Herpesvirus and Autophagy: “All Right, Everybody Be Cool, This Is a Robbery!”

Abstract
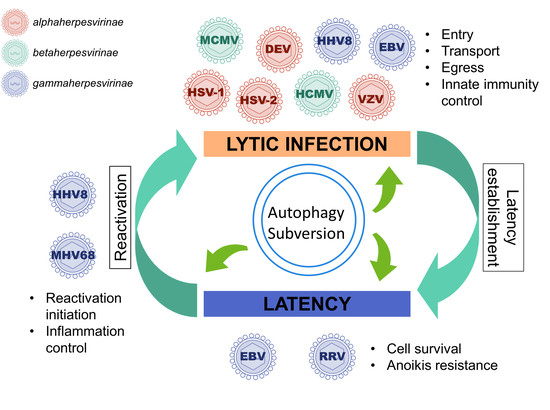
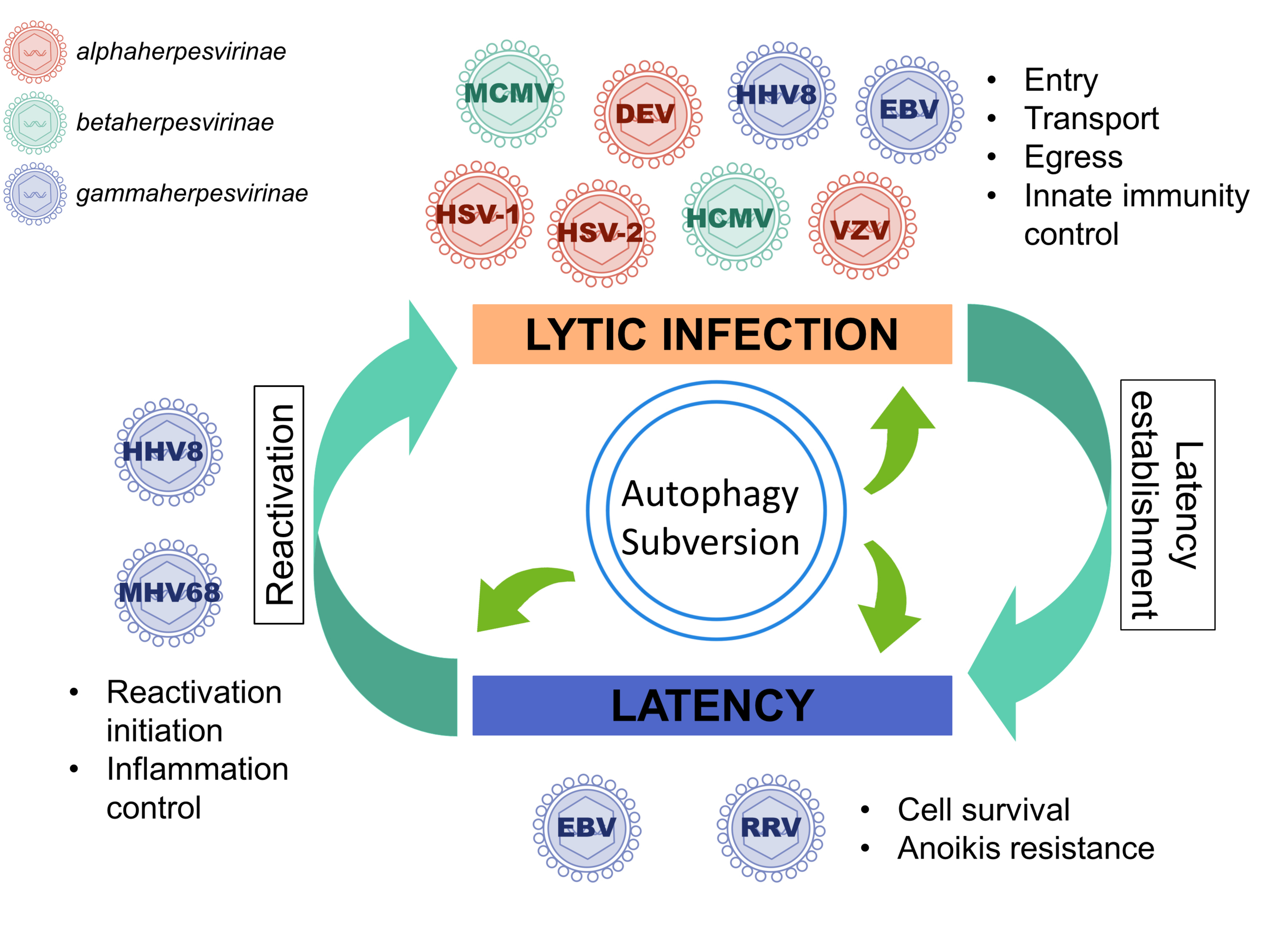
1. A Brief Introduction to Herpesviruses
2. Autophagy
2.1. Background
2.2. Physiological Cellular Roles, Defense Mechanism, Innate and Adaptive Immunity
2.3. Herpes Viral Escape
3. Autophagy Subversion by Herpesviruses
3.1. Autophagy Promotes Viral Production of Several Herpesviruses
3.2. During Lytic Cycle, EBV, and HHV8 Inhibit the Autophagic Flux and Exploit the Autophagic Vacuoles for Their Transport or Their Egress
3.3. Stimulation of Functional Autophagy and Lytic Cycle
3.4. Autophagy Can Be Enhanced during Latency to Favor Cell Survival and It Can Control Excessive Inflammation during Reactivation
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1802–1822. [Google Scholar]
- Dupont, L.; Reeves, M.B. Cytomegalovirus latency and reactivation: Recent insights into an age old problem. Rev. Med. Virol. 2016, 26, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.; Sinclair, J. Aspects of human cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 2008, 325, 297–313. [Google Scholar] [PubMed]
- Chen, H.S.; Lu, F.; Lieberman, P.M. Epigenetic regulation of EBV and KSHV latency. Curr. Opin. Virol. 2013, 3, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [PubMed]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R.; Mocarski, E.S. Viral and host control of cytomegalovirus maturation. Trends Microbiol. 2012, 20, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, E.; di Gaeta, S.; Torrisi, M.R.; Ward, P.L.; Roizman, B.; Campadelli-Fiume, G. Redistribution of microtubules and Golgi apparatus in herpes simplex virus-infected cells and their role in viral exocytosis. J. Virol. 1995, 69, 7472–7482. [Google Scholar] [PubMed]
- Campadelli, G.; Brandimarti, R.; di Lazzaro, C.; Ward, P.L.; Roizman, B.; Torrisi, M.R. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 1993, 90, 2798–2802. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Pellett, P.E. Spatial relationships between markers for secretory and endosomal machinery in human cytomegalovirus-infected cells versus those in uninfected cells. J. Virol. 2011, 85, 5864–5879. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Nowag, H.; Guhl, B.; Thriene, K.; Romao, S.; Ziegler, U.; Dengjel, J.; Munz, C. Macroautophagy proteins assist Epstein Barr virus production and get incorporated into the virus particles. EBioMedicine 2014, 1, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Jarosinski, K.W.; Jackson, W.; Carpenter, J.E.; Grose, C. Exocytosis of varicella-zoster virions involves a convergence of endosomal and autophagy pathways. J. Virol. 2016, 90, 8673–8685. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy revisited: A conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Van Noorden, R.; Ledford, H. Medicine Nobel for research on how cells ‘eat themselves’. Nature 2016, 538, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta 2013, 1833, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Molino, D.; Zemirli, N.; Codogno, P.; Morel, E. The journey of the autophagosome through mammalian cell organelles and membranes. J. Mol. Biol. 2017, 429, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Giordano, F.; Dupont, N.; Grasso, D.; Vaccaro, M.I.; Codogno, P.; Morel, E. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 2017, 36, 2018–2033. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Akioka, M.; Kondo-Kakuta, C.; Yamamoto, H.; Ohsumi, Y. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J. Cell Sci. 2013, 126, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Tavernarakis, N.; Kroemer, G. Can autophagy promote longevity? Nat. Cell Biol. 2010, 12, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Al Rawi, S.; Louvet-Vallee, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Allophagy: A macroautophagic process degrading spermatozoid-inherited organelles. Autophagy 2012, 8, 421–423. [Google Scholar] [CrossRef] [PubMed]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Lorin, S.; Hamai, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Zou, Z.; Sun, Q.; Luby-Phelps, K.; Cheng, P.; Hogan, R.N.; Gilpin, C.; Levine, B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007, 128, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Pareja, M.E.; Colombo, M.I. Autophagic clearance of bacterial pathogens: Molecular recognition of intracellular microorganisms. Front. Cell. Infect. Microbiol. 2013, 3, 54. [Google Scholar] [PubMed]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Judith, D.; Mostowy, S.; Bourai, M.; Gangneux, N.; Lelek, M.; Lucas-Hourani, M.; Cayet, N.; Jacob, Y.; Prevost, M.C.; Pierre, P.; et al. Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 2013, 14, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Munz, C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; English, L.; Rondeau, C.; Leib, D.; Lippe, R.; Desjardins, M. Inhibition of the host translation shutoff response by herpes simplex virus 1 triggers nuclear envelope-derived autophagy. J. Virol. 2013, 87, 3990–3997. [Google Scholar] [CrossRef] [PubMed]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Esclatine, A. Chapter 8: Modulation of autophagy by Herpesvirus proteins. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Hayat, M.A., Ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2015; pp. 145–158. [Google Scholar]
- Talloczy, Z.; Jiang, W.; Virgin, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Alexander, D.; Talloczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, D.R.; Wadhwani, N.R.; Longnecker, R.; Muller, W.J. Differential reliance on autophagy for protection from HSV encephalitis between newborns and adults. PLoS Pathog. 2015, 11, e1004580. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Seo, G.J.; Choi, Y.J.; Kwak, M.J.; Ge, J.; Rodgers, M.A.; Shi, M.; Leslie, B.J.; Hopfner, K.P.; Ha, T.; et al. Crosstalk between the cGAS DNA sensor and BECLIN-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe 2014, 15, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.B.; Horan, K.A.; Holm, C.K.; Stranks, A.J.; Mettenleiter, T.C.; Simon, A.K.; Jensen, S.B.; Rixon, F.J.; He, B.; Paludan, S.R. Activation of autophagy by alpha-herpesviruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on stimulator of IFN genes. J. Immunol. 2011, 187, 5268–5276. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.E.; Ward, S.L.; Mizushima, N.; Levine, B.; Leib, D.A. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 2007, 81, 12128–12134. [Google Scholar] [CrossRef] [PubMed]
- Yordy, B.; Iijima, N.; Huttner, A.; Leib, D.; Iwasaki, A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 2012, 12, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, P.A.; Leib, D.A. Herpes simplex virus gamma34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Lussignol, M.; Queval, C.; Bernet-Camard, M.F.; Cotte-Laffitte, J.; Beau, I.; Codogno, P.; Esclatine, A. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 2013, 87, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Mouna, L.; Hernandez, E.; Bonte, D.; Brost, R.; Amazit, L.; Delgui, L.R.; Brune, W.; Geballe, A.P.; Beau, I.; Esclatine, A. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy 2016, 12, 327–342. [Google Scholar] [CrossRef] [PubMed]
- Chaumorcel, M.; Lussignol, M.; Mouna, L.; Cavignac, Y.; Fahie, K.; Cotte-Laffitte, J.; Geballe, A.; Brune, W.; Beau, I.; Codogno, P.; et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 2012, 86, 2571–2584. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chang, B.; Brulois, K.F.; Castro, K.; Min, C.K.; Rodgers, M.A.; Shi, M.; Ge, J.; Feng, P.; Oh, B.H.; et al. Kaposi’s sarcoma-associated herpesvirus K7 modulates Rubicon-mediated inhibition of autophagosome maturation. J. Virol. 2013, 87, 12499–12503. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; Granato, M.; Pentassuglia, G.; Lacconi, V.; Gilardini Montani, M.S.; Gonnella, R.; Tafani, M.; Torrisi, M.R.; Faggioni, A.; Cirone, M. KSHV reduces autophagy in THP-1 cells and in differentiating monocytes by decreasing CAST/calpastatin and ATG5 expression. Autophagy 2016, 12, 2311–2325. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Lucania, G.; Bergamo, P.; Trivedi, P.; Frati, L.; Faggioni, A. Human herpesvirus 8 (HHV-8) inhibits monocyte differentiation into dendritic cells and impairs their immunostimulatory activity. Immunol. Lett. 2007, 113, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef] [PubMed]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Hou, L.; Tang, Y.D.; Liu, Y.; Wang, S.; Wang, J.; Shen, N.; An, T.; Tian, Z.; Cai, X. Pseudorabies virus infection inhibits autophagy in permissive cells in vitro. Sci. Rep. 2017, 7, 39964. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Moreau, K.; Segarra, A.; Tourbiez, D.; Travers, M.A.; Rubinsztein, D.C.; Renault, T. Autophagy plays an important role in protecting Pacific oysters from OsHV-1 and Vibrio aestuarianus infections. Autophagy 2015, 11, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Segarra, A.; Pepin, J.F.; Arzul, I.; Morga, B.; Faury, N.; Renault, T. Detection and description of a particular Ostreid herpesvirus 1 genotype associated with massive mortality outbreaks of Pacific oysters, Crassostrea gigas, in France in 2008. Virus Res. 2010, 153, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Talloczy, Z.; Virgin, H.W.; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Chaumorcel, M.; Souquere, S.; Pierron, G.; Codogno, P.; Esclatine, A. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 2008, 4, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Braggin, J.E.; Child, S.J.; Geballe, A.P. Essential role of protein kinase R antagonism by TRS1 in human cytomegalovirus replication. Virology 2016, 489, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Lampe, M.; Gunther, T.; Brune, W. The viral Bcl-2 homologs of Kaposi’s sarcoma-associated herpesvirus and Rhesus Rhadinovirus share an essential role for viral replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chang, B.; Lee, P.; Brulois, K.F.; Ge, J.; Shi, M.; Rodgers, M.A.; Feng, P.; Oh, B.H.; Liang, C.; et al. Identification of the essential role of viral Bcl-2 for Kaposi’s sarcoma-associated herpesvirus lytic replication. J. Virol. 2015, 89, 5308–5317. [Google Scholar] [CrossRef] [PubMed]
- Pujals, A.; Favre, L.; Pioche-Durieu, C.; Robert, A.; Meurice, G.; Le Gentil, M.; Chelouah, S.; Martin-Garcia, N.; Le Cam, E.; Guettier, C.; et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy 2015, 11, 2275–2287. [Google Scholar] [CrossRef] [PubMed]
- Siracusano, G.; Venuti, A.; Lombardo, D.; Mastino, A.; Esclatine, A.; Sciortino, M.T. Early activation of MyD88-mediated autophagy sustains HSV-1 replication in human monocytic THP-1 cells. Sci. Rep. 2016, 6, 31302. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Li, M.; Wang, K.; Wang, S.; Lu, Q.; Yan, J.; Mossman, K.L.; Lin, R.; Zheng, C. The herpes simplex virus 1-encoded envelope glycoprotein B activates NF-κB through the Toll-like receptor 2 and MyD88/TRAF6-dependent signaling pathway. PLoS ONE 2013, 8, e54586. [Google Scholar] [CrossRef] [PubMed]
- Petrovski, G.; Pasztor, K.; Orosz, L.; Albert, R.; Mencel, E.; Moe, M.C.; Kaarniranta, K.; Facsko, A.; Megyeri, K. Herpes simplex virus types 1 and 2 modulate autophagy in SIRC corneal cells. J. Biosci. 2014, 39, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Yakoub, A.M.; Shukla, D. Basal autophagy is required for herpes simplex virus-2 infection. Sci. Rep. 2015, 5, 12985. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.N.; Jackson, W.; Laird, D.T.; Culp, T.D.; Grose, C.; Haynes, J.I., 2nd; Benetti, L. Varicella-zoster virus infection induces autophagy in both cultured cells and human skin vesicles. J. Virol. 2009, 83, 5466–5476. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.C.; Zhao, L.L.; Li, S.Q.; Niu, Y.J.; Jiang, X.J.; Xu, L.J.; Lu, T.F.; Han, L.X.; Liu, S.W.; Chen, H.Y. Autophagy activated by duck enteritis virus infection positively affects its replication. J. Gen. Virol. 2017, 98, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Dhama, K.; Kumar, N.; Saminathan, M.; Tiwari, R.; Karthik, K.; Kumar, M.A.; Palanivelu, M.; Shabbir, M.Z.; Malik, Y.S.; Singh, R.K. Duck virus enteritis (duck plague)—A comprehensive update. Vet. Q. 2017, 37, 57–80. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.; Aitken, J.; Sutherland, J.S.; Nicholl, M.J.; Preston, V.G.; Preston, C.M. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J. Virol. 2011, 85, 4212–4221. [Google Scholar] [CrossRef] [PubMed]
- Belzile, J.P.; Sabalza, M.; Craig, M.; Clark, E.; Morello, C.S.; Spector, D.H. Trehalose, an mTOR-independent inducer of autophagy, inhibits human cytomegalovirus infection in multiple cell types. J. Virol. 2015, 90, 1259–1277. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Morishita, H.; Hama, Y.; Tsukamoto, S.; Matsui, T.; Toyota, Y.; Kodama, A.; Ishihara, T.; Mizushima, T.; Mizushima, N. An autophagic flux probe that releases an internal control. Mol. Cell 2016, 64, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Chen, L.W.; Wang, W.H.; Chang, P.J.; Chiu, Y.F.; Hung, C.C.; Lin, Y.J.; Liou, J.Y.; Tsai, W.J.; Hung, C.L.; et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J. Virol. 2014, 88, 12133–12145. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.; Colavita, F.; Ciccosanti, F.; Fimia, G.M.; Lieberman, P.M.; Mattia, E. Inhibition of autophagy in EBV-positive Burkitt’s lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. 2015, 6, e1876. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Santarelli, R.; Filardi, M.; Gonnella, R.; Farina, A.; Torrisi, M.R.; Faggioni, A.; Cirone, M. The activation of KSHV lytic cycle blocks autophagy in PEL cells. Autophagy 2015, 11, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.J.; Yang, Z.; Zhou, Y.; Wood, C. Enhancement of autophagy during lytic replication by the Kaposi’s sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 2010, 84, 7448–7458. [Google Scholar] [CrossRef] [PubMed]
- Gannage, M.; Dormann, D.; Albrecht, R.; Dengjel, J.; Torossi, T.; Ramer, P.C.; Lee, M.; Strowig, T.; Arrey, F.; Conenello, G.; et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009, 6, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Beale, R.; Wise, H.; Stuart, A.; Ravenhill, B.J.; Digard, P.; Randow, F. A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 2014, 15, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Faure, M. The p value of HPIV3-mediated autophagy inhibition. Cell Host Microbe 2014, 15, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.; Yamada, M.; Moninger, T.; Grose, C. Visualization and quantitation of abundant macroautophagy in virus-infected cells by confocal three-dimensional fluorescence imaging. J. Virol. Methods 2013, 193, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Zerboni, L.; Arvin, A.M.; Grose, C. Autophagic flux without a block differentiates varicella-zoster virus infection from herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.E.; Jackson, W.; Benetti, L.; Grose, C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J. Virol. 2011, 85, 9414–9424. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Grose, C. Autophagy and the effects of its inhibition on varicella-zoster virus glycoprotein biosynthesis and infectivity. J. Virol. 2014, 88, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed]
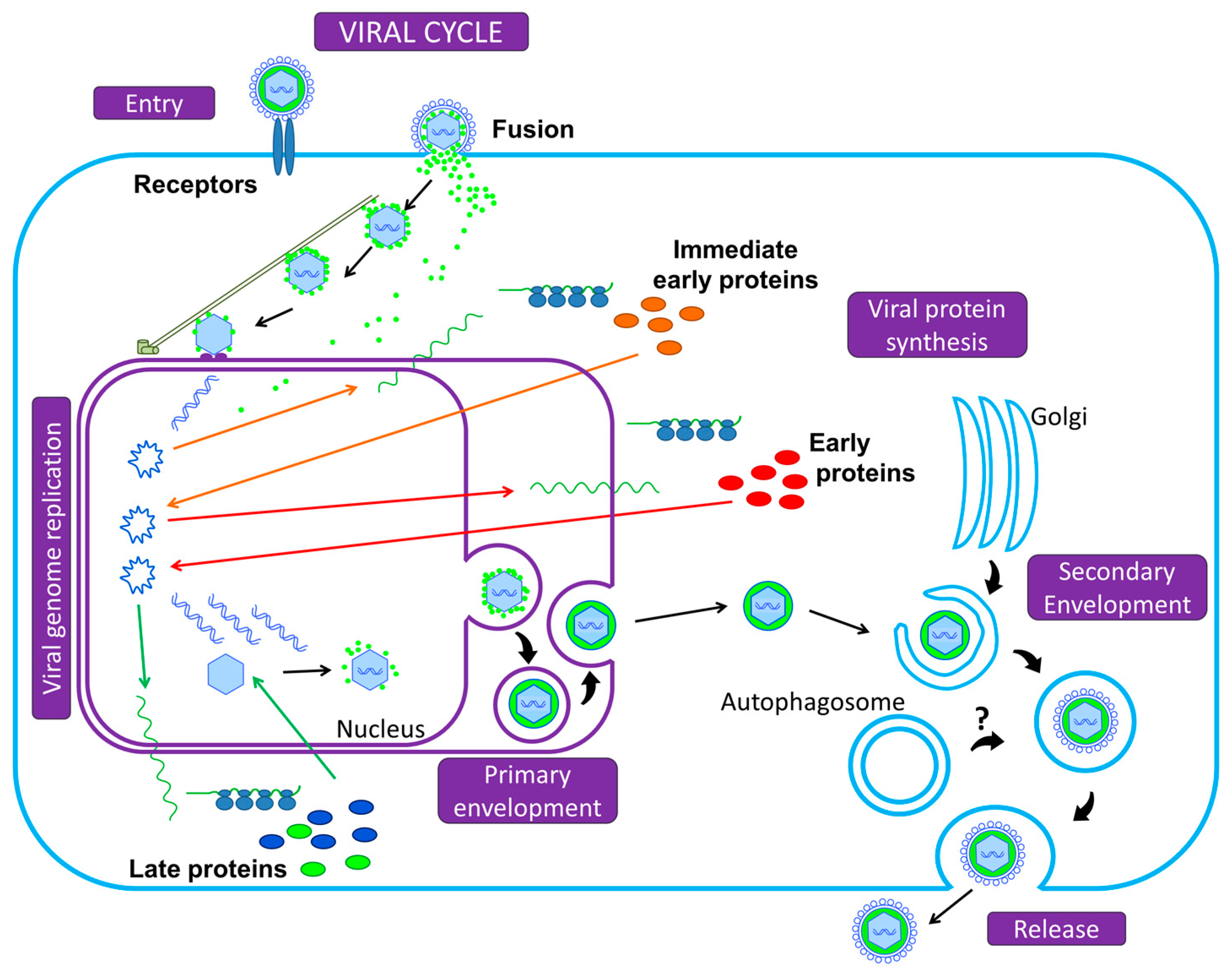
- Fliss, P.M.; Jowers, T.P.; Brinkmann, M.M.; Holstermann, B.; Mack, C.; Dickinson, P.; Hohenberg, H.; Ghazal, P.; Brune, W. Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 2012, 8, e1002517. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.; Sickmann, A.; Lembo, D.; Brune, W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc. Natil. Acad. Sci. USA 2008, 105, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Zhang, M.; Marshall, B.; Smith, S.; Covar, J.; Atherton, S. Interplay of autophagy and apoptosis during murine cytomegalovirus infection of RPE cells. Mol. Vis. 2014, 20, 1161–1173. [Google Scholar] [PubMed]
- Austin, P.J.; Flemington, E.; Yandava, C.N.; Strominger, J.L.; Speck, S.H. Complex transcription of the Epstein-Barr virus BamHI fragment H rightward open reading frame 1 (BHRF1) in latently and lytically infected B lymphocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 3678–3682. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Fotheringham, J.A.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2 induces autophagy to promote abnormal acinus formation. J. Virol. 2015, 89, 6940–6944. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Minamitani, T.; Samanta, M. Epstein-Barr virus latent membrane protein 2A contributes to anoikis resistance through ERK activation. J. Virol. 2013, 87, 8227–8234. [Google Scholar] [CrossRef] [PubMed]
- Ritthipichai, K.; Nan, Y.; Bossis, I.; Zhang, Y. Viral FLICE inhibitory protein of rhesus monkey rhadinovirus inhibits apoptosis by enhancing autophagosome formation. PLoS ONE 2012, 7, e39438. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Cyr, D.P.; Hill, R.J.; Lee, P.W.; McCormick, C. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012, 11, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Buck, M.D.; Desai, C.; Zhang, X.; Loginicheva, E.; Martinez, J.; Freeman, M.L.; Saitoh, T.; Akira, S.; Guan, J.L.; et al. Autophagy genes enhance Murine Gammaherpesvirus 68 reactivation from latency by preventing virus-induced systemic inflammation. Cell Host Microbe 2016, 19, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Steed, A.L.; Barton, E.S.; Tibbetts, S.A.; Popkin, D.L.; Lutzke, M.L.; Rochford, R.; Virgin, H.W., 4th. Gamma interferon blocks gammaherpesvirus reactivation from latency. J. Virol. 2006, 80, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Colbert, C.L.; Becker, N.; Wei, Y.; Levine, B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the γ-herpesvirus 68 Bcl-2 homolog M11. Autophagy 2008, 4, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Mei, Y.; Sanishvili, R.; Levine, B.; Colbert, C.L.; Sinha, S. Targeting γ-herpesvirus 68 Bcl-2-mediated down-regulation of autophagy. J. Biol. Chem. 2014, 289, 8029–8040. [Google Scholar] [CrossRef] [PubMed]
- E, X.; Hwang, S.; Oh, S.; Lee, J.S.; Jeong, J.H.; Gwack, Y.; Kowalik, T.F.; Sun, R.; Jung, J.U.; Liang, C. Viral Bcl-2-mediated evasion of autophagy aids chronic infection of γherpesvirus 68. PLoS Pathog. 2009, 5, e1000609. [Google Scholar] [CrossRef] [PubMed]
- Katzenell, S.; Leib, D.A. Herpes Simplex Virus and Interferon Signaling Induce Novel Autophagic Clusters in Sensory Neurons. J. Virol. 2016, 90, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed]
- Sin, S.H.; Roy, D.; Wang, L.; Staudt, M.R.; Fakhari, F.D.; Patel, D.D.; Henry, D.; Harrington, W.J., Jr.; Damania, B.A.; Dittmer, D.P. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 2007, 109, 2165–2173. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Name | Abbreviation | Host | Subversion of Autophagy | Ref. |
|---|---|---|---|---|---|
| Alphaherpesvirinae | Herpes simplex virus type 1 | HSV-1 | Human | Transient activation of autophagy in THP-1 cells via MyD88 adaptor protein, beneficial for viral entry | [67] |
| Herpes simplex virus type 2 | HSV-2 | Human | Basal autophagy promotes viral replication in fibroblasts | [70] | |
| Varicella-Zoster virus | VZV | Human | VZV stimulates complete autophagy in several cell types and that is necessary for efficient viral glycoprotein processing. Hijack of amphisomes for viral egress. | [71,86,87,90] [15] | |
| Duck enteritis virus | DEV | Waterfowl | Autophagy is stimulated at late time of infection to optimize viral production. | [72] | |
| Betaherpesvirinae | Human Cytomegalovirus | HCMV | Human | Infection stimulates autophagy and subsequently blocks autophagosome degradation. Autophagy proteins or membranes participate in viral propagation. | [49,50,74] |
| Murine Cytomegalovirus | MCMV | Mouse | The viral protein M45 targets NEMO to autophagosomes for selective degradation and therefore participates to the inhibition of innate immunity | [91] | |
| Gammaherpesvirinae | Epstein-Barr virus | EBV | Human | During lytic cycle: autophagic flux is blocked and autophagic vacuoles are hijacked by the virus for envelopment/egress During latency: autophagy stimulation by LMP1 and LMP2A favors cell survival | [14,78] [66,95,96] |
| Kaposi’s sarcoma-associated Herpesvirus | KSHV or HHV8 | Human | Evidence of viral particle transport in autophagosomes and positive role of autophagy during viral reactivation During latency: Autophagy inhibition blocks oncogene-induced senescence | [80,81] [99] | |
| Rhesus monkey Rhadinovirus | RRV | Rhesus monkey | During latency: vFLIP-induced autophagy protects cells from apoptosis | [98] | |
| Murid Herpesvirus 68 | MHV68 | Mouse and small rodents | Autophagy participates in the control of inflammation, allowing virus reactivation from latency | [100] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lussignol, M.; Esclatine, A. Herpesvirus and Autophagy: “All Right, Everybody Be Cool, This Is a Robbery!”. Viruses 2017, 9, 372. https://doi.org/10.3390/v9120372
Lussignol M, Esclatine A. Herpesvirus and Autophagy: “All Right, Everybody Be Cool, This Is a Robbery!”. Viruses. 2017; 9(12):372. https://doi.org/10.3390/v9120372
Chicago/Turabian StyleLussignol, Marion, and Audrey Esclatine. 2017. "Herpesvirus and Autophagy: “All Right, Everybody Be Cool, This Is a Robbery!”" Viruses 9, no. 12: 372. https://doi.org/10.3390/v9120372
APA StyleLussignol, M., & Esclatine, A. (2017). Herpesvirus and Autophagy: “All Right, Everybody Be Cool, This Is a Robbery!”. Viruses, 9(12), 372. https://doi.org/10.3390/v9120372