Gammaherpesviral Tegument Proteins, PML-Nuclear Bodies and the Ubiquitin-Proteasome System


Abstract
1. Gammaherpesviruses
2. The Ubiquitin-Proteasome System
3. PML Nuclear Bodies
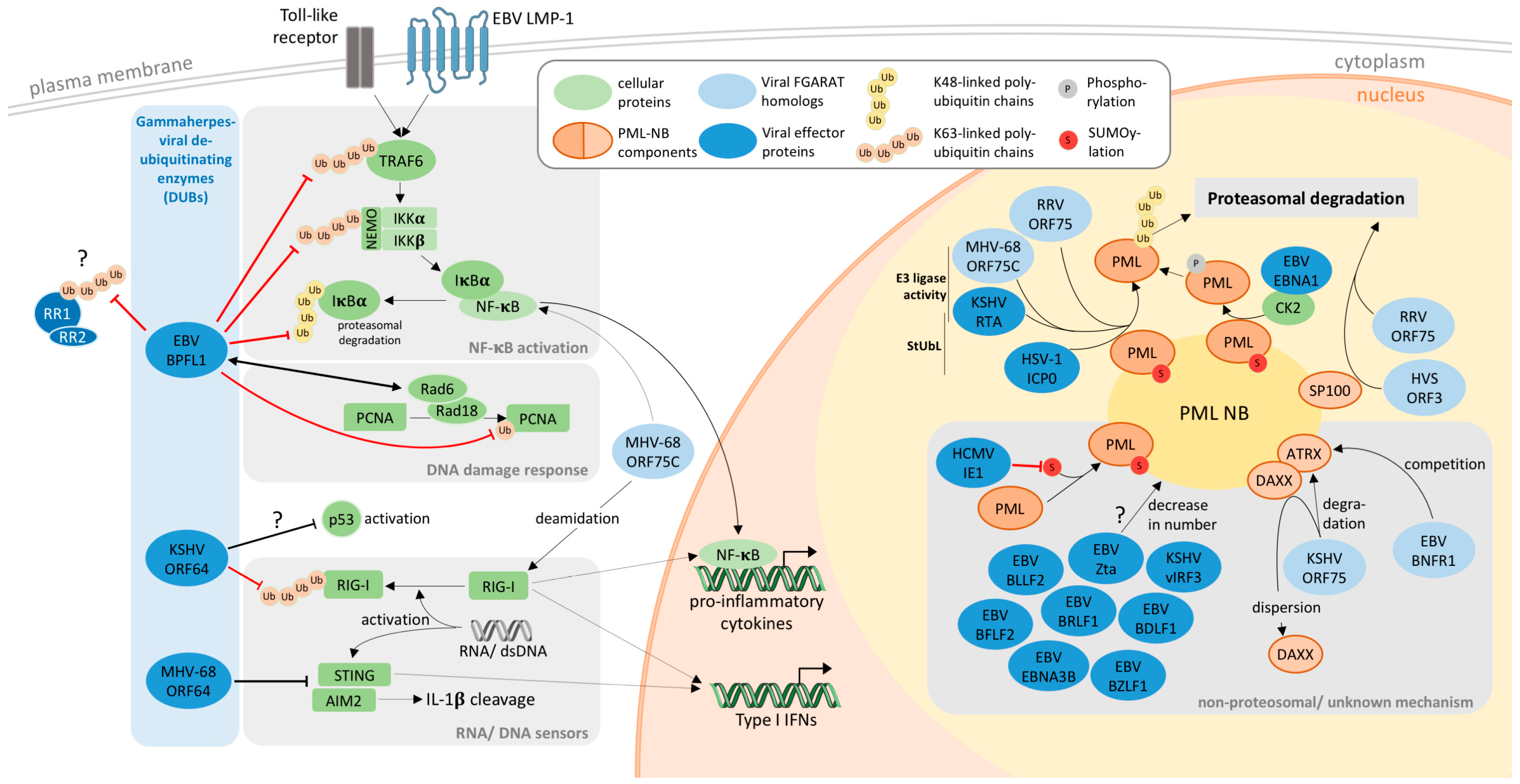
4. PML-NBs as Targets of Gamma-Herpesviral FGARATs
5. Gammaherpesviral DUBs
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gandhi, M.K. Epstein-Barr virus-associated lymphomas. Expert Rev. Anti. Infect. Ther. 2014. [Google Scholar] [CrossRef] [PubMed]
- Mariggio, G.; Koch, S.; Schulz, T.F. Kaposi sarcoma herpesvirus pathogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Dupin, N.; Grandadam, M.; Calvez, V.; Gorin, I.; Aubin, J.T.; Havard, S.; Lamy, F.; Leibowitch, M.; Huraux, J.M.; Escande, J.P.; et al. Herpesvirus-like DNA sequences in patients with mediterranean kaposi’s sarcoma. Lancet 1995, 345, 761–762. [Google Scholar] [CrossRef]
- Ganem, D. KSHV and the pathogenesis of kaposi sarcoma: Listening to human biology and medicine. J. Clin. Investig. 2010, 120, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Wood, W.A.; Lee, S.J.; Shea, T.C.; Naresh, K.N.; Kazembe, P.N.; Casper, C.; Hesseling, P.B.; Mitsuyasu, R.T. Meeting the challenge of hematologic malignancies in sub-Saharan Africa. Blood 2012, 119, 5078–5087. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Stamminger, T. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 2008, 1783, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Stamminger, T. Emerging role of PML nuclear bodies in innate immune signaling. J. Virol. 2016, 90, 5850–5854. [Google Scholar] [CrossRef] [PubMed]
- Korioth, F.; Maul, G.G.; Plachter, B.; Stamminger, T.; Frey, J. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp. Cell Res. 1996, 229, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Maul, G.G.; Everett, R.D. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 1994, 75, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Cuchet-Lourenco, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential sumo targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef] [PubMed]
- Cuchet-Lourenco, D.; Vanni, E.; Glass, M.; Orr, A.; Everett, R.D. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with pml isoform I and induces its sumo-independent degradation. J. Virol. 2012, 86, 11209–11222. [Google Scholar] [CrossRef] [PubMed]
- Schilling, E.M.; Scherer, M.; Reuter, N.; Schweininger, J.; Muller, Y.A.; Stamminger, T. The human cytomegalovirus IE1 protein antagonizes PML nuclear body-mediated intrinsic immunity via the inhibition of PML de novo SUMOylation. J. Virol. 2017, 91, e02049-16. [Google Scholar] [CrossRef] [PubMed]
- Adamson, A.L.; Kenney, S. Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 2001, 75, 2388–2399. [Google Scholar] [CrossRef] [PubMed]
- Bowling, B.L.; Adamson, A.L. Functional interactions between the Epstein-Barr virus BZLF1 protein and the promyelocytic leukemia protein. Virus Res. 2006, 117, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Zimmerman, N.; Chen, T.; Domagala, M.; Frappier, L. Genome-wide screen of three herpesviruses for protein subcellular localization and alteration of PML nuclear bodies. PLoS Pathog. 2008, 4, e1000100. [Google Scholar] [CrossRef] [PubMed]
- Scaglioni, P.P.; Yung, T.M.; Cai, L.F.; Erdjument-Bromage, H.; Kaufman, A.J.; Singh, B.; Teruya-Feldstein, J.; Tempst, P.; Pandolfi, P.P. A CK2-dependent mechanism for degradation of the pml tumor suppressor. Cell 2006, 126, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Sivachandran, N.; Cao, J.Y.; Frappier, L. Epstein-Barr virus nuclear antigen 1 Hijacks the host kinase CK2 to disrupt PML nuclear bodies. J. Virol. 2010, 84, 11113–11123. [Google Scholar] [CrossRef] [PubMed]
- Ling, P.D.; Tan, J.; Sewatanon, J.; Peng, R. Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J. Virol. 2008, 82, 8000–8012. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, M.; Gill, M.B.; Losing, J.B.; May, J.S.; Stevenson, P.G. Multiple functions for ORF75c in murid herpesvirus-4 infection. PLoS ONE 2008, 3, e2781. [Google Scholar] [CrossRef] [PubMed]
- Sewatanon, J.; Ling, P.D. Murine gammaherpesvirus 68 ORF75c contains ubiquitin E3 ligase activity and requires PML SUMOylation but not other known cellular PML regulators, CK2 and E6AP, to mediate pml degradation. Virology 2013, 440, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Ensser, A.; Pflanz, R.; Fleckenstein, B. Primary structure of the alcelaphine herpesvirus 1 genome. J. Virol. 1997, 71, 6517–6525. [Google Scholar] [PubMed]
- Full, F.; Reuter, N.; Zielke, K.; Stamminger, T.; Ensser, A. Herpesvirus saimiri antagonizes nuclear domain 10-instituted intrinsic immunity via an ORF3-mediated selective degradation of cellular protein Sp100. J. Virol. 2012, 86, 3541–3553. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Messick, T.E.; Lieberman, P.M. Disruption of host antiviral resistances by gammaherpesvirus tegument proteins with homology to the fgarat purine biosynthesis enzyme. Curr. Opin. Virol. 2015, 14, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Chan, L.; Gibeault, R.; Conn, K.; Dheekollu, J.; Domsic, J.; Marmorstein, R.; Schang, L.M.; Lieberman, P.M. Viral reprogramming of the Daxx histone H3.3 chaperone during early Epstein-Barr virus infection. J. Virol. 2014, 88, 14350–14363. [Google Scholar] [CrossRef] [PubMed]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Sturzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposi’s sarcoma associated herpesvirus tegument protein orf75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog. 2014, 10, e1003863. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Villar, L.; Campagna, M.; Lopitz-Otsoa, F.; Gallego, P.; Gonzalez-Santamaria, J.; Gonzalez, D.; Rodriguez, M.S.; Rivas, C. Covalent modification by SUMO is required for efficient disruption of PML oncogenic domains by kaposi’s sarcoma-associated herpesvirus latent protein LANA2. J. Gen. Virol. 2011, 92, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Villar, L.; Lopitz-Otsoa, F.; Gallego, P.; Munoz-Fontela, C.; Gonzalez-Santamaria, J.; Campagna, M.; Shou-Jiang, G.; Rodriguez, M.S.; Rivas, C. Kaposi’s sarcoma-associated herpesvirus protein LANA2 disrupts PML oncogenic domains and inhibits pml-mediated transcriptional repression of the survivin gene. J. Virol. 2009, 83, 8849–8858. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Kobayashi, K.; Kim, K.Y.; Pochampalli, M.; Izumiya, C.; Shevchenko, B.; Wang, D.H.; Huerta, S.B.; Martinez, A.; Campbell, M.; et al. Kaposi’s sarcoma-associated herpesvirus k-Rta exhibits SUMO-targeting ubiquitin ligase (STUBL) like activity and is essential for viral reactivation. PLoS Pathog. 2013, 9, e1003506. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.S.; Grosskopf, A.K.; Jungnickl, D.; Scholz, B.; Ensser, A. Viral FGARAT homolog ORF75 of rhesus monkey rhadinovirus effects proteasomal degradation of the ND10 components SP100 and PML. J. Virol. 2016, 90, 8013–8028. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Deng, Z.; Vladimirova, O.; Wiedmer, A.; Lu, F.; Lieberman, P.M.; Patel, D.J. Structural basis underlying viral hijacking of a histone chaperone complex. Nat. Commun. 2016, 7, 12707. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Neuhierl, B.; Baldwin, G.; Bannert, H.; Hub, B.; Mautner, J.; Behrends, U.; Delecluse, H.J. Epstein-Barr virus bnrf1 protein allows efficient transfer from the endosomal compartment to the nucleus of primary b lymphocytes. J. Virol. 2006, 80, 9435–9443. [Google Scholar] [CrossRef] [PubMed]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer 2007, 7, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Y.; Ahn, J.H.; Alcendor, D.J.; Jang, W.J.; Xiao, J.; Hayward, S.D.; Hayward, G.S. Origin-independent assembly of kaposi’s sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-k8 protein and cellular pml. J. Virol. 2001, 75, 1487–1506. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Y.; Wang, X.; Zhao, R.; Wang, Y. Modulation of global sumoylation by kaposi’s sarcoma-associated herpesvirus and its effects on viral gene expression. J. Med. Virol. 2017, 89, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- Rozen, R.; Sathish, N.; Li, Y.; Yuan, Y. Virion-wide protein interactions of kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008, 82, 4742–4750. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.H.; Zagha, E.; Serrano, O.K.; Ku, C.C.; Zerboni, L.; Baiker, A.; Santos, R.; Spengler, M.; Lynch, J.; Grose, C.; et al. Mutational analysis of the repeated open reading frames, ORFS 63 and 70 and ORFS 64 and 69, of varicella-zoster virus. J. Virol. 2001, 75, 8224–8239. [Google Scholar] [CrossRef] [PubMed]
- Gredmark-Russ, S.; Isaacson, M.K.; Kattenhorn, L.; Cheung, E.J.; Watson, N.; Ploegh, H.L. A gammaherpesvirus ubiquitin-specific protease is involved in the establishment of murine gammaherpesvirus 68 infection. J. Virol. 2009, 83, 10644–10652. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.M.; Wang, L.; Damania, B. Kaposi’s sarcoma-associated herpesvirus encodes a viral deubiquitinase. J. Virol. 2009, 83, 10224–10233. [Google Scholar] [CrossRef] [PubMed]
- Gredmark, S.; Schlieker, C.; Quesada, V.; Spooner, E.; Ploegh, H.L. A functional ubiquitin-specific protease embedded in the large tegument protein (ORF64) of murine gammaherpesvirus 68 is active during the course of infection. J. Virol. 2007, 81, 10300–10309. [Google Scholar] [CrossRef] [PubMed]
- Kattenhorn, L.M.; Korbel, G.A.; Kessler, B.M.; Spooner, E.; Ploegh, H.L. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family herpesviridae. Mol. Cell 2005, 19, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.T.; Oh, S.E.; Lee, Y.O.; Gibson, W.; Ahn, J.H. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J. Virol. 2009, 83, 12046–12056. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, M.; Braem, S.G.; de Jong, A.; Delagic, N.; Peeters, J.G.; Boer, I.G.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef] [PubMed]
- Gastaldello, S.; Chen, X.; Callegari, S.; Masucci, M.G. Caspase-1 promotes epstein-barr virus replication by targeting the large tegument protein deneddylase to the nucleus of productively infected cells. PLoS Pathog. 2013, 9, e1003664. [Google Scholar] [CrossRef] [PubMed]
- Schlieker, C.; Korbel, G.A.; Kattenhorn, L.M.; Ploegh, H.L. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 2005, 79, 15582–15585. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Loveland, A.N.; Kattenhorn, L.M.; Ploegh, H.L.; Gibson, W. High-molecular-weight protein (PUL48) of human cytomegalovirus is a competent deubiquitinating protease: Mutant viruses altered in its active-site cysteine or histidine are viable. J. Virol. 2006, 80, 6003–6012. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qian, C.; Cao, X. Post-translational modification control of innate immunity. Immunity 2016, 45, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhao, J.; Song, S.; He, X.; Minassian, A.; Zhou, Y.; Zhang, J.; Brulois, K.; Wang, Y.; Cabo, J.; et al. Viral pseudo-enzymes activate RIG-I via deamidation to evade cytokine production. Mol. Cell 2015, 58, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Schattgen, S.A.; Pisitkun, P.; Jorgensen, J.P.; Hilterbrand, A.T.; Wang, L.J.; West, J.A.; Hansen, K.; Horan, K.A.; Jakobsen, M.R.; et al. Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J. Immunol. 2015, 194, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Murata, T.; Kanda, T.; Isomura, H.; Narita, Y.; Sugimoto, A.; Kawashima, D.; Tsurumi, T. Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-κB signaling during productive replication. J. Virol. 2013, 87, 4060–4070. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.B.; Ning, S.; Bentz, G.L.; Dufour, F.; Gershburg, E.; Shackelford, J.; Langelier, Y.; Pagano, J.S. The Epstein-Barr virus (EBV) deubiquitinating enzyme bplf1 reduces EBV ribonucleotide reductase activity. J. Virol. 2009, 83, 4345–4353. [Google Scholar] [CrossRef] [PubMed]
- Sewatanon, J.; Ling, P.D. Murine gammaherpesvirus 68 encodes a second PML-modifying protein. J. Virol. 2014, 88, 3591–3597. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Whitehurst, C.B.; Pagano, J.S. The rad6/18 ubiquitin complex interacts with the Epstein-Barr virus deubiquitinating enzyme, BPLF1, and contributes to virus infectivity. J. Virol. 2014, 88, 6411–6422. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.B.; Vaziri, C.; Shackelford, J.; Pagano, J.S. Epstein-Barr virus BPLF1 deubiquitinates PCNA and attenuates polymerase η recruitment to DNA damage sites. J. Virol. 2012, 86, 8097–8106. [Google Scholar] [CrossRef] [PubMed]
- Dyson, O.F.; Pagano, J.S.; Whitehurst, C.B. The translesion polymerase pol η is required for efficient Epstein-Barr virus infectivity and is regulated by the viral deubiquitinating enzyme BPLF1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, P.; Konrad, A.; Jochmann, R.; Lausen, B.; Holz, P.; Naschberger, E.; Neipel, F.; Britzen-Laurent, N.; Sturzl, M. Structural proteins of kaposi’s sarcoma-associated herpesvirus antagonize p53-mediated apoptosis. Oncogene 2015, 34, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Gastaldello, S.; Hildebrand, S.; Faridani, O.; Callegari, S.; Palmkvist, M.; Di Guglielmo, C.; Masucci, M.G. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-ring ligases. Nat. Cell Biol. 2010, 12, 351–361. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Virus | Protein | PML-NB Target | Mechanism | Reference |
|---|---|---|---|---|
| Epstein-Barr virus | BNRF1 | ATRX, DAXX | disruption of DAXX/ATRX binding | [27,28] |
| Epstein-Barr virus | EBNA1 | PML | proteasomal degradation | [20] |
| Epstein-Barr virus | BZLF1 | PML | redistribution | [16,17] |
| Kaposi’s sarcoma-associated herpesvirus | ORF75 | ATRX, DAXX | unknown, redistribution | [29] |
| Kaposi’s sarcoma-associated herpesvirus | vIRF3 | PML | redistribution | [30,31] |
| Kaposi’s sarcoma-associated herpesvirus | RTA | PML | proteasomal degradation | [32] |
| Rhesus Rhadinovirus | ORF75 | SP100, PML | proteasomal degradation | [33] |
| Herpesvirus saimiri | ORF3 | SP100 | proteasomal degradation | [25] |
| Murine herpesvirus 68 | ORF75C | PML | proteasomal degradation | [21,22,23] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Full, F.; Hahn, A.S.; Großkopf, A.K.; Ensser, A. Gammaherpesviral Tegument Proteins, PML-Nuclear Bodies and the Ubiquitin-Proteasome System. Viruses 2017, 9, 308. https://doi.org/10.3390/v9100308
Full F, Hahn AS, Großkopf AK, Ensser A. Gammaherpesviral Tegument Proteins, PML-Nuclear Bodies and the Ubiquitin-Proteasome System. Viruses. 2017; 9(10):308. https://doi.org/10.3390/v9100308
Chicago/Turabian StyleFull, Florian, Alexander S. Hahn, Anna K. Großkopf, and Armin Ensser. 2017. "Gammaherpesviral Tegument Proteins, PML-Nuclear Bodies and the Ubiquitin-Proteasome System" Viruses 9, no. 10: 308. https://doi.org/10.3390/v9100308
APA StyleFull, F., Hahn, A. S., Großkopf, A. K., & Ensser, A. (2017). Gammaherpesviral Tegument Proteins, PML-Nuclear Bodies and the Ubiquitin-Proteasome System. Viruses, 9(10), 308. https://doi.org/10.3390/v9100308