland, Brisbane, Australia, 2011. [Google Scholar]
- Humphery-Smith, I.; Cybinski, D.H.; Byrnes, K.A.; St George, T.D. Seroepidemiology of arboviruses among seabirds and island residents of the Great Barrier Reef and Coral Sea. Epidemiol. Infect. 1991, 107, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Gauci, P.J.; McAllister, J.; Mitchell, I.R.; Boyle, D.B.; Bulach, D.M.; Weir, R.P.; Melville, L.F.; Gubala, A.J. Genomic characterisation of three mapputta group viruses, a serogroup of Australian and papua new guinean bunyaviruses associated with human disease. PLoS ONE 2015, 10, e0116561. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Aquino, V.H.; Moreli, M.L.; Moraes Figueiredo, L.T. Analysis of oropouche virus L protein amino acid sequence showed the presence of an additional conserved region that could harbour an important role for the polymerase activity. Arch. Virol. 2003, 148, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.; Poch, O.; Delarue, M.; Bishop, D.H.L.; Bouloy, M. Rift valley fever virus L segment: Correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 1994, 75, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar] [PubMed]
- Reguera, J.; Weber, F.; Cusack, S. Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 2010, 6, e1001101. [Google Scholar] [CrossRef] [PubMed]
- Honig, J.E.; Osborne, J.C.; Nichol, S.T. Crimean–Congo hemorrhagic fever virus genome L RNA segment and encoded protein. Virology 2004, 321, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, R.; Williams, S.H.; Sameroff, S.; Sanchez Leon, M.; Jain, K.; Lipkin, W.I. Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J. Virol. 2014, 88, 11480–11492. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, S.; Holzer, B.; Bridgen, A.; McMullan, G.; Quinn, D.G.; Baron, M.D. Dugbe virus ovarian tumour domain interferes with ubiquitin/ISG15-regulated innate immune cell signalling. J. Gen. Virol. 2013, 94, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Clerx, J.P.M.; Casals, J.; Bishop, D.H.L. Structural characteristics of Nairoviruses (Genus Nairovirus, Bunyaviridae). J. Gen. Virol. 1981, 55, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Lasecka, L.; Baron, M.D. The molecular biology of nairoviruses, an emerging group of tick-borne arboviruses. Arch. Virol. 2014, 159, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.J.; Vincent, M.J.; Nichol, S.T. Characterization of the glycoproteins of Crimean-Congo hemorrhagic fever virus. J. Virol. 2002, 76, 7263–7275. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, É.; Vincent, M.J.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus glycoprotein processing by the endoprotease SKI-1/S1P is critical for virus infectivity. J. Virol. 2007, 81, 13271–13276. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.J.; Sanchez, A.J.; Erickson, B.R.; Basak, A.; Chretien, M.; Seidah, N.G.; Nichol, S.T. Crimean-congo hemorrhagic fever virus glycoprotein proteolytic processing by subtilase SKI-1. J. Virol. 2003, 77, 8640–8649. [Google Scholar] [CrossRef] [PubMed]
- Altamura, L.A.; Bertolotti-Ciarlet, A.; Teigler, J.; Paragas, J.; Schmaljohn, C.S.; Doms, R.W. Identification of a novel C-terminal cleavage of Crimean-Congo hemorrhagic fever virus PreG(N) that leads to generation of an NSM protein. J. Virol. 2007, 81, 6632–6642. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Y.; Duckers, H.J.; Sullivan, N.J.; Sanchez, A.; Nabel, E.G.; Nabel, G.J. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000, 6, 886. [Google Scholar] [PubMed]
- Sanchez, A.J.; Vincent, M.J.; Erickson, B.R.; Nichol, S.T. Crimean-congo hemorrhagic fever virus glycoprotein precursor is cleaved by Furin-like and SKI-1 proteases to generate a novel 38-kilodalton glycoprotein. J. Virol. 2006, 80, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.D.; Surtees, R.; Walter, C.T.; Ariza, A.; Bergeron, É.; Nichol, S.T.; Hiscox, J.A.; Edwards, T.A.; Barr, J.N. Structure, function, and evolution of the Crimean-Congo hemorrhagic fever virus nucleocapsid protein. J. Virol. 2012, 86, 10914–10923. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lou, Z.; Rao, Z.; Wang, W.; Ji, W.; Zhou, H.; Yang, C.; Deng, M.; Deng, F.; Wang, H.; et al. Crimean-Congo hemorrhagic fever virus nucleoprotein reveals endonuclease activity in bunyaviruses. Proc. Natl. Acad. Sci. USA 2012, 109, 5046–5051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dutta, S.; Karlberg, H.; Devignot, S.; Weber, F.; Hao, Q.; Tan, Y.J.; Mirazimi, A.; Kotaka, M. Structure of Crimean-Congo hemorrhagic fever virus nucleoprotein: Superhelical homo-oligomers and the role of caspase-3 cleavage. J. Virol. 2012, 86, 12294–12303. [Google Scholar] [CrossRef] [PubMed]
- Honig, J.E.; Osborne, J.C.; Nichol, S.T. The high genetic variation of viruses of the genus Nairovirus reflects the diversity of their predominant tick hosts. Virology 2004, 318, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Lowbridge, C.P.; Doggett, S.L.; Graves, S. Bug Breakfast in the Bulletin. Tickborne diseases. N. S. W. Public Health Bull. 2011, 22, 237. [Google Scholar] [CrossRef] [PubMed]
- Barker, S.C.; Walker, A.R.; Campelo, D. A list of the 70 species of Australian ticks; diagnostic guides to and species accounts of Ixodes holocyclus (paralysis tick), Ixodes cornuatus (southern paralysis tick) and Rhipicephalus australis (Australian cattle tick); and consideration of the place of Australia in the evolution of ticks with comments on four controversial ideas. Int. J. Parasitol. 2014, 44, 941–953. [Google Scholar] [PubMed]
- Barker, S.C. The Australian paralysis tick may be the missing link in the transmission of Hendra virus from bats to horses to humans. Med. Hypotheses 2003, 60, 481–483. [Google Scholar] [CrossRef]
- Gofton, A.W.; Oskam, C.L.; Paparini, A.; Greay, T.L.; Ryan, U.; Irwin, P.; Lo, N.; Wei, H.; McCarl, V.; Beninati, T.; et al. Inhibition of the endosymbiont “Candidatus Midichloria mitochondrii” during 16S rRNA gene profiling reveals potential pathogens in Ixodes ticks from Australia. Parasites Vectors 2015, 8, 345. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced arbovirus surveillance with deep sequencing: Identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef] [PubMed]
- St. George, T.D.; Standeast, H.A.; Doherty, R.L.; Carley, J.G.; Fillipich, C.; Brandsma, J. The isolation of saumarez reef virus, a new flavivirus, from bird ticks ornithodoros capensis and ixodes eudyptidis in Australia. Aust. J. Exp. Biol. Med. 1977, 55, 493. [Google Scholar] [CrossRef]
- Gauci, P.J.; McAllister, J.; Mitchell, I.R.; George, T.D.S.; Cybinski, D.H.; Davis, S.S.; Gubala, A.J.; St George, T.D. Hunter island group phlebovirus in ticks, Australia. Emerg. Infect. Dis. 2015, 21, 2246–2248. [Google Scholar] [CrossRef] [PubMed]
- Bixing, H.; Firth, C.; Watterson, D.; Allcock, R.; Colmant, A.M.G.; Hobson-Peters, J.; Kirkland, P.; Hewitson, G.; McMahon, J.; Hall-Mendelin, S.; et al. Genetic characterization of archived bunyaviruses and their potential for emergence in Australia. Emerg. Infect. Dis. 2016, 22, 833–840. [Google Scholar]
- Huang, B.; Allcock, R.; Warrilow, D. Newly characterized arboviruses of northern Australia. Virol. Rep. 2016, 6, 11–17. [Google Scholar] [CrossRef]
- McAllister, J.; Gauci, P.J.; Mitchell, I.R.; Gubala, A.J.; Boyle, D.B.; Bulach, D.M.; Weir, R.P.; Melville, L.F.; Davis, S.S. Genomic characterisation of Almpiwar virus, Harrison Dam virus and Walkabout Creek virus; three novel rhabdoviruses from northern Australia. Virol. Rep. 2014, 3, 1–17. [Google Scholar] [CrossRef]
- Estrada-Peña, A.; de la Fuente, J. Review: The ecology of ticks and epidemiology of tick-borne viral diseases. Antivir. Res. 2014, 108, 104–128. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Román, R.; Díaz-Martín, V.; de la Fuente, J.; Pérez-Sánchez, R. Soft Ticks as Pathogen Vectors: Distribution, Surveillance and Control, Parasitology; Shah, M.M., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment | Region | Length (nt/aa) for Indicated Viruses | ||||||
|---|---|---|---|---|---|---|---|---|
| VINHV | DGKV | AMV | AHV | TUNV | SAPV | a CCHFV | ||
| L | 5′UTR | 53 | 57 | 41 | 35 | 35 | 96 | 76 |
| L ORF | 11,847/3948 | 11,847/3948 | 11,925/3974 | 11,868/3955 | 11,868/3955 | 11,871/3956 | 11,837/3945 | |
| 3′UTR | 233 | 106 | 249 | 214 | 400 | 218 | 194 | |
| segment total | 12,133 | 12,010 | 12,215 | 12,117 | 12,303 | 12,185 | 12,108 | |
| M | 5′UTR | 41 b | 42 | 14 | 30 | 30 | 14 | 92 |
| GPC ORF | 4245/1414 | 4239/1412 | 4203/1400 | 4200/1399 | 4167/1388 | 4341/1446 | 5055/1684 | |
| 3′UTR | 187 | 159 | 224 | 472 | 609 | 434 | 219 | |
| segment total | 4473 | 4440 | 4441 | 4702 | 4806 | 4789 | 5366 | |
| S | 5′UTR | 58 | 50 | 51 | 54 | 56 | 54 | 55 |
| NP ORF | 1500/499 | 1500/499 | 1497/498 | 1497/498 | 1497/498 | 1476/491 | 1448/482 | |
| 3′UTR | 171 | 221 | 212 | 226 | 315 | 137 | 168 | |
| segment total | 1729 | 1771 | 1760 | 1777 | 1868 | 1668 | 1672 | |
| Virus | VINHV | DGKV | AHV | TUNV | AMV |
|---|---|---|---|---|---|
| VINHV | |||||
| DGKV | 72.0 | ||||
| AHV | 62.1 | 59.3 | |||
| TUNV | 62.1 | 59.9 | 95.4 | ||
| AMV | 60.6 | 63.4 | 76.0 | 75.8 | |
| SAPV | 54.4 | 52.9 | 52.4 | 52.4 | 52.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gauci, P.J.; McAllister, J.; Mitchell, I.R.; Cybinski, D.; St George, T.; Gubala, A.J. Genomic Characterisation of Vinegar Hill Virus, An Australian Nairovirus Isolated in 1983 from Argas Robertsi Ticks Collected from Cattle Egrets. Viruses 2017, 9, 373. https://doi.org/10.3390/v9120373
Gauci PJ, McAllister J, Mitchell IR, Cybinski D, St George T, Gubala AJ. Genomic Characterisation of Vinegar Hill Virus, An Australian Nairovirus Isolated in 1983 from Argas Robertsi Ticks Collected from Cattle Egrets. Viruses. 2017; 9(12):373. https://doi.org/10.3390/v9120373
Chicago/Turabian StyleGauci, Penelope J., Jane McAllister, Ian R. Mitchell, Daisy Cybinski, Toby St George, and Aneta J. Gubala. 2017. "Genomic Characterisation of Vinegar Hill Virus, An Australian Nairovirus Isolated in 1983 from Argas Robertsi Ticks Collected from Cattle Egrets" Viruses 9, no. 12: 373. https://doi.org/10.3390/v9120373
APA StyleGauci, P. J., McAllister, J., Mitchell, I. R., Cybinski, D., St George, T., & Gubala, A. J. (2017). Genomic Characterisation of Vinegar Hill Virus, An Australian Nairovirus Isolated in 1983 from Argas Robertsi Ticks Collected from Cattle Egrets. Viruses, 9(12), 373. https://doi.org/10.3390/v9120373