EBV and Apoptosis: The Viral Master Regulator of Cell Fate?



Abstract
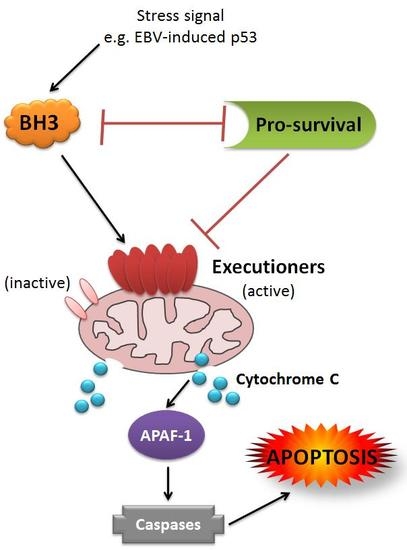
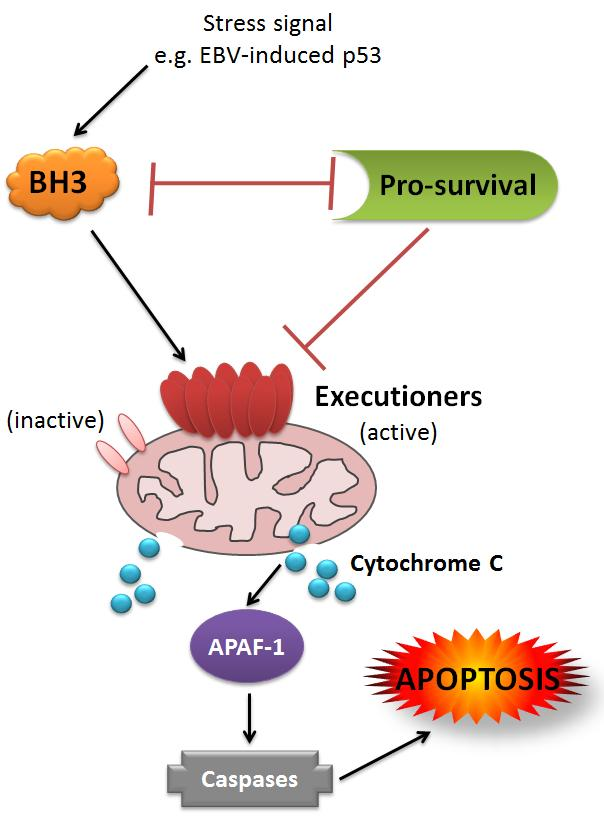{kind=link}
{kind=link}
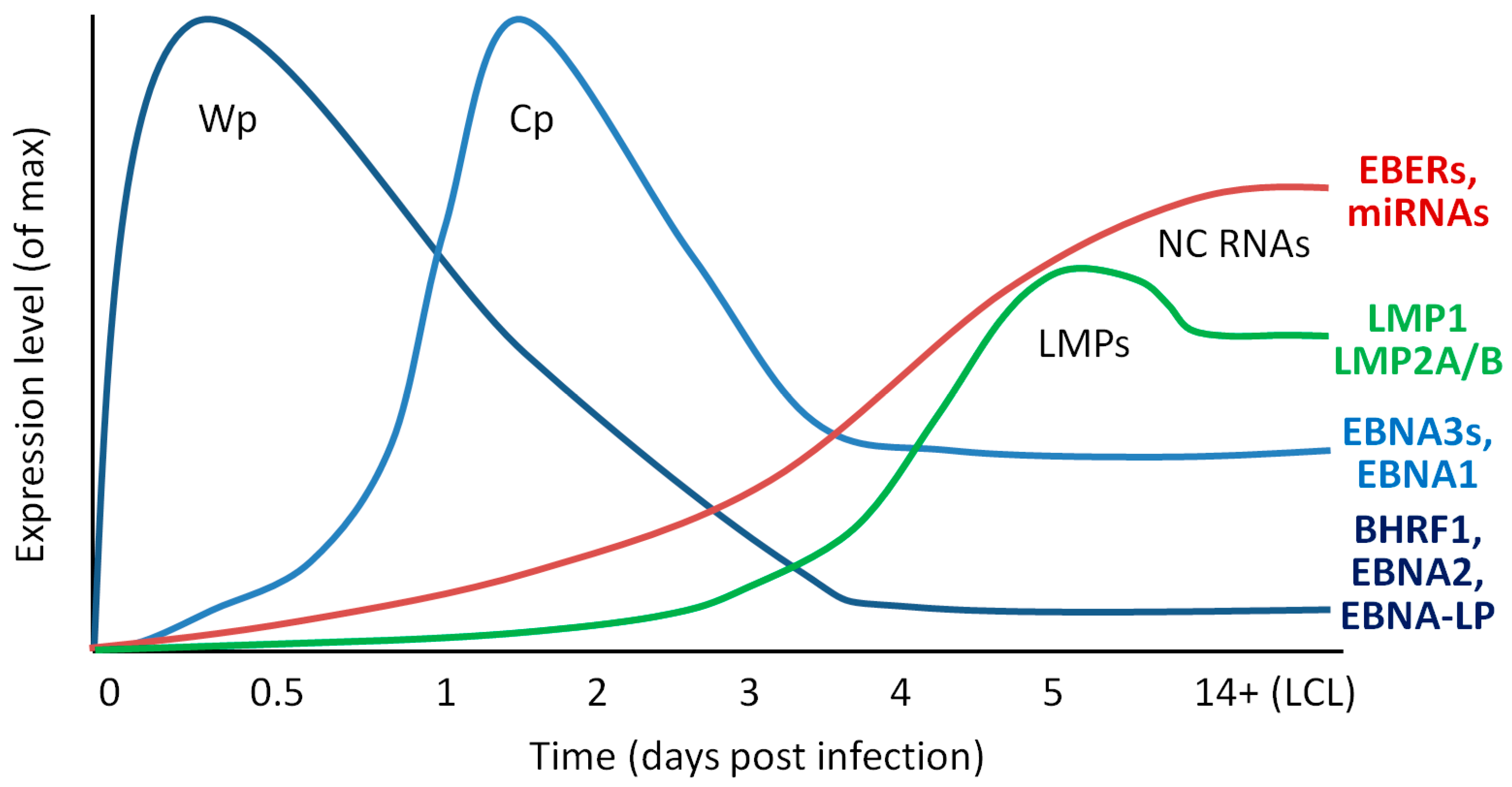{kind=link}
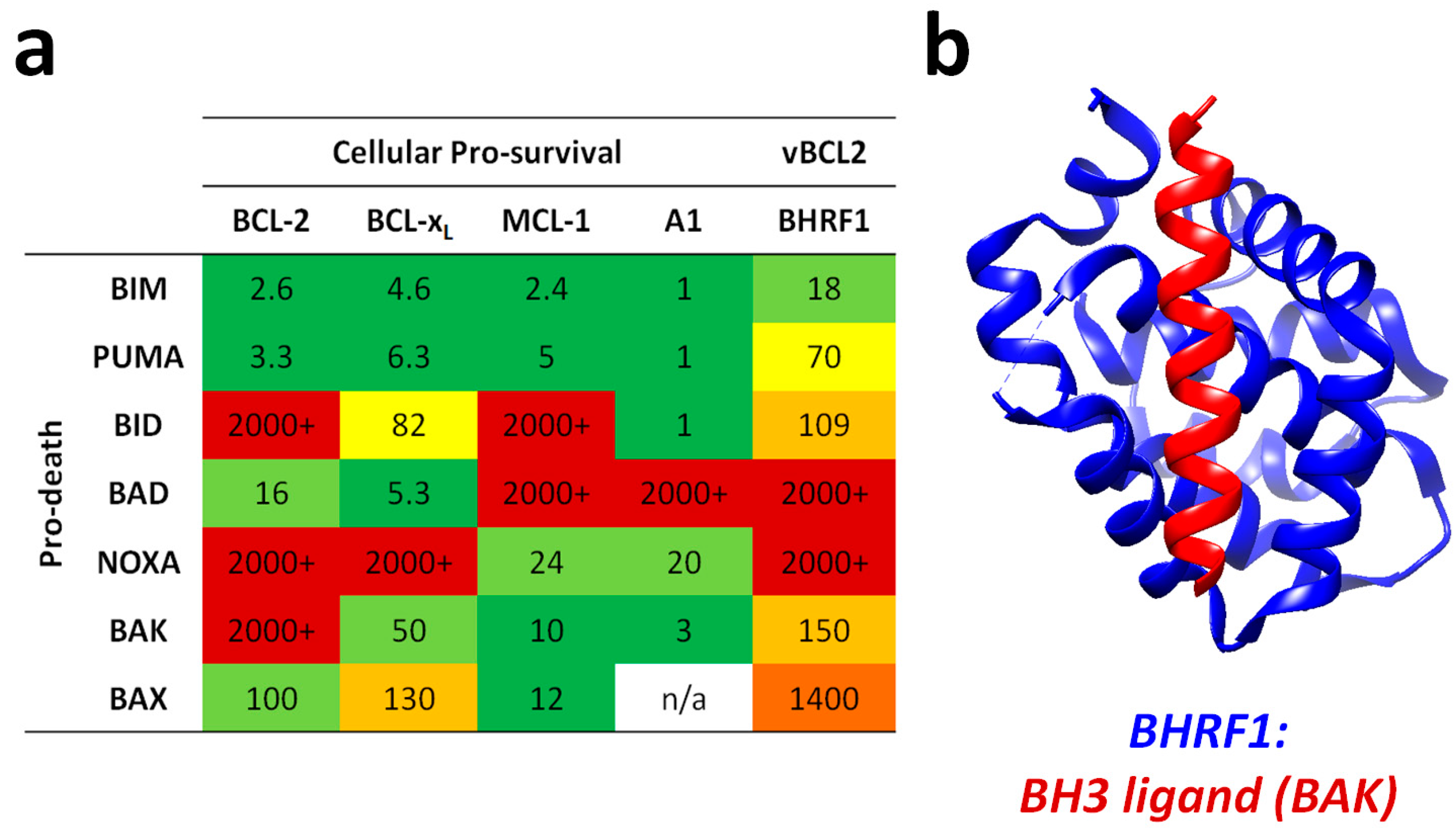{kind=link}
{kind=link}
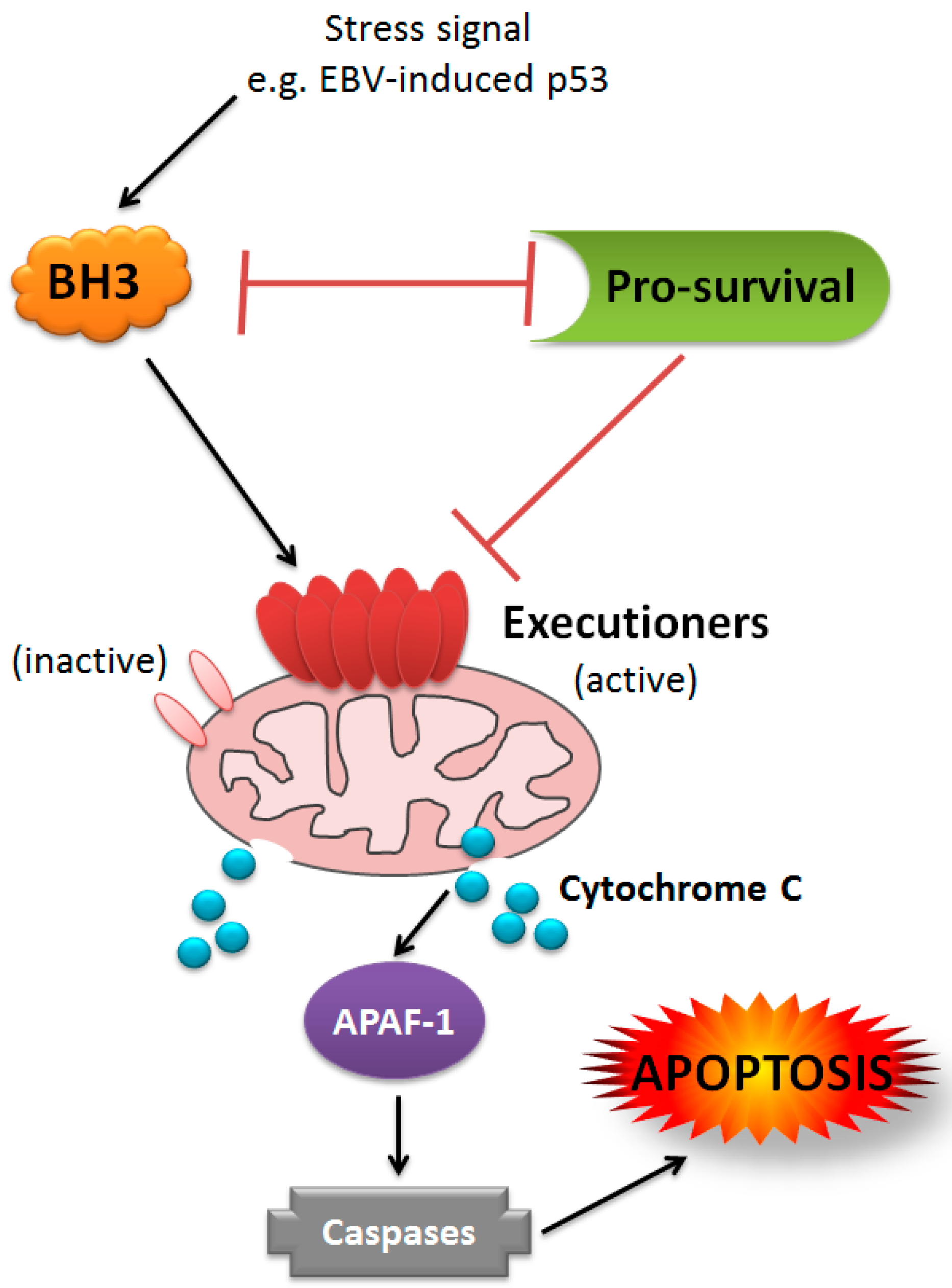
1. Introduction
2. ‘Transforming’ Cell Death: Latent Genes
2.1. Dynamics of Early Infection
2.2. EBNA-2 and EBNA-LP
2.3. EBNA-3A, -3B and -3C
2.4. EBNA-1
2.5. LMP1
2.6. LMP2A and LMP2B
2.7. BHRF1 and BALF1
2.8. Non-Coding RNAs
2.8.1. EBERs
2.8.2. BHRF1 microRNAs
2.8.3. BART microRNAs
3. Lytic Cycle Genes and Transformation
4. Cooperative Cell Death Inhibition by EBV
4.1. Counteracting the DNA Damage Response
4.2. Cooperation in Transformation
4.3. Cooperative Inhibition of Apoptosis in Malignancy
5. Burkitt Lymphoma (BL)
5.1. c-MYC
5.2. The Contribution of EBV
5.3. Restricted EBV Latency in BL
5.3.1. Latency I
5.3.2. Wp-Restricted Latency
6. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rickinson, A.A.E.K. Fields Virology, 4th ed.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2001; Volume 2, p. 3063. [Google Scholar]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Epstein, M.A.; Barr, Y.M.; Achong, B.G. A Second Virus-Carrying Tissue Culture Strain (Eb2) of Lymphoblasts from Burkitt’s Lymphoma. Pathol. Biol. (Paris) 1964, 12, 1233–1234. [Google Scholar]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr virus: An important vaccine target for cancer prevention. Sci. Transl. Med. 2011, 3, 107fs7. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D. A sarcoma involving the jaws in African children. Br. J. Surg. 1958, 46, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Burkitt, D.P.; Wright, D.H. Burkitt’s Lymphoma; Churchill Livingstone: London, UK, 1970. [Google Scholar]
- Burkitt, D. A Children’s Cancer with Geographical Limitations. Cancer Prog. 1963, 92, 102–113. [Google Scholar] [PubMed]
- Burkitt, D.P. Observations on the geography of malignant lymphoma. East Afr. Med. J. 1961, 38, 511–514. [Google Scholar] [PubMed]
- Haddow, A.J. An Improved Map for the Study of Burkitt’s Lymphoma Syndrome in Africa. East Afr. Med. J. 1963, 40, 429–432. [Google Scholar] [PubMed]
- Haddow, A.J. Age Incidence in Burkitt’s Lymphoma Syndrome. East Afr. Med. J. 1964, 41, 1–6. [Google Scholar] [PubMed]
- Kelly, G.L.; Rickinson, A.B. Burkitt lymphoma: Revisiting the pathogenesis of a virus-associated malignancy. Hematol. Am. Soc. Hematol. Educ. Progr. 2007, 2007, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Crawford, D.H.; Rickinson, A.; Johannessen, I.O. Cancer Virus: The Story of Epstein-Barr Virus; OUP Oxford: Oxford, UK, 2014. [Google Scholar]
- Pope, J.H.; Horne, M.K.; Scott, W. Transformation of foetal human keukocytes in vitro by filtrates of a human leukaemic cell line containing herpes-like virus. Int. J. Cancer 1968, 3, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Henle, W.; Diehl, V.; Kohn, G.; Zur Hausen, H.; Henle, G. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science 1967, 157, 1064–1065. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Adams, A.; Bjursell, G.; Bornkamm, G.W.; Kaschka-Dierich, C.; Jehn, U. Covalently closed circular duplex DNA of Epstein-Barr virus in a human lymphoid cell line. J. Mol. Biol. 1976, 102, 511–530. [Google Scholar] [CrossRef]
- Hammerschmidt, W.; Sugden, B. Replication of Epstein-Barr viral DNA. Cold Spring Harb. Perspect. Biol. 2013, 5, a013029. [Google Scholar] [CrossRef] [PubMed]
- Rowe, D.T.; Rowe, M.; Evan, G.I.; Wallace, L.E.; Farrell, P.J.; Rickinson, A.B. Restricted expression of EBV latent genes and T-lymphocyte-detected membrane antigen in Burkitt’s lymphoma cells. EMBO J. 1986, 5, 2599–2607. [Google Scholar] [PubMed]
- Rowe, M.; Rowe, D.T.; Gregory, C.D.; Young, L.S.; Farrell, P.J.; Rupani, H.; Rickinson, A.B. Differences in B cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt’s lymphoma cells. EMBO J. 1987, 6, 2743–2751. [Google Scholar] [PubMed]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: The Wp/BHRF1 link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Kieff, E. Epstein-Barr virus BHRF1 micro- and stable RNAs during latency III and after induction of replication. J. Virol. 2007, 81, 9967–9975. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Schafer, A.; Lu, S.; Bilello, J.P.; Desrosiers, R.C.; Edwards, R.; Raab-Traub, N.; Cullen, B.R. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006, 2, e23. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.D.; Gottlieb, E.; Lerner, M.R.; Steitz, J.A. Striking similarities are exhibited by two small Epstein-Barr virus-encoded ribonucleic acids and the adenovirus-associated ribonucleic acids VAI and VAII. Mol. Cell. Biol. 1981, 1, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, R.; Fitzsimmons, L.; Thomas, W.A.; Kelly, G.L.; Rowe, M.; Bell, A.I. Quantitative studies of Epstein-Barr virus-encoded microRNAs provide novel insights into their regulation. J. Virol. 2011, 85, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Birkenbach, M.; Kieff, E. Early events in Epstein-Barr virus infection of human B lymphocytes. Virology 1991, 181, 595–608. [Google Scholar] [CrossRef]
- Austin, P.J.; Flemington, E.; Yandava, C.N.; Strominger, J.L.; Speck, S.H. Complex transcription of the Epstein-Barr virus BamHI fragment H rightward open reading frame 1 (BHRF1) in latently and lytically infected B lymphocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 3678–3682. [Google Scholar] [CrossRef] [PubMed]
- Abbot, S.D.; Rowe, M.; Cadwallader, K.; Ricksten, A.; Gordon, J.; Wang, F.; Rymo, L.; Rickinson, A.B. Epstein-Barr virus nuclear antigen 2 induces expression of the virus-encoded latent membrane protein. J. Virol. 1990, 64, 2126–2134. [Google Scholar] [PubMed]
- Zimber-Strobl, U.; Kremmer, E.; Grasser, F.; Marschall, G.; Laux, G.; Bornkamm, G.W. The Epstein-Barr virus nuclear antigen 2 interacts with an EBNA2 responsive cis-element of the terminal protein 1 gene promoter. EMBO J. 1993, 12, 167–175. [Google Scholar] [PubMed]
- Schlager, S.; Speck, S.H.; Woisetschlager, M. Transcription of the Epstein-Barr virus nuclear antigen 1 (EBNA1) gene occurs before induction of the BCR2 (Cp) EBNA gene promoter during the initial stages of infection in B cells. J. Virol. 1996, 70, 3561–3570. [Google Scholar] [PubMed]
- Shannon-Lowe, C.; Baldwin, G.; Feederle, R.; Bell, A.; Rickinson, A.; Delecluse, H.J. Epstein-Barr virus-induced B-cell transformation: Quantitating events from virus binding to cell outgrowth. J. Gen. Virol. 2005, 86, 3009–3019. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, K.; Haar, J.; Tsai, M.H.; Poirey, R.; Feederle, R.; Delecluse, H.J. A Viral microRNA Cluster Regulates the Expression of PTEN, p27 and of a bcl-2 Homolog. PLoS Pathog. 2016, 12, e1005405. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Tourigny, J.P.; Forte, E.; Salinas, R.E.; Dave, S.S.; Luftig, M.A. Analysis of Epstein-Barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-κB activation. J. Virol. 2012, 86, 11096–11106. [Google Scholar] [CrossRef] [PubMed]
- Tierney, R.J.; Shannon-Lowe, C.D.; Fitzsimmons, L.; Bell, A.I.; Rowe, M. Unexpected patterns of Epstein-Barr virus transcription revealed by a High throughput PCR array for absolute quantification of viral mRNA. Virology 2015, 474, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Hofelmayr, H.; Strobl, L.J.; Marschall, G.; Bornkamm, G.W.; Zimber-Strobl, U. Activated Notch1 can transiently substitute for EBNA2 in the maintenance of proliferation of LMP1-expressing immortalized B cells. J. Virol. 2001, 75, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Strobl, L.J.; Hofelmayr, H.; Marschall, G.; Brielmeier, M.; Bornkamm, G.W.; Zimber-Strobl, U. Activated Notch1 modulates gene expression in B cells similarly to Epstein-Barr viral nuclear antigen 2. J. Virol. 2000, 74, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Taniguchi, Y.; Tamura, K.; Minoguchi, S.; Fukuhara, T.; Strobl, L.J.; Zimber-Strobl, U.; Bornkamm, G.W.; Honjo, T. Functional replacement of the intracellular region of the Notch1 receptor by Epstein-Barr virus nuclear antigen 2. J. Virol. 1998, 72, 6034–6039. [Google Scholar] [PubMed]
- Grossman, S.R.; Johannsen, E.; Tong, X.; Yalamanchili, R.; Kieff, E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 7568–7572. [Google Scholar] [CrossRef] [PubMed]
- Yalamanchili, R.; Tong, X.; Grossman, S.; Johannsen, E.; Mosialos, G.; Kieff, E. Genetic and biochemical evidence that EBNA 2 interaction with a 63-kDa cellular GTG-binding protein is essential for B lymphocyte growth transformation by EBV. Virology 1994, 204, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Zimber-Strobl, U.; Strobl, L.J.; Meitinger, C.; Hinrichs, R.; Sakai, T.; Furukawa, T.; Honjo, T.; Bornkamm, G.W. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J. 1994, 13, 4973–4982. [Google Scholar] [PubMed]
- Henkel, T.; Ling, P.D.; Hayward, S.D.; Peterson, M.G. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 1994, 265, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Tierney, R.; Nagra, J.; Hutchings, I.; Shannon-Lowe, C.; Altmann, M.; Hammerschmidt, W.; Rickinson, A.; Bell, A. Epstein-Barr virus exploits BSAP/Pax5 to achieve the B-cell specificity of its growth-transforming program. J. Virol. 2007, 81, 10092–10100. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, K.H.; Weidner, M.; Osborne, B.A.; Hayward, S.D. Epstein-Barr virus EBNA2 blocks Nur77-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2002, 99, 11878–11883. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, K.H.; Farrell, C.J.; Ling, P.D.; Kempkes, B.; Park, J.H.; Hayward, S.D. EBNA2 is required for protection of latently Epstein-Barr virus-infected B cells against specific apoptotic stimuli. J. Virol. 2004, 78, 12694–12697. [Google Scholar] [CrossRef] [PubMed]
- Godoi, P.H.; Wilkie-Grantham, R.P.; Hishiki, A.; Sano, R.; Matsuzawa, Y.; Yanagi, H.; Munte, C.E.; Chen, Y.; Yao, Y.; Marassi, F.M.; et al. Orphan Nuclear Receptor NR4A1 Binds a Novel Protein Interaction Site on Anti-apoptotic B Cell Lymphoma Gene 2 Family Proteins. J. Biol. Chem. 2016, 291, 14072–14084. [Google Scholar] [CrossRef] [PubMed]
- Pegman, P.M.; Smith, S.M.; D’Souza, B.N.; Loughran, S.T.; Maier, S.; Kempkes, B.; Cahill, P.A.; Simmons, M.J.; Gelinas, C.; Walls, D. Epstein-Barr virus nuclear antigen 2 trans-activates the cellular antiapoptotic bfl-1 gene by a CBF1/RBPJ kappa-dependent pathway. J. Virol. 2006, 80, 8133–8144. [Google Scholar] [CrossRef] [PubMed]
- Campion, E.M.; Hakimjavadi, R.; Loughran, S.T.; Phelan, S.; Smith, S.M.; D’Souza, B.N.; Tierney, R.J.; Bell, A.I.; Cahill, P.A.; Walls, D. Repression of the proapoptotic cellular BIK/NBK gene by Epstein-Barr virus antagonizes transforming growth factor beta1-induced B-cell apoptosis. J. Virol. 2014, 88, 5001–5013. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J.; Palmero, I.; Peters, G.; Farrell, P.J. EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO J. 1994, 13, 3321–3328. [Google Scholar] [PubMed]
- Mannick, J.B.; Cohen, J.I.; Birkenbach, M.; Marchini, A.; Kieff, E. The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J. Virol. 1991, 65, 6826–6837. [Google Scholar] [PubMed]
- Hammerschmidt, W.; Sugden, B. Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature 1989, 340, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Tierney, R.J.; Kao, K.Y.; Nagra, J.K.; Rickinson, A.B. Epstein-Barr virus BamHI W repeat number limits EBNA2/EBNA-LP coexpression in newly infected B cells and the efficiency of B-cell transformation: A rationale for the multiple W repeats in wild-type virus strains. J. Virol. 2011, 85, 12362–12375. [Google Scholar] [CrossRef] [PubMed]
- Kashuba, E.; Yurchenko, M.; Szirak, K.; Stahl, J.; Klein, G.; Szekely, L. Epstein-Barr virus-encoded EBNA-5 binds to Epstein-Barr virus-induced Fte1/S3a protein. Exp. Cell Res. 2005, 303, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Kashuba, E.; Mattsson, K.; Pokrovskaja, K.; Kiss, C.; Protopopova, M.; Ehlin-Henriksson, B.; Klein, G.; Szekely, L. EBV-encoded EBNA-5 associates with P14ARF in extranucleolar inclusions and prolongs the survival of P14ARF-expressing cells. Int. J. Cancer 2003, 105, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, G.; Nakajima, K.; Kawaguchi, Y.; Yamanashi, Y.; Hirai, K. Epstein-Barr virus (EBV) nuclear antigen leader protein (EBNA-LP) forms complexes with a cellular anti-apoptosis protein Bcl-2 or its EBV counterpart BHRF1 through HS1-associated protein X-1. Microbiol. Immunol. 2003, 47, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J.; Bazot, Q.; White, R.E. The EBNA3 Family: Two Oncoproteins and a Tumour Suppressor that Are Central to the Biology of EBV in B Cells. Curr. Top. Microbiol. Immunol. 2015, 391, 61–117. [Google Scholar] [PubMed]
- Le Roux, A.; Kerdiles, B.; Walls, D.; Dedieu, J.F.; Perricaudet, M. The Epstein-Barr virus determined nuclear antigens EBNA-3A, -3B, and -3C repress EBNA-2-mediated transactivation of the viral terminal protein 1 gene promoter. Virology 1994, 205, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Yenamandra, S.P.; Sompallae, R.; Klein, G.; Kashuba, E. Comparative analysis of the Epstein-Barr virus encoded nuclear proteins of EBNA-3 family. Comput. Biol. Med. 2009, 39, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Hertle, M.L.; Popp, C.; Petermann, S.; Maier, S.; Kremmer, E.; Lang, R.; Mages, J.; Kempkes, B. Differential gene expression patterns of EBV infected EBNA-3A positive and negative human B lymphocytes. PLoS Pathog. 2009, 5, e1000506. [Google Scholar] [CrossRef] [PubMed]
- Skalska, L.; White, R.E.; Franz, M.; Ruhmann, M.; Allday, M.J. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. 2010, 6, e1000951. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, B.; Robertson, E.; Kieff, E. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 1993, 67, 2014–2025. [Google Scholar] [PubMed]
- Tomkinson, B.; Kieff, E. Use of second-site homologous recombination to demonstrate that Epstein-Barr virus nuclear protein 3B is not important for lymphocyte infection or growth transformation in vitro. J. Virol. 1992, 66, 2893–2903. [Google Scholar] [PubMed]
- Chen, A.; Divisconte, M.; Jiang, X.; Quink, C.; Wang, F. Epstein-Barr virus with the latent infection nuclear antigen 3B completely deleted is still competent for B-cell growth transformation in vitro. J. Virol. 2005, 79, 4506–4509. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Parker, G.A.; Watanatanasup, E.; White, R.E.; Allday, M.J. BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res. 2012, 40, 7233–7246. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [PubMed]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Zhao, B.; Johannsen, E.; Kieff, E.; Zou, J.; Takada, K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc. Natl. Acad. Sci. USA 2011, 108, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Wu, Y.; Ishikawa, S.; Kanda, T.; Iwakiri, D.; Takada, K. Epstein-Barr virus nuclear protein EBNA3C is required for cell cycle progression and growth maintenance of lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 2006, 103, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Skalska, L.; White, R.E.; Parker, G.A.; Turro, E.; Sinclair, A.J.; Paschos, K.; Allday, M.J. Induction of p16(INK4a) is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. 2013, 9, e1003187. [Google Scholar] [CrossRef]
- Jiang, S.; Willox, B.; Zhou, H.; Holthaus, A.M.; Wang, A.; Shi, T.T.; Maruo, S.; Kharchenko, P.V.; Johannsen, E.C.; Kieff, E.; et al. Epstein-Barr virus nuclear antigen 3C binds to BATF/IRF4 or SPI1/IRF4 composite sites and recruits Sin3A to repress CDKN2A. Proc. Natl. Acad. Sci. USA 2014, 111, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Guo, Y.; Xiao, B.; Banerjee, S.; Saha, A.; Lu, J.; Glisovic, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS Pathog. 2011, 7, e1002418. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 2009, 388, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Murakami, M.; Kumar, P.; Bajaj, B.; Sims, K.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2. J. Virol. 2009, 83, 4652–4669. [Google Scholar] [CrossRef] [PubMed]
- McClellan, M.J.; Wood, C.D.; Ojeniyi, O.; Cooper, T.J.; Kanhere, A.; Arvey, A.; Webb, H.M.; Palermo, R.D.; Harth-Hertle, M.L.; Kempkes, B.; et al. Modulation of enhancer looping and differential gene targeting by Epstein-Barr virus transcription factors directs cellular reprogramming. PLoS Pathog. 2013, 9, e1003636. [Google Scholar] [CrossRef] [PubMed]
- Sears, J.; Ujihara, M.; Wong, S.; Ott, C.; Middeldorp, J.; Aiyar, A. The amino terminus of Epstein-Barr Virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J. Virol. 2004, 78, 11487–11505. [Google Scholar] [CrossRef] [PubMed]
- Mackey, D.; Sugden, B. Applications of oriP plasmids and their mode of replication. Methods Enzymol. 1999, 306, 308–328. [Google Scholar] [PubMed]
- Leight, E.R.; Sugden, B. EBNA-1: A protein pivotal to latent infection by Epstein-Barr virus. Rev. Med. Virol. 2000, 10, 83–100. [Google Scholar] [CrossRef]
- Frappier, L. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses 2012, 4, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Sompallae, R.; Masucci, M.G. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 2009, 28, 3997–4008. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Murakami, M.; Verma, S.C.; Cai, Q.L.; Haldar, S.; Kaul, R.; Wasik, M.A.; Middeldorp, J.; Robertson, E.S. Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 2011, 410, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Wood, V.H.; O’Neil, J.D.; Wei, W.; Stewart, S.E.; Dawson, C.W.; Young, L.S. Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFbeta signaling pathways. Oncogene 2007, 26, 4135–4147. [Google Scholar] [CrossRef] [PubMed]
- Canaan, A.; Haviv, I.; Urban, A.E.; Schulz, V.P.; Hartman, S.; Zhang, Z.; Palejev, D.; Deisseroth, A.B.; Lacy, J.; Snyder, M.; et al. EBNA1 regulates cellular gene expression by binding cellular promoters. Proc. Natl. Acad. Sci. USA 2009, 106, 22421–22426. [Google Scholar] [CrossRef] [PubMed]
- Dresang, L.R.; Vereide, D.T.; Sugden, B. Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: Defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J. Virol. 2009, 83, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Wikramasinghe, P.; Norseen, J.; Tsai, K.; Wang, P.; Showe, L.; Davuluri, R.V.; Lieberman, P.M. Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1). Virol. J. 2010, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Kaye, K.M.; Izumi, K.M.; Kieff, E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 9150–9154. [Google Scholar] [CrossRef] [PubMed]
- Dirmeier, U.; Neuhierl, B.; Kilger, E.; Reisbach, G.; Sandberg, M.L.; Hammerschmidt, W. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by epstein-barr virus. Cancer Res. 2003, 63, 2982–2989. [Google Scholar] [PubMed]
- Rastelli, J.; Homig-Holzel, C.; Seagal, J.; Muller, W.; Hermann, A.C.; Rajewsky, K.; Zimber-Strobl, U. LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood 2008, 111, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Uchida, J.; Yasui, T.; Takaoka-Shichijo, Y.; Muraoka, M.; Kulwichit, W.; Raab-Traub, N.; Kikutani, H. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 1999, 286, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.M.; Kieff, E.D. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc. Natl. Acad. Sci. USA 1997, 94, 12592–12597. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.M.; Kaye, K.M.; Kieff, E.D. The Epstein-Barr virus LMP1 amino acid sequence that engages tumor necrosis factor receptor associated factors is critical for primary B lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA 1997, 94, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Mosialos, G.; Birkenbach, M.; Yalamanchili, R.; VanArsdale, T.; Ware, C.; Kieff, E. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell 1995, 80, 389–399. [Google Scholar] [CrossRef]
- Luftig, M.; Prinarakis, E.; Yasui, T.; Tsichritzis, T.; Cahir-McFarland, E.; Inoue, J.; Nakano, H.; Mak, T.W.; Yeh, W.C.; Li, X.; et al. Epstein-Barr virus latent membrane protein 1 activation of NF-kappaB through IRAK1 and TRAF6. Proc. Natl. Acad. Sci. USA 2003, 100, 15595–15600. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.H.; Marquitz, A.R.; Raab-Traub, N. Changes in expression induced by Epstein-Barr Virus LMP1-CTAR1: Potential role of bcl3. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Gewurz, B.E.; Mar, J.C.; Padi, M.; Zhao, B.; Shinners, N.P.; Takasaki, K.; Bedoya, E.; Zou, J.Y.; Cahir-McFarland, E.; Quackenbush, J.; et al. Canonical NF-κB activation is essential for Epstein-Barr virus latent membrane protein 1 TES2/CTAR2 gene regulation. J. Virol. 2011, 85, 6764–6773. [Google Scholar] [CrossRef] [PubMed]
- Ersing, I.; Bernhardt, K.; Gewurz, B.E. NF-kappaB and IRF7 pathway activation by Epstein-Barr virus Latent Membrane Protein 1. Viruses 2013, 5, 1587–1606. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 modulates distinctive NF-κB pathways through C-terminus-activating region 1 to regulate epidermal growth factor receptor expression. J. Virol. 2010, 84, 6605–6614. [Google Scholar] [CrossRef] [PubMed]
- Pratt, Z.L.; Zhang, J.; Sugden, B. The latent membrane protein 1 (LMP1) oncogene of Epstein-Barr virus can simultaneously induce and inhibit apoptosis in B cells. J. Virol. 2012, 86, 4380–4393. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, B.; Rowe, M.; Walls, D. The bfl-1 gene is transcriptionally upregulated by the Epstein-Barr virus LMP1, and its expression promotes the survival of a Burkitt’s lymphoma cell line. J. Virol. 2000, 74, 6652–6658. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, B.N.; Edelstein, L.C.; Pegman, P.M.; Smith, S.M.; Loughran, S.T.; Clarke, A.; Mehl, A.; Rowe, M.; Gelinas, C.; Walls, D. Nuclear factor κB -dependent activation of the antiapoptotic bfl-1 gene by the Epstein-Barr virus latent membrane protein 1 and activated CD40 receptor. J. Virol. 2004, 78, 1800–1816. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Rowe, M.; Gregory, C.; Croom-Carter, D.; Wang, F.; Longnecker, R.; Kieff, E.; Rickinson, A. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell 1991, 65, 1107–1115. [Google Scholar] [CrossRef]
- Wang, S.; Rowe, M.; Lundgren, E. Expression of the Epstein Barr virus transforming protein LMP1 causes a rapid and transient stimulation of the Bcl-2 homologue Mcl-1 levels in B-cell lines. Cancer Res. 1996, 56, 4610–4613. [Google Scholar] [PubMed]
- Tsai, S.C.; Lin, S.J.; Lin, C.J.; Chou, Y.C.; Lin, J.H.; Yeh, T.H.; Chen, M.R.; Huang, L.M.; Lu, M.Y.; Huang, Y.C.; et al. Autocrine CCL3 and CCL4 induced by the oncoprotein LMP1 promote Epstein-Barr virus-triggered B cell proliferation. J. Virol. 2013, 87, 9041–9052. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Barrera, L.A.; Ersing, I.; Willox, B.; Schmidt, S.C.; Greenfeld, H.; Zhou, H.; Mollo, S.B.; Shi, T.T.; Takasaki, K.; et al. The NF-κB genomic landscape in lymphoblastoid B cells. Cell Rep. 2014, 8, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Walsh, M.J.; Bernhardt, K.; Ashbaugh, C.W.; Trudeau, S.J.; Ashbaugh, I.Y.; Jiang, S.; Jiang, C.; Zhao, B.; Root, D.E.; et al. CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 2017, 21, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Le Clorennec, C.; Ouk, T.S.; Youlyouz-Marfak, I.; Panteix, S.; Martin, C.C.; Rastelli, J.; Adriaenssens, E.; Zimber-Strobl, U.; Coll, J.; Feuillard, J.; et al. Molecular basis of cytotoxicity of Epstein-Barr virus (EBV) latent membrane protein 1 (LMP1) in EBV latency III B cells: LMP1 induces type II ligand-independent autoactivation of CD95/Fas with caspase 8-mediated apoptosis. J. Virol. 2008, 82, 6721–6733. [Google Scholar] [CrossRef] [PubMed]
- Le Clorennec, C.; Youlyouz-Marfak, I.; Adriaenssens, E.; Coll, J.; Bornkamm, G.W.; Feuillard, J. EBV latency III immortalization program sensitizes B cells to induction of CD95-mediated apoptosis via LMP1: Role of NF-κB, STAT1, and p53. Blood 2006, 107, 2070–2078. [Google Scholar] [CrossRef] [PubMed]
- Laux, G.; Dugrillon, F.; Eckert, C.; Adam, B.; Zimber-Strobl, U.; Bornkamm, G.W. Identification and characterization of an Epstein-Barr virus nuclear antigen 2-responsive cis element in the bidirectional promoter region of latent membrane protein and terminal protein 2 genes. J. Virol. 1994, 68, 6947–6958. [Google Scholar] [PubMed]
- Laux, G.; Perricaudet, M.; Farrell, P.J. A spliced Epstein-Barr virus gene expressed in immortalized lymphocytes is created by circularization of the linear viral genome. EMBO J. 1988, 7, 769–774. [Google Scholar] [PubMed]
- Sample, J.; Liebowitz, D.; Kieff, E. Two related Epstein-Barr virus membrane proteins are encoded by separate genes. J. Virol. 1989, 63, 933–937. [Google Scholar] [PubMed]
- Kim, O.J.; Yates, J.L. Mutants of Epstein-Barr virus with a selective marker disrupting the TP gene transform B cells and replicate normally in culture. J. Virol. 1993, 67, 7634–7640. [Google Scholar] [PubMed]
- Longnecker, R.; Miller, C.L.; Tomkinson, B.; Miao, X.Q.; Kieff, E. Deletion of DNA encoding the first five transmembrane domains of Epstein-Barr virus latent membrane proteins 2A and 2B. J. Virol. 1993, 67, 5068–5074. [Google Scholar] [PubMed]
- Longnecker, R.; Miller, C.L.; Miao, X.Q.; Tomkinson, B.; Kieff, E. The last seven transmembrane and carboxy-terminal cytoplasmic domains of Epstein-Barr virus latent membrane protein 2 (LMP2) are dispensable for lymphocyte infection and growth transformation in vitro. J. Virol. 1993, 67, 2006–2013. [Google Scholar] [PubMed]
- Speck, P.; Kline, K.A.; Cheresh, P.; Longnecker, R. Epstein-Barr virus lacking latent membrane protein 2 immortalizes B cells with efficiency indistinguishable from that of wild-type virus. J. Gen. Virol. 1999, 80, 2193–2203. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Longnecker, R. Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway. J. Virol. 2007, 81, 9299–9306. [Google Scholar] [CrossRef] [PubMed]
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar] [PubMed]
- Mancao, C.; Altmann, M.; Jungnickel, B.; Hammerschmidt, W. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood 2005, 106, 4339–4344. [Google Scholar] [CrossRef] [PubMed]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Longnecker, R. LMP2A survival and developmental signals are transmitted through Btk-dependent and Btk-independent pathways. Virology 2001, 291, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.; Swart, R.; Katzman, R.B.; Ikeda, M.; Ikeda, A.; Longnecker, R.; Dykstra, M.L.; Pierce, S.K. The effects of the Epstein-Barr virus latent membrane protein 2A on B cell function. Int. Rev. Immunol. 2001, 20, 805–835. [Google Scholar] [CrossRef] [PubMed]
- Swart, R.; Ruf, I.K.; Sample, J.; Longnecker, R. Latent membrane protein 2A-mediated effects on the phosphatidylinositol 3-Kinase/Akt pathway. J. Virol. 2000, 74, 10838–10845. [Google Scholar] [CrossRef] [PubMed]
- Laux, G.; Economou, A.; Farrell, P.J. The terminal protein gene 2 of Epstein-Barr virus is transcribed from a bidirectional latent promoter region. J. Gen. Virol. 1989, 70, 3079–3084. [Google Scholar] [CrossRef] [PubMed]
- Rovedo, M.; Longnecker, R. Epstein-Barr virus latent membrane protein 2A preferentially signals through the Src family kinase Lyn. J. Virol. 2008, 82, 8520–8528. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Hinds, M.G. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013, 4, e909. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S.; Bahar, M.W.; Abrescia, N.G.; McVey, C.E.; Bartlett, N.W.; Chen, R.A.; Stuart, D.I.; Grimes, J.M.; Smith, G.L. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 2007, 88, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, M.; Zhai, D.; Jin, C.; Aleshin, A.E.; Stec, B.; Reed, J.C.; Liddington, R.C. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 2007, 16, 118–124. [Google Scholar] [CrossRef] [PubMed]
- DiPerna, G.; Stack, J.; Bowie, A.G.; Boyd, A.; Kotwal, G.; Zhang, Z.; Arvikar, S.; Latz, E.; Fitzgerald, K.A.; Marshall, W.L. Poxvirus protein N1L targets the I-κB kinase complex, inhibits signaling to NF-κB by the tumor necrosis factor superfamily of receptors, and inhibits NF-κB and IRF3 signaling by toll-like receptors. J. Biol. Chem. 2004, 279, 36570–36578. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.C.; Bahar, M.W.; Cooray, S.; Chen, R.A.; Whalen, D.M.; Abrescia, N.G.; Alderton, D.; Owens, R.J.; Stuart, D.I.; Smith, G.L.; et al. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-κB rather than apoptosis. PLoS Pathog. 2008, 4, e1000128. [Google Scholar] [CrossRef] [PubMed]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.L.; Yim, C.; Gustafson, E.; Graf, T.; Sage, D.R.; Hanify, K.; Williams, L.; Fingeroth, J.; Finberg, R.W. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J. Virol. 1999, 73, 5181–5185. [Google Scholar] [PubMed]
- Henderson, S.; Huen, D.; Rowe, M.; Dawson, C.; Johnson, G.; Rickinson, A. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. USA 1993, 90, 8479–8483. [Google Scholar] [CrossRef] [PubMed]
- Foight, G.W.; Keating, A.E. Locating Herpesvirus Bcl-2 Homologs in the Specificity Landscape of Anti-Apoptotic Bcl-2 Proteins. J. Mol. Biol. 2015, 427, 2468–2490. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.; Colman, P.M. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, A.M.; Letai, A. BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death Differ. 2008, 15, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Desbien, A.L.; Kappler, J.W.; Marrack, P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. USA 2009, 106, 5663–5668. [Google Scholar] [CrossRef] [PubMed]
- Fanidi, A.; Hancock, D.C.; Littlewood, T.D. Suppression of c-Myc-induced apoptosis by the Epstein-Barr virus gene product BHRF1. J. Virol. 1998, 72, 8392–8395. [Google Scholar] [PubMed]
- Foghsgaard, L.; Jaattela, M. The ability of BHRF1 to inhibit apoptosis is dependent on stimulus and cell type. J. Virol. 1997, 71, 7509–7517. [Google Scholar] [PubMed]
- Kawanishi, M.; Tada-Oikawa, S.; Kawanishi, S. Epstein-Barr virus BHRF1 functions downstream of Bid cleavage and upstream of mitochondrial dysfunction to inhibit TRAIL-induced apoptosis in BJAB cells. Biochem. Biophys. Res. Commun. 2002, 297, 682–687. [Google Scholar] [CrossRef]
- McCarthy, N.J.; Hazlewood, S.A.; Huen, D.S.; Rickinson, A.B.; Williams, G.T. The Epstein-Barr virus gene BHRF1, a homologue of the cellular oncogene Bcl-2, inhibits apoptosis induced by gamma radiation and chemotherapeutic drugs. Adv. Exp. Med. Biol. 1996, 406, 83–97. [Google Scholar] [PubMed]
- Watanabe, A.; Maruo, S.; Ito, T.; Ito, M.; Katsumura, K.R.; Takada, K. Epstein-Barr virus-encoded Bcl-2 homologue functions as a survival factor in Wp-restricted Burkitt lymphoma cell line P3HR-1. J. Virol. 2010, 84, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Yee, J.; White, R.E.; Anderton, E.; Allday, M.J. Latent Epstein-Barr Virus Can Inhibit Apoptosis in B Cells by Blocking the Induction of NOXA Expression. PLoS ONE 2011, 6, e28506. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.G.; Song, Q.; Cross, S.; Misko, I.; Lees-Miller, S.P.; Lavin, M.F. Resistance to etoposide-induced apoptosis in a Burkitt’s lymphoma cell line. Int. J. Cancer 1998, 77, 755–762. [Google Scholar] [CrossRef]
- Bellows, D.S.; Howell, M.; Pearson, C.; Hazlewood, S.A.; Hardwick, J.M. Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J. Virol. 2002, 76, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes. Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Meusburger, S.; Hawkins, C.J.; Riglar, D.T.; Lee, E.F.; Fairlie, W.D.; Huang, D.C.; Adams, J.M. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. USA 2008, 105, 18081–18087. [Google Scholar] [CrossRef] [PubMed]
- Smits, C.; Czabotar, P.E.; Hinds, M.G.; Day, C.L. Structural plasticity underpins promiscuous binding of the prosurvival protein A1. Structure 2008, 16, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Lerner, M.R.; Andrews, N.C.; Miller, G.; Steitz, J.A. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1981, 78, 805–809. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Adland, E.; Bell, A.I.; Delecluse, H.J.; Rickinson, A.B.; Rowe, M. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: Viral genome expression, genome maintenance, and genome amplification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Pfuhl, T.; Motsch, N.; Barth, S.; Nicholls, J.; Grasser, F.; Meister, G. Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J. Virol. 2009, 83, 3333–3341. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Chen, G.H.; Chen, Y.H.; Liu, C.Y.; Chang, K.P.; Chang, Y.S.; Chen, H.C. Characterization of Epstein-Barr virus miRNAome in nasopharyngeal carcinoma by deep sequencing. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Sullivan, C.S.; Ganem, D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 2006, 12, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.H.; Marquitz, A.R.; Raab-Traub, N. Epstein-Barr virus BART microRNAs are produced from a large intron prior to splicing. J. Virol. 2008, 82, 9094–9106. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Tomkinson, B.; Kieff, E. Recombinant Epstein-Barr virus with small RNA (EBER) genes deleted transforms lymphocytes and replicates in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Huneycutt, B.S.; Reiss, C.S.; Kieff, E. Epstein-Barr virus-encoded small RNAs (EBERs) do not modulate interferon effects in infected lymphocytes. J. Virol. 1992, 66, 5133–5136. [Google Scholar] [PubMed]
- Yajima, M.; Kanda, T.; Takada, K. Critical role of Epstein-Barr Virus (EBV)-encoded RNA in efficient EBV-induced B-lymphocyte growth transformation. J. Virol. 2005, 79, 4298–4307. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Maruo, S.; Yajima, M.; Kanda, T.; Takada, K. Epstein-Barr virus (EBV)-encoded RNA 2 (EBER2) but not EBER1 plays a critical role in EBV-induced B-cell growth transformation. J. Virol. 2007, 81, 11236–11245. [Google Scholar] [CrossRef] [PubMed]
- Gregorovic, G.; Bosshard, R.; Karstegl, C.E.; White, R.E.; Pattle, S.; Chiang, A.K.; Dittrich-Breiholz, O.; Kracht, M.; Russ, R.; Farrell, P.J. Cellular gene expression that correlates with EBER expression in Epstein-Barr Virus-infected lymphoblastoid cell lines. J. Virol. 2011, 85, 3535–3545. [Google Scholar] [CrossRef] [PubMed]
- Benetti, R.; Del Sal, G.; Monte, M.; Paroni, G.; Brancolini, C.; Schneider, C. The death substrate Gas2 binds m-calpain and increases susceptibility to p53-dependent apoptosis. EMBO J. 2001, 20, 2702–2714. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.T.; Bolderson, E.; Adams, M.N.; Baird, A.M.; Zhang, S.D.; Gately, K.A.; Umezawa, K.; O’Byrne, K.J.; Richard, D.J. Activation and cleavage of SASH1 by caspase-3 mediates an apoptotic response. Cell Death Dis. 2016, 7, e2469. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; O’Hara, A.; Araujo, I.; Barreto, J.; Carvalho, E.; Sapucaia, J.B.; Ramos, J.C.; Luz, E.; Pedroso, C.; Manrique, M.; et al. EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1–3. Cancer Res. 2008, 68, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar] [CrossRef] [PubMed]
- Pratt, Z.L.; Kuzembayeva, M.; Sengupta, S.; Sugden, B. The microRNAs of Epstein-Barr Virus are expressed at dramatically differing levels among cell lines. Virology 2009, 386, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Haar, J.; Bernhardt, K.; Linnstaedt, S.D.; Bannert, H.; Lips, H.; Cullen, B.R.; Delecluse, H.J. The members of an Epstein-Barr virus microRNA cluster cooperate to transform B lymphocytes. J. Virol. 2011, 85, 9801–9810. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Linnstaedt, S.D.; Bannert, H.; Lips, H.; Bencun, M.; Cullen, B.R.; Delecluse, H.J. A viral microRNA cluster strongly potentiates the transforming properties of a human herpesvirus. PLoS Pathog. 2011, 7, e1001294. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Moosmann, A.; Gromminger, S.; Walz, N.; Grundhoff, A.; Hammerschmidt, W. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.; Linnstaedt, S.D.; Esoda, C.; Krisko, J.F.; Martinez-Torres, F.; Delecluse, H.J.; Cullen, B.R.; Garcia, J.V. A cluster of virus-encoded microRNAs accelerates acute systemic Epstein-Barr virus infection but does not significantly enhance virus-induced oncogenesis in vivo. J. Virol. 2013, 87, 5437–5446. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.H.; Lekprasert, P.; Mukherjee, N.; Skalsky, R.L.; Corcoran, D.L.; Cullen, B.R.; Ohler, U. MicroRNA target site identification by integrating sequence and binding information. Nat. Methods 2013, 10, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Cullen, B.R. EBV Noncoding RNAs. Curr. Top. Microbiol. Immunol. 2015, 391, 181–217. [Google Scholar] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Warnatz, H.J.; Schmidt, D.; Manke, T.; Piccini, I.; Sultan, M.; Borodina, T.; Balzereit, D.; Wruck, W.; Soldatov, A.; Vingron, M.; et al. The BTB and CNC homology 1 (BACH1) target genes are involved in the oxidative stress response and in control of the cell cycle. J. Biol. Chem. 2011, 286, 23521–23532. [Google Scholar] [CrossRef] [PubMed]
- Castellini, L.; Moon, E.J.; Razorenova, O.V.; Krieg, A.J.; von Eyben, R.; Giaccia, A.J. KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res. 2017, 45, 3674–3692. [Google Scholar] [CrossRef] [PubMed]
- Gilligan, K.J.; Rajadurai, P.; Lin, J.C.; Busson, P.; Abdel-Hamid, M.; Prasad, U.; Tursz, T.; Raab-Traub, N. Expression of the Epstein-Barr virus BamHI A fragment in nasopharyngeal carcinoma: Evidence for a viral protein expressed in vivo. J. Virol. 1991, 65, 6252–6259. [Google Scholar] [PubMed]
- Hitt, M.M.; Allday, M.J.; Hara, T.; Karran, L.; Jones, M.D.; Busson, P.; Tursz, T.; Ernberg, I.; Griffin, B.E. EBV gene expression in an NPC-related tumour. EMBO J. 1989, 8, 2639–2651. [Google Scholar] [PubMed]
- Sadler, R.H.; Raab-Traub, N. Structural analyses of the Epstein-Barr virus BamHI A transcripts. J. Virol. 1995, 69, 1132–1141. [Google Scholar] [PubMed]
- Smith, P.R.; Gao, Y.; Karran, L.; Jones, M.D.; Snudden, D.; Griffin, B.E. Complex nature of the major viral polyadenylated transcripts in Epstein-Barr virus-associated tumors. J. Virol. 1993, 67, 3217–3225. [Google Scholar] [PubMed]
- Zhang, J.; Chen, H.; Weinmaster, G.; Hayward, S.D. Epstein-Barr virus BamHi-a rightward transcript-encoded RPMS protein interacts with the CBF1-associated corepressor CIR to negatively regulate the activity of EBNA2 and NotchIC. J. Virol. 2001, 75, 2946–2956. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.R.; de Jesus, O.; Turner, D.; Hollyoake, M.; Karstegl, C.E.; Griffin, B.E.; Karran, L.; Wang, Y.; Hayward, S.D.; Farrell, P.J. Structure and coding content of CST (BART) family RNAs of Epstein-Barr virus. J. Virol. 2000, 74, 3082–3092. [Google Scholar] [CrossRef] [PubMed]
- Kusano, S.; Raab-Traub, N. An Epstein-Barr virus protein interacts with Notch. J. Virol. 2001, 75, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Al-Mozaini, M.; Bodelon, G.; Karstegl, C.E.; Jin, B.; Al-Ahdal, M.; Farrell, P.J. Epstein-Barr virus BART gene expression. J. Gen. Virol. 2009, 90, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, J.; Brink, A.A.; Vervoort, M.B.; van Zijp, M.J.; Meijer, C.J.; van den Brule, A.J.; Middeldorp, J.M. In vivo transcription of the Epstein-Barr virus (EBV) BamHI-A region without associated in vivo BARF0 protein expression in multiple EBV-associated disorders. J. Gen. Virol. 2003, 84, 2647–2659. [Google Scholar] [CrossRef] [PubMed]
- Bornkamm, G.W.; Delius, H.; Zimber, U.; Hudewentz, J.; Epstein, M.A. Comparison of Epstein-Barr virus strains of different origin by analysis of the viral DNAs. J. Virol. 1980, 35, 603–618. [Google Scholar] [PubMed]
- Raab-Traub, N.; Dambaugh, T.; Kieff, E. DNA of Epstein-Barr virus VIII: B95–8, the previous prototype, is an unusual deletion derivative. Cell 1980, 22, 257–267. [Google Scholar] [CrossRef]
- Vereide, D.T.; Seto, E.; Chiu, Y.F.; Hayes, M.; Tagawa, T.; Grundhoff, A.; Hammerschmidt, W.; Sugden, B. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene 2014, 33, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Dolken, L.; Malterer, G.; Erhard, F.; Kothe, S.; Friedel, C.C.; Suffert, G.; Marcinowski, L.; Motsch, N.; Barth, S.; Beitzinger, M.; et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human gamma-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 2010, 7, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; Ock, J.; Lee, H.J.; Lee, Y.J.; Kwon, B.M.; Hong, S.H. Early growth response protein 1 upregulation and nuclear translocation by 2’-benzoyloxycinnamaldehyde induces prostate cancer cell death. Cancer Lett. 2013, 329, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Cartron, P.F.; Er, E.; Oliver, L.; Juin, P.; Armstrong, L.C.; Bornstein, P.; Mihara, K.; Manon, S.; Vallette, F.M. TOM22, a core component of the mitochondria outer membrane protein translocation pore, is a mitochondrial receptor for the proapoptotic protein Bax. Cell Death Differ. 2007, 14, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.J.; Rabinowitz, G.S.; Yario, T.A.; Luna, J.M.; Darnell, R.B.; Steitz, J.A. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012, 31, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Xu, G.; Deng, N.; Taylor, C.; Zhu, D.; Flemington, E.K. Quantitative and qualitative RNA-Seq-based evaluation of Epstein-Barr virus transcription in type I latency Burkitt’s lymphoma cells. J. Virol. 2010, 84, 13053–13058. [Google Scholar] [CrossRef] [PubMed]
- Moss, W.N.; Steitz, J.A. Genome-wide analyses of Epstein-Barr virus reveal conserved RNA structures and a novel stable intronic sequence RNA. BMC Genom. 2013, 14, 543. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [PubMed]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B.; et al. Down-regulation of BLIMP1alpha by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: Implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011, 117, 5907–5917. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J. Epigenetic control of Epstein-Barr virus transcription—Relevance to viral life cycle? Front. Genet. 2013, 4, 161. [Google Scholar] [CrossRef] [PubMed]
- Niller, H.H.; Wolf, H.; Minarovits, J. Epigenetic dysregulation of the host cell genome in Epstein-Barr virus-associated neoplasia. Semin. Cancer Biol. 2009, 19, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Kalla, M.; Schmeinck, A.; Bergbauer, M.; Pich, D.; Hammerschmidt, W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. USA 2010, 107, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog. 2012, 8, e1002902. [Google Scholar] [CrossRef] [PubMed]
- Gruffat, H.; Sergeant, A. Characterization of the DNA-binding site repertoire for the Epstein-Barr virus transcription factor R. Nucleic Acids Res. 1994, 22, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Gutsch, D.E.; Marcu, K.B.; Kenney, S.C. The Epstein-Barr virus BRLF1 gene product transactivates the murine and human c-myc promoters. Cell. Mol. Biol. 1994, 40, 747–760. [Google Scholar] [PubMed]
- Ragoczy, T.; Miller, G. Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. J. Virol. 2001, 75, 5240–5251. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, W.; Sugden, B. Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 1988, 55, 427–433. [Google Scholar] [CrossRef]
- Kawanishi, M. Epstein-Barr virus induces fragmentation of chromosomal DNA during lytic infection. J. Virol. 1993, 67, 7654–7658. [Google Scholar] [PubMed]
- Morrison, T.E.; Kenney, S.C. BZLF1, an Epstein-Barr virus immediate-early protein, induces p65 nuclear translocation while inhibiting p65 transcriptional function. Virology 2004, 328, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Thomas, W.A.; Haigh, T.A.; Fitzsimmons, L.; Long, H.M.; Hislop, A.D.; Taylor, G.S.; Rowe, M. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathog. 2011, 7, e1002455. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.R.; Quinn, L.L.; Rowe, M.; Zuo, J. Induction of the Lytic Cycle Sensitizes Epstein-Barr Virus-Infected B Cells to NK Cell Killing That Is Counteracted by Virus-Mediated NK Cell Evasion Mechanisms in the Late Lytic Cycle. J. Virol. 2015, 90, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Inman, G.J.; Binne, U.K.; Parker, G.A.; Farrell, P.J.; Allday, M.J. Activators of the Epstein-Barr Virus Lytic Program Concomitantly Induce Apoptosis, but Lytic Gene Expression Protects from Cell Death. J. Virol. 2001, 75, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Oussaief, L.; Hippocrate, A.; Clybouw, C.; Rampanou, A.; Ramirez, V.; Desgranges, C.; Vazquez, A.; Khelifa, R.; Joab, I. Activation of the lytic program of the Epstein-Barr virus in Burkitt’s lymphoma cells leads to a two steps downregulation of expression of the proapoptotic protein BimEL, one of which is EBV-late-gene expression dependent. Virology 2009, 387, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Iwakiri, D.; Yamamoto, K.; Maruo, S.; Kanda, T.; Takada, K. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J. Virol. 2007, 81, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampe, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar] [PubMed]
- Kim, H.; Choi, H.; Lee, S.K. Epstein-Barr Virus MicroRNA miR-BART20–5p Suppresses Lytic Induction by Inhibiting BAD-Mediated caspase-3-Dependent Apoptosis. J. Virol. 2015, 90, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Cahir-McFarland, E.; Zhao, B.; Kieff, E. Virus and cell RNAs expressed during Epstein-Barr virus replication. J. Virol. 2006, 80, 2548–2565. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Price, A.M.; McFadden, K.; Yan, C.M.; Luftig, M.A. Mitogen-induced B-cell proliferation activates Chk2-dependent G1/S cell cycle arrest. PLoS ONE 2014, 9, e87299. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed]
- McFadden, K.; Hafez, A.Y.; Kishton, R.; Messinger, J.E.; Nikitin, P.A.; Rathmell, J.C.; Luftig, M.A. Metabolic stress is a barrier to Epstein-Barr virus-mediated B-cell immortalization. Proc. Natl. Acad. Sci. USA 2016, 113, E782–E790. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J.; Sinclair, A.; Parker, G.; Crawford, D.H.; Farrell, P.J. Epstein-Barr virus efficiently immortalizes human B cells without neutralizing the function of p53. EMBO J. 1995, 14, 1382–1391. [Google Scholar] [PubMed]
- Bernasconi, M.; Ueda, S.; Krukowski, P.; Bornhauser, B.C.; Ladell, K.; Dorner, M.; Sigrist, J.A.; Campidelli, C.; Aslandogmus, R.; Alessi, D.; et al. Early gene expression changes by Epstein-Barr virus infection of B-cells indicate CDKs and survivin as therapeutic targets for post-transplant lymphoproliferative diseases. Int. J. Cancer 2013, 133, 2341–2350. [Google Scholar] [CrossRef] [PubMed]
- Szekely, L.; Pokrovskaja, K.; Jiang, W.Q.; Selivanova, G.; Lowbeer, M.; Ringertz, N.; Wiman, K.G.; Klein, G. Resting B-cells, EBV-infected B-blasts and established lymphoblastoid cell lines differ in their Rb, p53 and EBNA-5 expression patterns. Oncogene 1995, 10, 1869–1874. [Google Scholar] [PubMed]
- Levine, A.J. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: P53. Virology 2009, 384, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Luftig, M.A. MDM2-dependent inhibition of p53 is required for Epstein-Barr virus B-cell growth transformation and infected-cell survival. J. Virol. 2009, 83, 2491–2499. [Google Scholar] [CrossRef] [PubMed]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Yang, K.; El-Naccache, D.W.; Sun, Z.; Robertson, E.S. EBNA3C regulates p53 through induction of Aurora kinase B. Oncotarget 2015, 6, 5788–5803. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Bamidele, A.; Murakami, M.; Robertson, E.S. EBNA3C attenuates the function of p53 through interaction with inhibitor of growth family proteins 4 and 5. J. Virol. 2011, 85, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Kashuba, E.; Yurchenko, M.; Yenamandra, S.P.; Snopok, B.; Szekely, L.; Bercovich, B.; Ciechanover, A.; Klein, G. Epstein-Barr virus-encoded EBNA-5 forms trimolecular protein complexes with MDM2 and p53 and inhibits the transactivating function of p53. Int. J. Cancer 2011, 128, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.L.; Strasser, A. The essential role of evasion from cell death in cancer. Adv. Cancer Res. 2011, 111, 39–96. [Google Scholar] [PubMed]
- Gregory, C.D.; Dive, C.; Henderson, S.; Smith, C.A.; Williams, G.T.; Gordon, J.; Rickinson, A.B. Activation of Epstein-Barr virus latent genes protects human B cells from death by apoptosis. Nature 1991, 349, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Henderson, E.; Miller, G.; Robinson, J.; Heston, L. Efficiency of transformation of lymphocytes by Epstein-Barr virus. Virology 1977, 76, 152–163. [Google Scholar] [CrossRef]
- Sugden, B.; Mark, W. Clonal transformation of adult human leukocytes by Epstein-Barr virus. J. Virol. 1977, 23, 503–508. [Google Scholar] [PubMed]
- Kanda, T.; Furuse, Y.; Oshitani, H.; Kiyono, T. Highly Efficient CRISPR/Cas9-Mediated Cloning and Functional Characterization of Gastric Cancer-Derived Epstein-Barr Virus Strains. J. Virol. 2016, 90, 4383–4393. [Google Scholar] [CrossRef] [PubMed]
- Harris-Arnold, A.; Arnold, C.P.; Schaffert, S.; Hatton, O.; Krams, S.M.; Esquivel, C.O.; Martinez, O.M. Epstein-Barr virus modulates host cell microRNA-194 to promote IL-10 production and B lymphoma cell survival. Am. J. Transplant. 2015, 15, 2814–2824. [Google Scholar] [CrossRef] [PubMed]
- Hatton, O.; Lambert, S.L.; Phillips, L.K.; Vaysberg, M.; Natkunam, Y.; Esquivel, C.O.; Krams, S.M.; Martinez, O.M. Syk-induced phosphatidylinositol-3-kinase activation in Epstein-Barr virus posttransplant lymphoproliferative disorder. Am. J. Transplant. 2013, 13, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, M.R.; Reineke, T.; Rince, P.; Schuffler, P.; El Mchichi, B.; Fabre, M.; Jacquemin, E.; Durrbach, A.; Samuel, D.; Joab, I.; et al. Epstein-Barr virus infection and altered control of apoptotic pathways in posttransplant lymphoproliferative disorders. Pathobiology 2013, 80, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Magrath, I. Epidemiology: Clues to the pathogenesis of Burkitt lymphoma. Br. J. Haematol. 2012, 156, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Levine, P.H.; Kamaraju, L.S.; Connelly, R.R.; Berard, C.W.; Dorfman, R.F.; Magrath, I.; Easton, J.M. The American Burkitt’s Lymphoma Registry: Eight years’ experience. Cancer 1982, 49, 1016–1022. [Google Scholar] [CrossRef]
- Araujo, I.; Foss, H.D.; Bittencourt, A.; Hummel, M.; Demel, G.; Mendonca, N.; Herbst, H.; Stein, H. Expression of Epstein-Barr virus-gene products in Burkitt’s lymphoma in Northeast Brazil. Blood 1996, 87, 5279–5286. [Google Scholar] [PubMed]
- Queiroga, E.M.; Gualco, G.; Weiss, L.M.; Dittmer, D.P.; Araujo, I.; Klumb, C.E.; Harrington, W.J., Jr.; Bacchi, C.E. Burkitt lymphoma in Brazil is characterized by geographically distinct clinicopathologic features. Am. J. Clin. Pathol. 2008, 130, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Manolov, G.; Manolova, Y. Marker band in one chromosome 14 from Burkitt lymphomas. Nature 1972, 237, 33–34. [Google Scholar] [CrossRef] [PubMed]
- Zech, L.; Haglund, U.; Nilsson, K.; Klein, G. Characteristic chromosomal abnormalities in biopsies and lymphoid-cell lines from patients with Burkitt and non-Burkitt lymphomas. Int. J. Cancer 1976, 17, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Gerondakis, S.; Webb, E.; Corcoran, L.M.; Cory, S. Cellular myc oncogene is altered by chromosome translocation to an immunoglobulin locus in murine plasmacytomas and is rearranged similarly in human Burkitt lymphomas. Proc. Natl. Acad. Sci. USA 1983, 80, 1982–1986. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Harris, A.W.; Pinkert, C.A.; Corcoran, L.M.; Alexander, W.S.; Cory, S.; Palmiter, R.D.; Brinster, R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 1985, 318, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.V. The role of c-myc in cellular growth control. Oncogene 1999, 18, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Pelengaris, S.; Khan, M.; Evan, G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Gaidano, G.; Ballerini, P.; Gong, J.Z.; Inghirami, G.; Neri, A.; Newcomb, E.W.; Magrath, I.T.; Knowles, D.M.; Dalla-Favera, R. p53 mutations in human lymphoid malignancies: Association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 1991, 88, 5413–5417. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J.; Allan, G.J.; Shanahan, F.; Vousden, K.H.; Crook, T. p53 is frequently mutated in Burkitt’s lymphoma cell lines. EMBO J. 1991, 10, 2879–2887. [Google Scholar] [PubMed]
- Vousden, K.H.; Crook, T.; Farrell, P.J. Biological activities of p53 mutants in Burkitt’s lymphoma cells. J. Gen. Virol. 1993, 74, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Cherney, B.W.; Bhatia, K.G.; Sgadari, C.; Gutierrez, M.I.; Mostowski, H.; Pike, S.E.; Gupta, G.; Magrath, I.T.; Tosato, G. Role of the p53 tumor suppressor gene in the tumorigenicity of Burkitt’s lymphoma cells. Cancer Res. 1997, 57, 2508–2515. [Google Scholar] [PubMed]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.S.; Klangby, U.; Wiman, K.G. p14ARF homozygous deletion or MDM2 overexpression in Burkitt lymphoma lines carrying wild type p53. Oncogene 2001, 20, 2171–2177. [Google Scholar] [CrossRef] [PubMed]
- Egle, A.; Harris, A.W.; Bouillet, P.; Cory, S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 6164–6169. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Woo, D.; Roussel, M.F.; Cleveland, J.L. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol. 2001, 21, 5063–5070. [Google Scholar] [CrossRef] [PubMed]
- Maclean, K.H.; Keller, U.B.; Rodriguez-Galindo, C.; Nilsson, J.A.; Cleveland, J.L. c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol. Cell. Biol. 2003, 23, 7256–7270. [Google Scholar] [CrossRef] [PubMed]
- Juin, P.; Hunt, A.; Littlewood, T.; Griffiths, B.; Swigart, L.B.; Korsmeyer, S.; Evan, G. c-Myc functionally cooperates with Bax to induce apoptosis. Mol. Cell. Biol. 2002, 22, 6158–6169. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.O.; Ricci, M.S.; Miyashita, T.; Dicker, D.T.; Jin, Z.; Reed, J.C.; El-Deiry, W.S. Bax is a transcriptional target and mediator of c-myc-induced apoptosis. Cancer Res. 2000, 60, 6318–6325. [Google Scholar] [PubMed]
- Michalak, E.M.; Jansen, E.S.; Happo, L.; Cragg, M.S.; Tai, L.; Smyth, G.K.; Strasser, A.; Adams, J.M.; Scott, C.L. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009, 16, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Happo, L.; Cragg, M.S.; Phipson, B.; Haga, J.M.; Jansen, E.S.; Herold, M.J.; Dewson, G.; Michalak, E.M.; Vandenberg, C.J.; Smyth, G.K.; et al. Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood 2010, 116, 5256–5267. [Google Scholar] [CrossRef] [PubMed]
- Garrison, S.P.; Jeffers, J.R.; Yang, C.; Nilsson, J.A.; Hall, M.A.; Rehg, J.E.; Yue, W.; Yu, J.; Zhang, L.; Onciu, M.; et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol. Cell. Biol. 2008, 28, 5391–5402. [Google Scholar] [CrossRef] [PubMed]
- Piazza, R.; Magistroni, V.; Mogavero, A.; Andreoni, F.; Ambrogio, C.; Chiarle, R.; Mologni, L.; Bachmann, P.S.; Lock, R.B.; Collini, P.; et al. Epigenetic silencing of the proapoptotic gene BIM in anaplastic large cell lymphoma through an MeCP2/SIN3a deacetylating complex. Neoplasia 2013, 15, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S.; et al. Distinct Viral and Mutational Spectrum of Endemic Burkitt Lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Juopperi, T. The great MYC escape in tumorigenesis. Cancer Cell 2005, 8, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef] [PubMed]
- Hemann, M.T.; Bric, A.; Teruya-Feldstein, J.; Herbst, A.; Nilsson, J.A.; Cordon-Cardo, C.; Cleveland, J.L.; Tansey, W.P.; Lowe, S.W. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 2005, 436, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Kaymaz, Y.; Oduor, C.I.; Yu, H.; Otieno, J.A.; Ong’echa, J.M.; Moormann, A.M.; Bailey, J.A. Comprehensive Transcriptome and Mutational Profiling of Endemic Burkitt Lymphoma Reveals EBV Type-Specific Differences. Mol. Cancer Res. 2017, 15, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.; Fitzsimmons, L.; Bell, A.I. Epstein-Barr virus and Burkitt lymphoma. Chin. J. Cancer 2014, 33, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.; Komano, J.; Sugden, B. Epstein-Barr virus provides a survival factor to Burkitt’s lymphomas. Proc. Natl. Acad. Sci. USA 2003, 100, 14269–14274. [Google Scholar] [CrossRef] [PubMed]
- Nasimuzzaman, M.; Kuroda, M.; Dohno, S.; Yamamoto, T.; Iwatsuki, K.; Matsuzaki, S.; Mohammad, R.; Kumita, W.; Mizuguchi, H.; Hayakawa, T.; et al. Eradication of Epstein-Barr virus episome and associated inhibition of infected tumor cell growth by adenovirus vector-mediated transduction of dominant-negative EBNA1. Mol. Ther. 2005, 11, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Amato, T.; Abate, F.; Piccaluga, P.; Iacono, M.; Fallerini, C.; Renieri, A.; De Falco, G.; Ambrosio, M.R.; Mourmouras, V.; Ogwang, M.; et al. Clonality Analysis of Immunoglobulin Gene Rearrangement by Next-Generation Sequencing in Endemic Burkitt Lymphoma Suggests Antigen Drive Activation of BCR as Opposed to Sporadic Burkitt Lymphoma. Am. J. Clin. Pathol. 2016, 145, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Bellan, C.; Lazzi, S.; Hummel, M.; Palummo, N.; de Santi, M.; Amato, T.; Nyagol, J.; Sabattini, E.; Lazure, T.; Pileri, S.A.; et al. Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphomas. Blood 2005, 106, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Capello, D.; Scandurra, M.; Poretti, G.; Rancoita, P.M.; Mian, M.; Gloghini, A.; Deambrogi, C.; Martini, M.; Rossi, D.; Greiner, T.C.; et al. Genome wide DNA-profiling of HIV-related B-cell lymphomas. Br. J. Haematol. 2010, 148, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Shiramizu, B.; McGrath, M.S. Molecular pathogenesis of AIDS-associated non-Hodgkin’s lymphoma. Hematol. Oncol. Clin. N. Am. 1991, 5, 323–330. [Google Scholar]
- Pelicci, P.G.; Knowles, D.M., 2nd; Magrath, I.; Dalla-Favera, R. Chromosomal breakpoints and structural alterations of the c-myc locus differ in endemic and sporadic forms of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1986, 83, 2984–2988. [Google Scholar] [CrossRef] [PubMed]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Tanabe-Tochikura, A.; Kuroiwa, Y.; Takada, K. Isolation of Epstein-Barr virus (EBV)-negative cell clones from the EBV-positive Burkitt’s lymphoma (BL) line Akata: Malignant phenotypes of BL cells are dependent on EBV. J. Virol. 1994, 68, 6069–6073. [Google Scholar] [PubMed]
- Komano, J.; Sugiura, M.; Takada, K. Epstein-Barr virus contributes to the malignant phenotype and to apoptosis resistance in Burkitt’s lymphoma cell line Akata. J. Virol. 1998, 72, 9150–9156. [Google Scholar] [PubMed]
- Chodosh, J.; Holder, V.P.; Gan, Y.J.; Belgaumi, A.; Sample, J.; Sixbey, J.W. Eradication of latent Epstein-Barr virus by hydroxyurea alters the growth-transformed cell phenotype. J. Infect. Dis. 1998, 177, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Kirchmaier, A.L.; Sugden, B. Dominant-negative inhibitors of EBNA-1 of Epstein-Barr virus. J. Virol. 1997, 71, 1766–1775. [Google Scholar] [PubMed]
- Vereide, D.; Sugden, B. Proof for EBV’s sustaining role in Burkitt’s lymphomas. Semin. Cancer Biol. 2009, 19, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Vereide, D.T.; Sugden, B. Lymphomas differ in their dependence on Epstein-Barr virus. Blood 2011, 117, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, L.; Boyce, A.J.; Wei, W.; Chang, C.; Croom-Carter, D.; Tierney, R.J.; Herold, M.J.; Bell, A.I.; Strasser, A.; Kelly, G.L.; et al. Coordinated repression of BIM and PUMA by Epstein-Barr virus latent genes maintains the survival of Burkitt lymphoma cells. Cell Death Differ. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ruf, I.K.; Rhyne, P.W.; Yang, H.; Borza, C.M.; Hutt-Fletcher, L.M.; Cleveland, J.L.; Sample, J.T. Epstein-barr virus regulates c-MYC, apoptosis, and tumorigenicity in Burkitt lymphoma. Mol. Cell. Biol. 1999, 19, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Komano, J.; Maruo, S.; Kurozumi, K.; Oda, T.; Takada, K. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata. J. Virol. 1999, 73, 9827–9831. [Google Scholar] [PubMed]
- Ruf, I.K.; Rhyne, P.W.; Yang, C.; Cleveland, J.L.; Sample, J.T. Epstein-Barr virus small RNAs potentiate tumorigenicity of Burkitt lymphoma cells independently of an effect on apoptosis. J. Virol. 2000, 74, 10223–10228. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Longnecker, R. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J. Virol. 2004, 78, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Amick, A.C.; Longnecker, R. Epstein-Barr virus LMP2A bypasses p53 inactivation in a MYC model of lymphomagenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 17945–17950. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Swanson-Mungerson, M.; Amick, A.C.; Longnecker, R. Epstein-Barr virus in Burkitt’s lymphoma: A role for latent membrane protein 2A. Cell Cycle 2010, 9, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.I.; Groves, K.; Kelly, G.L.; Croom-Carter, D.; Hui, E.; Chan, A.T.; Rickinson, A.B. Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt’s lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays. J. Gen. Virol. 2006, 87, 2885–2890. [Google Scholar] [CrossRef] [PubMed]
- Tao, Q.; Robertson, K.D.; Manns, A.; Hildesheim, A.; Ambinder, R.F. Epstein-Barr virus (EBV) in endemic Burkitt’s lymphoma: Molecular analysis of primary tumor tissue. Blood 1998, 91, 1373–1381. [Google Scholar] [PubMed]
- Xue, S.A.; Labrecque, L.G.; Lu, Q.L.; Ong, S.K.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; Griffin, B.E. Promiscuous expression of Epstein-Barr virus genes in Burkitt’s lymphoma from the central African country Malawi. Int. J. Cancer 2002, 99, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.Y.; Siu, K.L.; Kok, K.H.; Lung, R.W.; Tsang, C.M.; To, K.F.; Kwong, D.L.; Tsao, S.W.; Jin, D.Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Marquitz, A.R.; Mathur, A.; Nam, C.S.; Raab-Traub, N. The Epstein-Barr Virus BART microRNAs target the pro-apoptotic protein Bim. Virology 2011, 412, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Pimienta, G.; Fok, V.; Haslip, M.; Nagy, M.; Takyar, S.; Steitz, J.A. Proteomics and Transcriptomics of BJAB Cells Expressing the Epstein-Barr Virus Noncoding RNAs EBER1 and EBER2. PLoS ONE 2015, 10, e0124638. [Google Scholar] [CrossRef] [PubMed]
- Coloff, J.L.; Mason, E.F.; Altman, B.J.; Gerriets, V.A.; Liu, T.; Nichols, A.N.; Zhao, Y.; Wofford, J.A.; Jacobs, S.R.; Ilkayeva, O.; et al. Akt requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J. Biol. Chem. 2011, 286, 5921–5933. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Guo, B.; Kang, J.; Deng, X.; Fan, Y.; Zhang, X.; Ai, K. Downregulation of Smurf2 ubiquitin ligase in pancreatic cancer cells reversed TGF-beta-induced tumor formation. Tumour Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; Carter, M.J.; O’Brien, D.I.; Clark, L.J.; Yu, J.; Michalak, E.M.; Happo, L.; Cragg, M.S.; Inman, G.J. Transforming Growth Factor-β directly induces PUMA during the rapid induction of apoptosis in Myc-driven B-cell lymphomas. J. Biol. Chem. 2013, 288, 5198–5219. [Google Scholar] [CrossRef] [PubMed]
- Flavell, J.R.; Baumforth, K.R.; Wood, V.H.; Davies, G.L.; Wei, W.; Reynolds, G.M.; Morgan, S.; Boyce, A.; Kelly, G.L.; Young, L.S.; et al. Down-regulation of the TGF-beta target gene, PTPRK, by the Epstein-Barr virus encoded EBNA1 contributes to the growth and survival of Hodgkin lymphoma cells. Blood 2008, 111, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.L.; Milner, A.E.; Baldwin, G.S.; Bell, A.I.; Rickinson, A.B. Three restricted forms of Epstein-Barr virus latency counteracting apoptosis in c-myc-expressing Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 14935–14940. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.; Bell, A.; Rickinson, A. Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat. Med. 2002, 8, 1098–1104. [Google Scholar] [CrossRef] [PubMed]
- Obexer, P.; Hagenbuchner, J.; Rupp, M.; Salvador, C.; Holzner, M.; Deutsch, M.; Porto, V.; Kofler, R.; Unterkircher, T.; Ausserlechner, M.J. p16INK4A sensitizes human leukemia cells to FAS- and glucocorticoid-induced apoptosis via induction of BBC3/Puma and repression of MCL1 and BCL2. J. Biol. Chem. 2009, 284, 30933–30940. [Google Scholar] [CrossRef] [PubMed]
- Garibal, J.; Hollville, E.; Bell, A.I.; Kelly, G.L.; Renouf, B.; Kawaguchi, Y.; Rickinson, A.B.; Wiels, J. Truncated form of the Epstein-Barr virus protein EBNA-LP protects against caspase-dependent apoptosis by inhibiting protein phosphatase 2A. J. Virol. 2007, 81, 7598–7607. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar]
- Ribeiro, J.; Oliveira, C.; Malta, M.; Sousa, H. Epstein-Barr virus gene expression and latency pattern in gastric carcinomas: A systematic review. Future Oncol. 2017, 13, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lin, Z.; Wu, Y.; Dong, J.; Zhao, B.; Cheng, Y.; Huang, P.; Xu, L.; Xia, T.; Xiong, D.; et al. Comprehensive profiling of EBV gene expression in nasopharyngeal carcinoma through paired-end transcriptome sequencing. Front. Med. 2016, 10, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Sun, P.; Zhang, Y.; Yan, L.; Luo, B. Expression of c-myc and PCNA in Epstein-Barr virus-associated gastric carcinoma. Exp. Ther. Med. 2013, 5, 1030–1034. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zur Hausen, A.; Brink, A.A.; Craanen, M.E.; Middeldorp, J.M.; Meijer, C.J.; van den Brule, A.J. Unique transcription pattern of Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: Expression of the transforming BARF1 gene. Cancer Res. 2000, 60, 2745–2748. [Google Scholar] [PubMed]
- Cabras, G.; Decaussin, G.; Zeng, Y.; Djennaoui, D.; Melouli, H.; Broully, P.; Bouguermouh, A.M.; Ooka, T. Epstein-Barr virus encoded BALF1 gene is transcribed in Burkitt’s lymphoma cell lines and in nasopharyngeal carcinoma’s biopsies. J. Clin. Virol. 2005, 34, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Strockbine, L.D.; Cohen, J.I.; Farrah, T.; Lyman, S.D.; Wagener, F.; DuBose, R.F.; Armitage, R.J.; Spriggs, M.K. The Epstein-Barr virus BARF1 gene encodes a novel, soluble colony-stimulating factor-1 receptor. J. Virol. 1998, 72, 4015–4021. [Google Scholar] [PubMed]
- Hoebe, E.K.; Le Large, T.Y.; Tarbouriech, N.; Oosterhoff, D.; De Gruijl, T.D.; Middeldorp, J.M.; Greijer, A.E. Epstein-Barr virus-encoded BARF1 protein is a decoy receptor for macrophage colony stimulating factor and interferes with macrophage differentiation and activation. Viral. Immunol. 2012, 25, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, R. Cross-talk between Epstein-Barr virus and microenvironment in the pathogenesis of lymphomas. Semin. Cancer Biol. 2015, 34, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Concha, M.; Wang, X.; Cao, S.; Baddoo, M.; Fewell, C.; Lin, Z.; Hulme, W.; Hedges, D.; McBride, J.; Flemington, E.K. Identification of new viral genes and transcript isoforms during Epstein-Barr virus reactivation using RNA-Seq. J. Virol. 2012, 86, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.P.; Haigh, T.A.; Taylor, G.S.; Long, H.M.; Lee, S.P.; Shannon-Lowe, C.; O’Connor, S.; Bollard, C.M.; Iqbal, J.; Chan, W.C.; et al. A novel latent membrane 2 transcript expressed in Epstein-Barr virus-positive NK- and T-cell lymphoproliferative disease encodes a target for cellular immunotherapy. Blood 2010, 116, 3695–3704. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.S.; Chan, C.P.; Kok, K.H.; Jin, D.Y. Mutagenesis and Genome Engineering of Epstein-Barr Virus in Cultured Human Cells by CRISPR/Cas9. Methods Mol. Biol. 2017, 1498, 23–31. [Google Scholar] [PubMed]
- Pujals, A.; Favre, L.; Pioche-Durieu, C.; Robert, A.; Meurice, G.; Le Gentil, M.; Chelouah, S.; Martin-Garcia, N.; Le Cam, E.; Guettier, C.; et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy 2015, 11, 2275–2287. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 2008, 27, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Brune, W.; Andoniou, C.E. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses 2017, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Veyer, D.L.; Carrara, G.; Maluquer de Motes, C.; Smith, G.L. Vaccinia virus evasion of regulated cell death. Immunol. Lett. 2017, 186, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Kofahi, H.M.; Taylor, N.G.; Hirasawa, K.; Grant, M.D.; Russell, R.S. Hepatitis C Virus Infection of Cultured Human Hepatoma Cells Causes Apoptosis and Pyroptosis in Both Infected and Bystander Cells. Sci. Rep. 2016, 6, 37433. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fitzsimmons, L.; Kelly, G.L. EBV and Apoptosis: The Viral Master Regulator of Cell Fate? Viruses 2017, 9, 339. https://doi.org/10.3390/v9110339
Fitzsimmons L, Kelly GL. EBV and Apoptosis: The Viral Master Regulator of Cell Fate? Viruses. 2017; 9(11):339. https://doi.org/10.3390/v9110339
Chicago/Turabian StyleFitzsimmons, Leah, and Gemma L. Kelly. 2017. "EBV and Apoptosis: The Viral Master Regulator of Cell Fate?" Viruses 9, no. 11: 339. https://doi.org/10.3390/v9110339
APA StyleFitzsimmons, L., & Kelly, G. L. (2017). EBV and Apoptosis: The Viral Master Regulator of Cell Fate? Viruses, 9(11), 339. https://doi.org/10.3390/v9110339