Preclinical Testing of an Oncolytic Parvovirus: Standard Protoparvovirus H-1PV Efficiently Induces Osteosarcoma Cell Lysis In Vitro

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Mammalian Cell Culture
2.3. Viruses and Virus Production
2.4. Detection of Infectious H-1PV Particles
2.5. Western Blot Analysis
2.6. Microscopy
2.7. Viral DNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.8. Cell Viability and Cell Death Assessment
2.9. Flow Cytometric Characterization of the Cell-Cycle Distribution of Cells and of the Sub-G1 Apoptotic Cell Population
3. Results
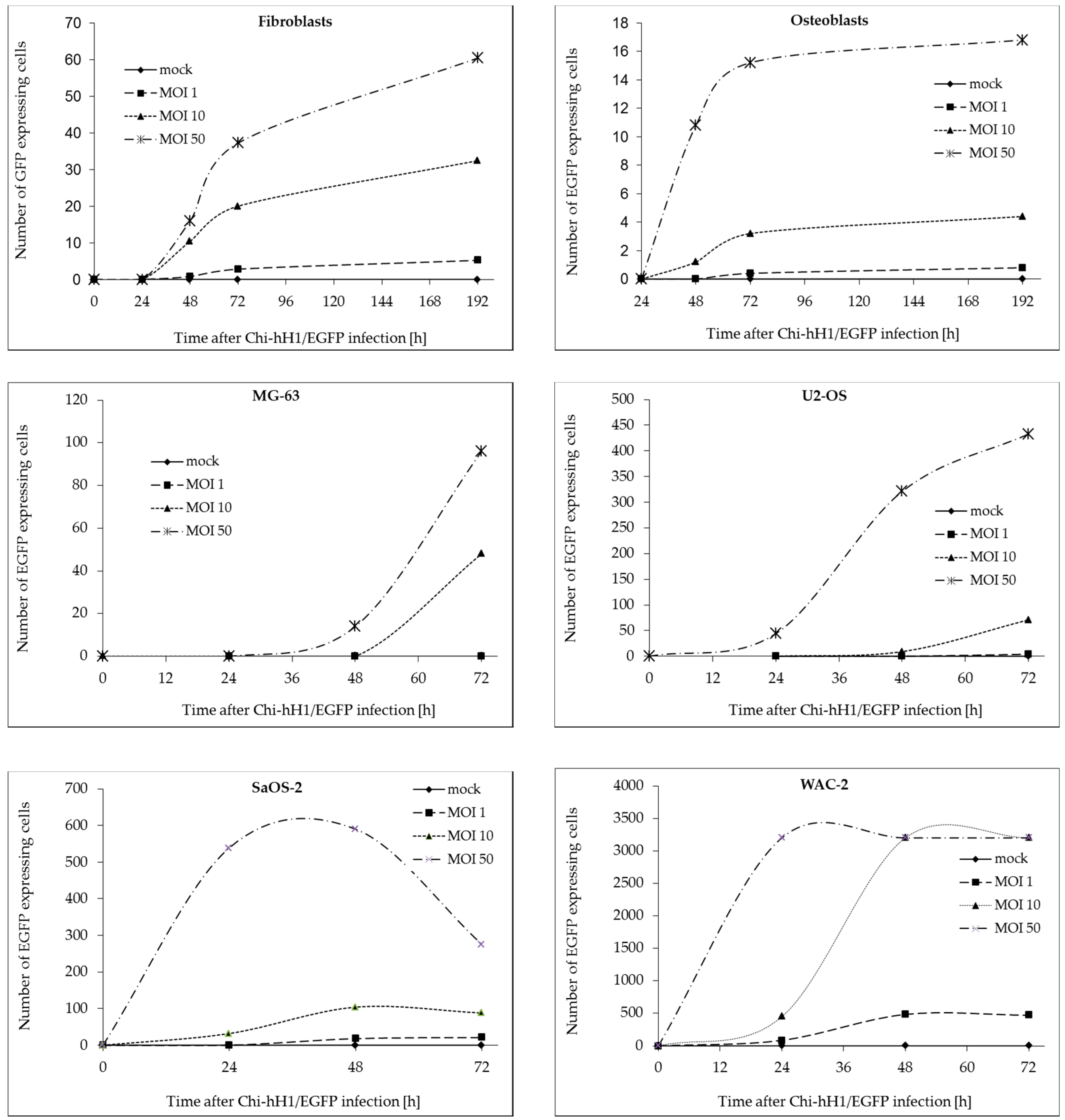
3.1. Protoparvovirus H-1PV Enters and Transduces Non-Transformed and Transformed Mesenchymal Cells
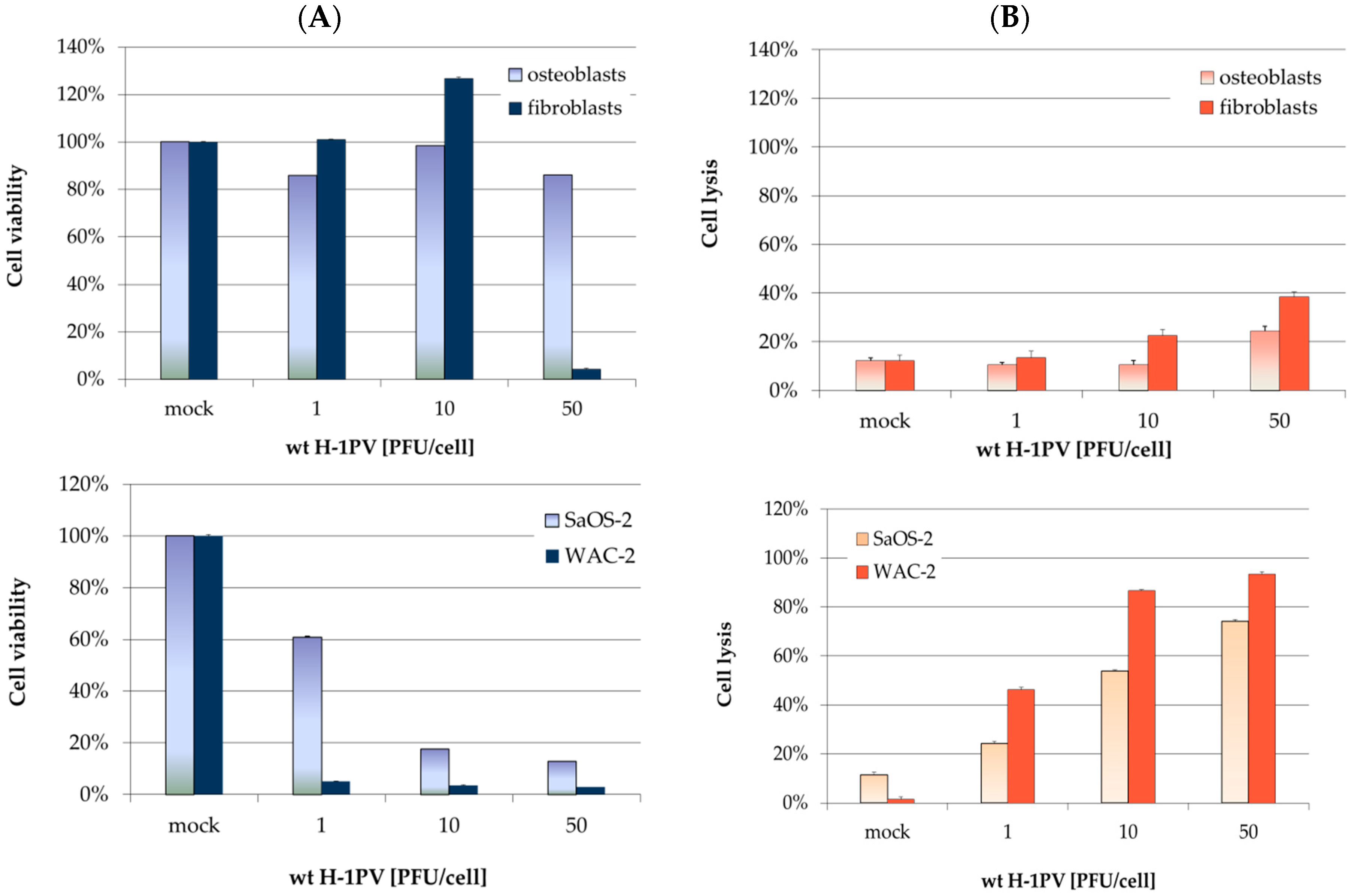
3.2. H-1PV Infection of Non-Transformed Mesenchymal Cells Induces Antiproliferative Effects and Toxicity Only at High Virus Doses
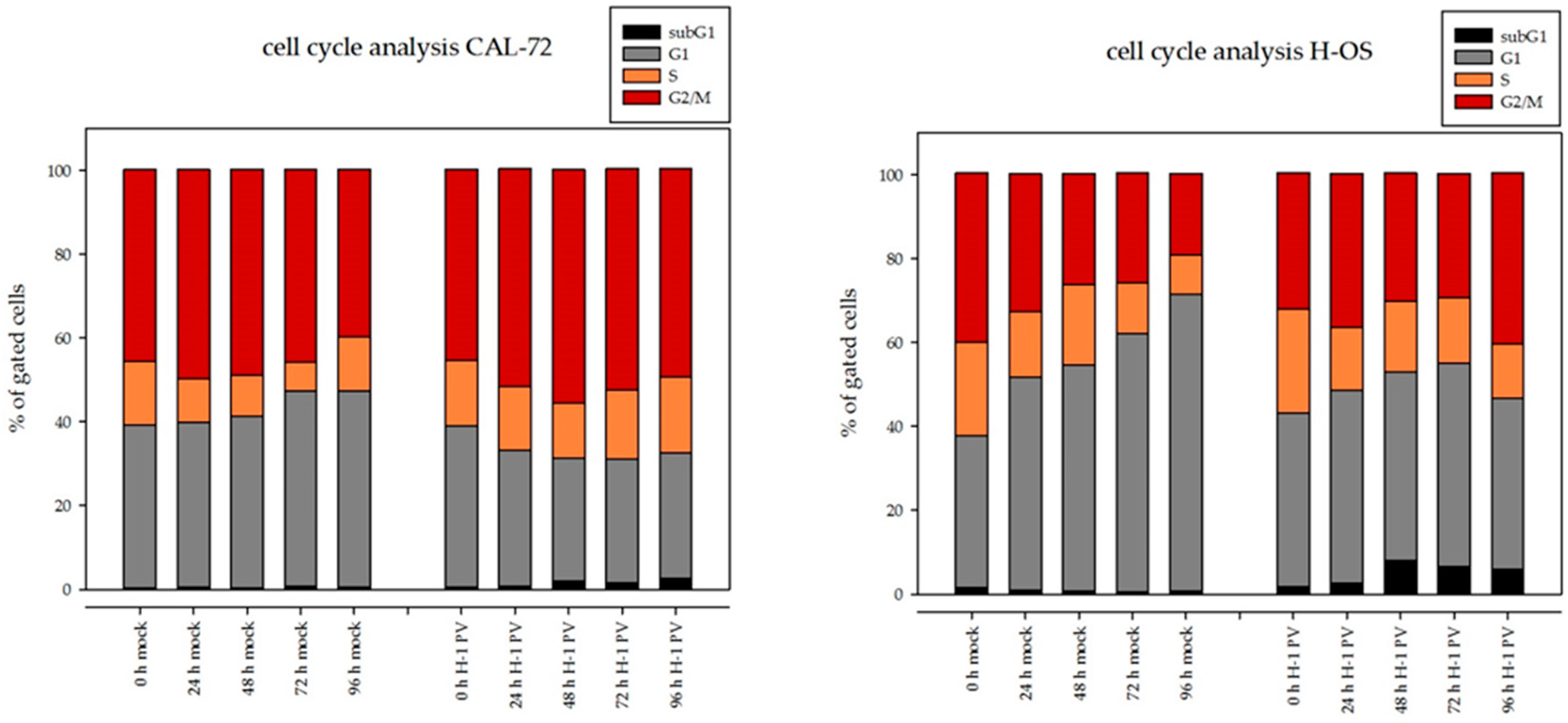
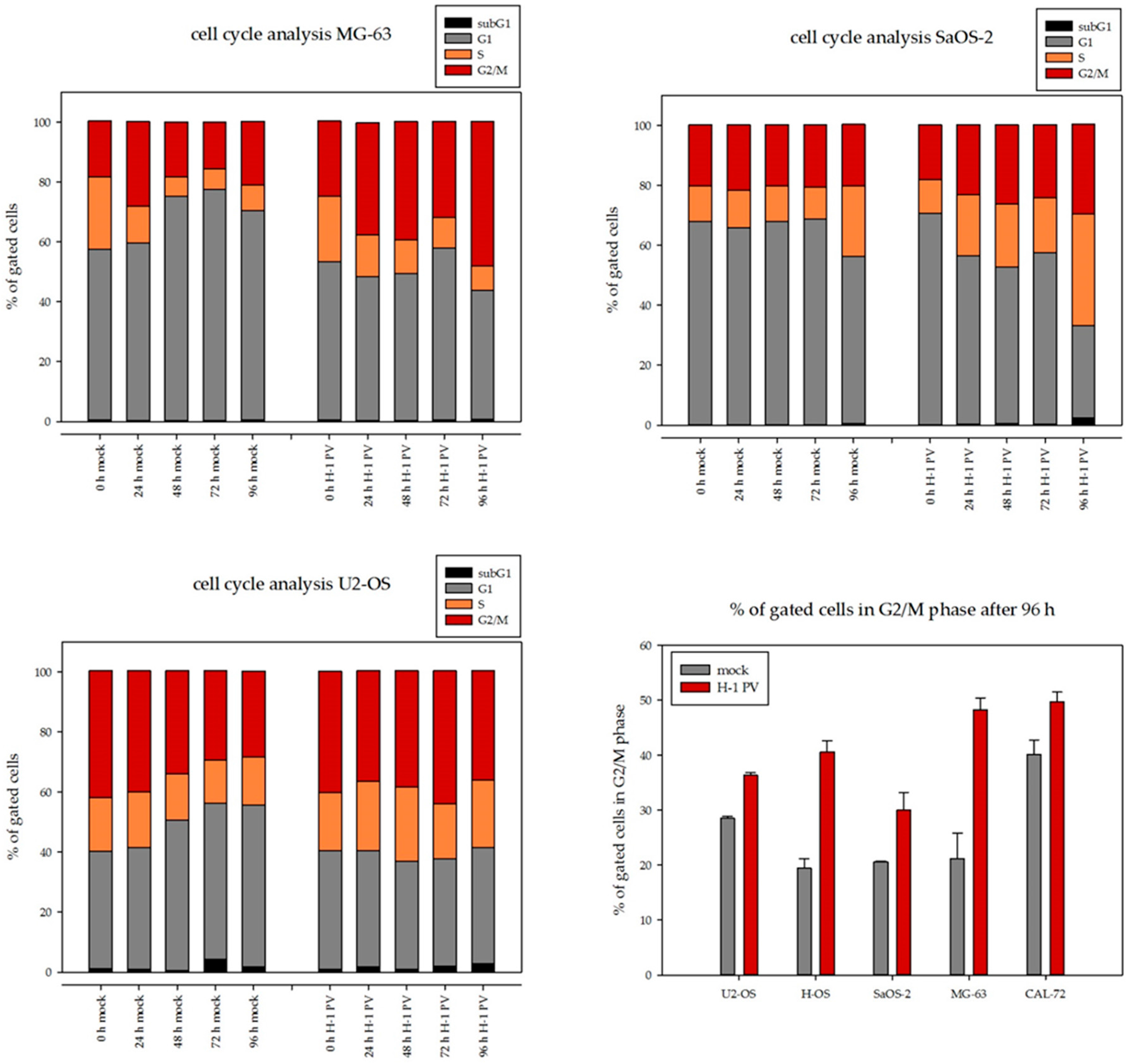
3.3. H-1PV Viral Protein Expression Induces Cell Cycle Arrest in G2/M in Osteosarcoma Cells
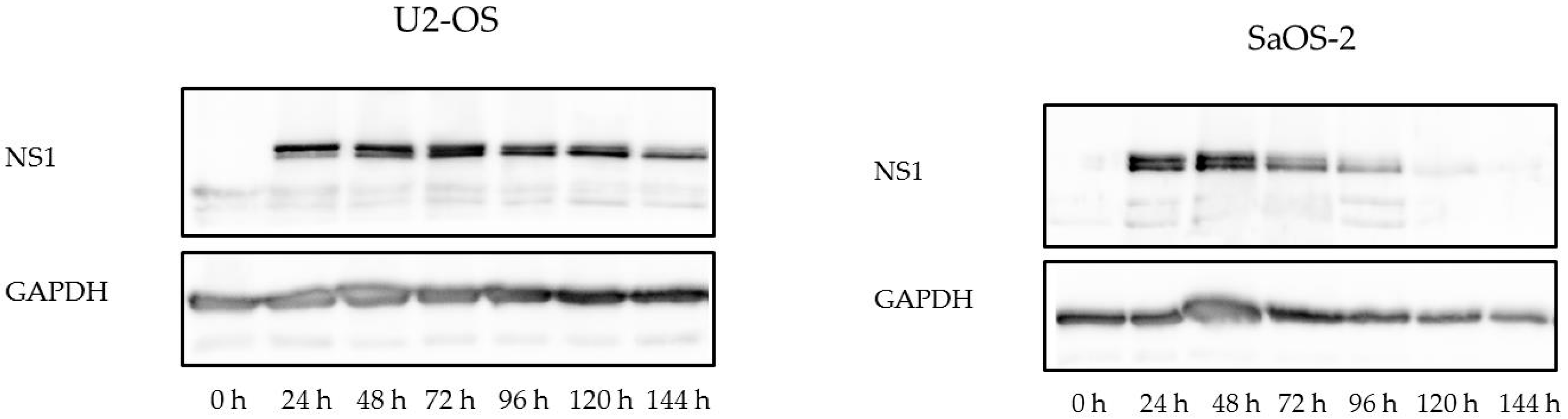
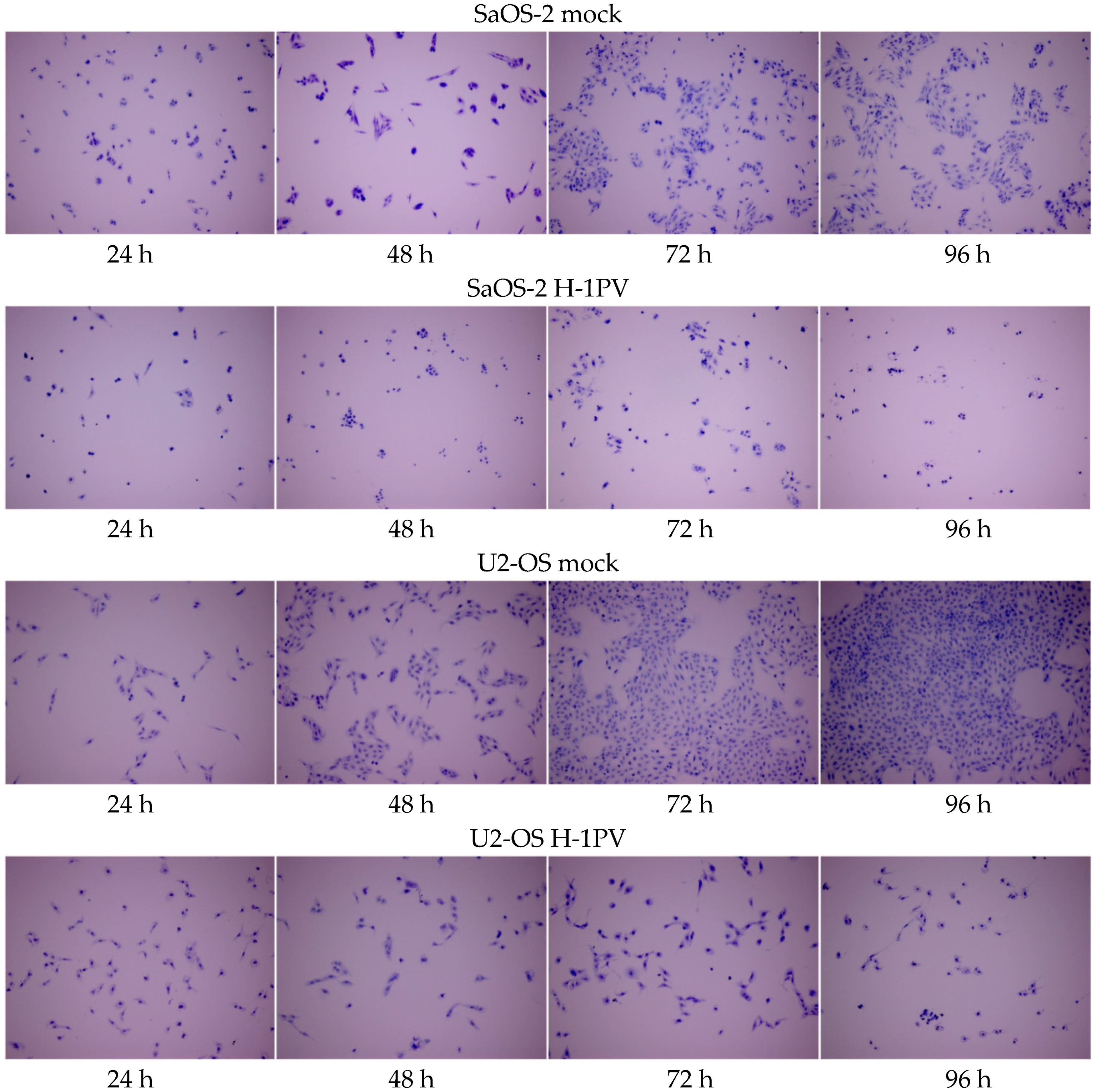
3.4. Osteosarcoma Cells Undergo Lytic H-1PV Infection
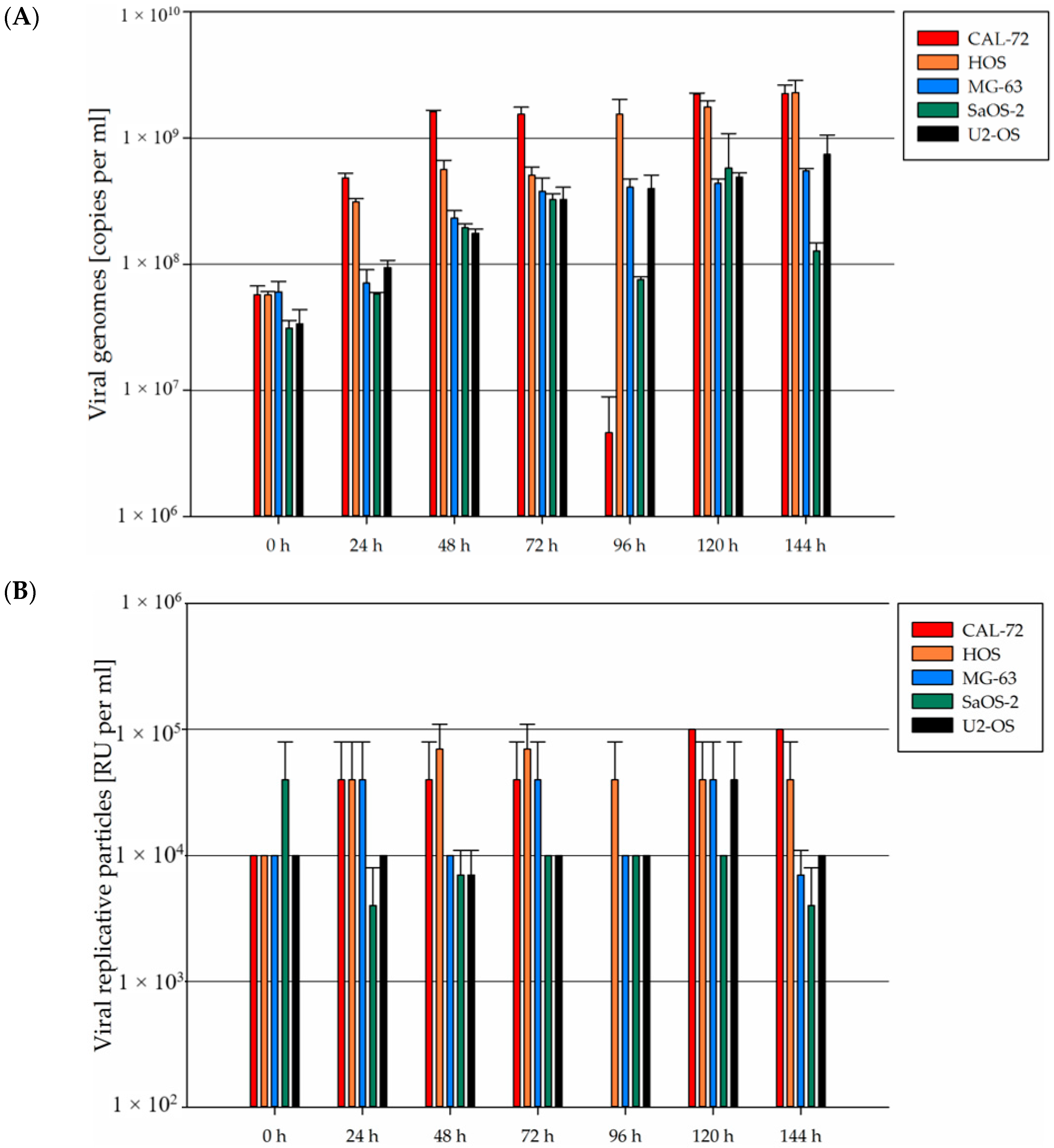
3.5. In Osteosarcoma Cells, H-1PV Infection Does Not Lead to Efficient Production of Infectious Viral Progeny
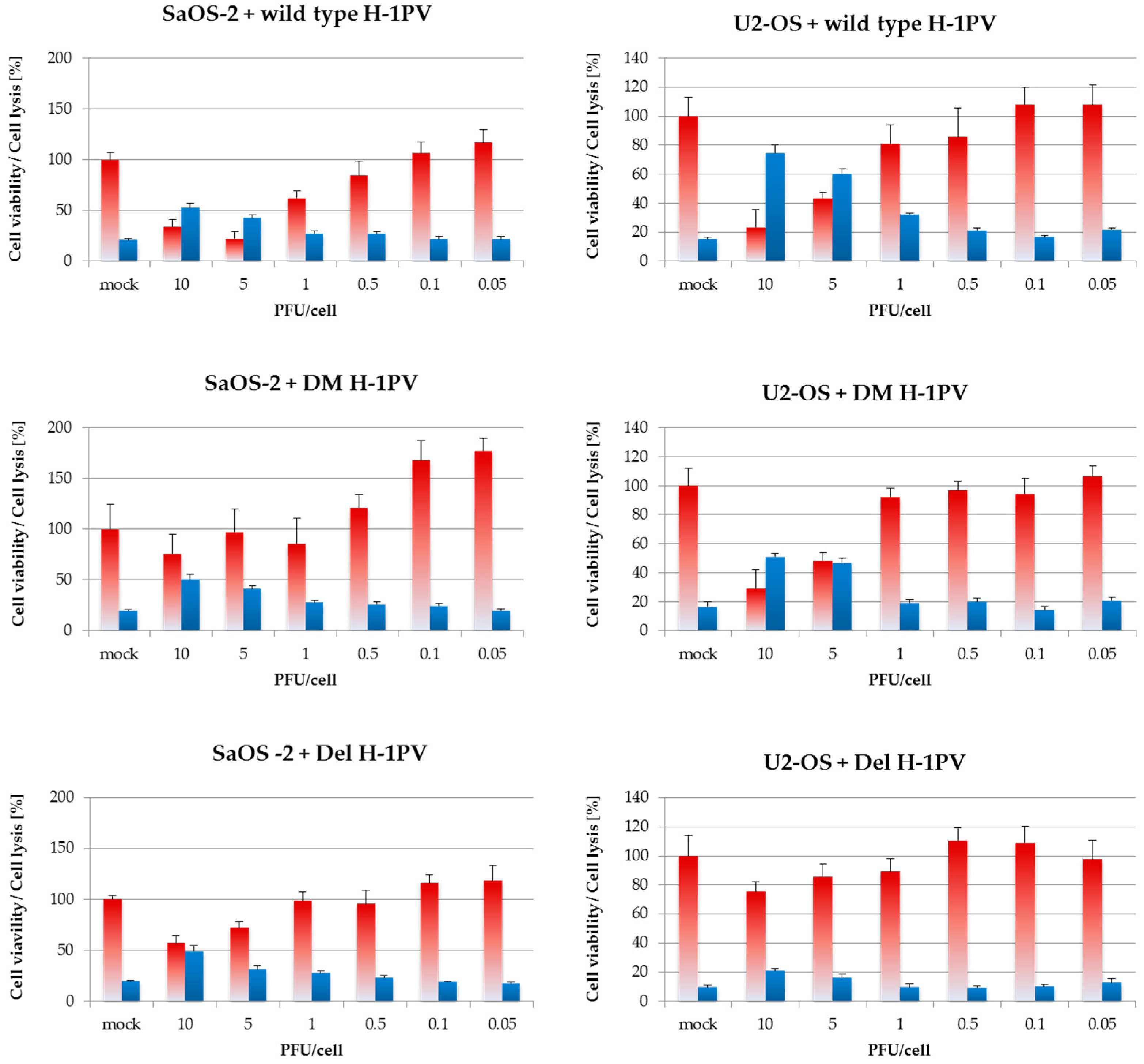
3.6. Wild-Type H-1PV Is More Cytotoxic Towards Osteosarcoma Cells Than H-1PV-Derived Fitness Mutants
4. Discussion
4.1. Safety Profile of Wild-Type H-1PV in Human Mesenchymal Cells
4.2. Osteosarcoma Cells Are Semi-Permissive to Wild Type H-1PV Infection
4.3. Fitness Mutants Do Not Show Increased Cytotoxicity Towards Osteosarcoma Cells In Vitro
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Jaffe, N. The Epidemiology of Osteosarcoma. Cancer Treat Res. 2009, 152, 3–13. [Google Scholar] [PubMed]
- Isakoff, M.S.; Bielack, S.S.; Meltzer, P.; Gorlick, R. Osteosarcoma: Current treatment and a collaborative pathway to success. J. Clin. Oncol. 2015, 33, 3029–3035. [Google Scholar] [CrossRef] [PubMed]
- Trama, A.; Botta, L.; Foschi, R.; Ferrari, A.; Stiller, C.; Desandes, E.; Maule, M.M.; Merletti, F.; Gatta, G. Survival of European adolescents and young adults diagnosed with cancer in 2000-07: Population-based data from EUROCARE-5. Lancet Oncol. 2016, 17, 896–906. [Google Scholar] [CrossRef]
- Anderson, M.E. Update on Survival in Osteosarcoma. Orthop. Clin. 2016, 47, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Aljubran, A.H.; Griffin, A.; Pintilie, M.; Blackstein, M. Osteosarcoma in adolescents and adults: Survival analysis with and without lung metastases. Ann. Oncol. 2009, 20, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival—A report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Abarrategi, A.; Tornin, J.; Martinez-Cruzado, L.; Hamilton, A.; Martinez-Campos, E.; Rodrigo, J.P.; Gonzalez, M.V.; Baldini, N.; Garcia-Castro, J.; Rodriguez, R. Osteosarcoma: Cells-of-origin, cancer stem cells, and targeted therapies. Stem Cells Int. 2016, 2016, 3631764. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.; Nieva, J.; Borad, M.J.; Breitbach, C.J. Oncolytic viruses: Perspectives on clinical development. Curr. Opin. Virol. 2015, 13, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Liu, G.; Wong-Staal, F. Oncolytic virotherapy as a personalized cancer vaccine. Int. J. Cancer 2008, 123, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.R.; Relph, K.; Harrington, K.; Melcher, A.; Pandha, H. Cancer immunotherapy via combining oncolytic virotherapy with chemotherapy: Recent advances. Oncolytic Virother. 2016, 5, 1–13. [Google Scholar] [PubMed]
- Morton, C.L.; Houghton, P.J.; Kolb, E.A.; Gorlick, R.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Keir, S.T.; Wu, J.; Smith, M.A. Initial testing of the replication competent Seneca Valley virus (NTX-010) by the pediatric preclinical testing program. Pediatr. Blood Cancer 2010, 55, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Ketola, A.; Hinkkanen, A.; Yongabi, F.; Furu, P.; Määttä, A.M.; Liimatainen, T.; Pirinen, R.; Björn, M.; Hakkarainen, T.; Mäkinen, K.; et al. Oncolytic Semliki forest virus vector as a novel candidate against unresectable osteosarcoma. Cancer Res. 2008, 68, 8342–8350. [Google Scholar] [CrossRef] [PubMed]
- Witlox, A.M.; van Beusechem, V.W.; Molenaar, B.; Bras, H.; Schaap, G.R.; Alemany, R.; Curiel, D.T.; Pinedo, H.M.; Wuisman, P.I.; Gerritsen, W.R. Conditionally replicative adenovirus with tropism expanded towards integrins inhibits osteosarcoma tumor growth in vitro and in vivo. Clin. Cancer Res. 2004, 10, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jung, C.; Liu, Y.H.; Bae, K.H.; Zhang, Y.P.; Zhang, H.J.; VanderPutten, D.; Jeng, M.H.; Gardner, T.A.; Kao, C. Anti-tumor efficacy of a transcriptional replication-competent adenovirus, Ad-OC-E1a, for osteosarcoma pulmonary metastasis. J. Gene Med. 2006, 8, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Graat, H.C.A.; van Beusechem, V.W.; Schagen, F.H.E.; Witlox, M.A.; Kleinerman, E.S.; Helder, M.N.; Gerritsen, W.R.; Kaspers, G.J.L.; Wuisman, P.I. Intravenous administration of the conditionally replicative adenovirus Ad5-Delta24RGD induces regression of osteosarcoma lung metastases. Mol. Cancer 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Velez, N.; Xipell, E.; Vera, B.; de la Rocha, A.A.; Zalacain, M.; Marrodan, L.; Gonzalez-Huarriz, M.; Toledo, G.; Cascallo, M.; Alemany, R.; et al. The oncolytic adenovirus vcn-01 as therapeutic approach against pediatric osteosarcoma. Clin. Cancer Res. 2016, 22, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Kolb, E.A.; Sampson, V.; Stabley, D.; Walter, A.; Sol-Church, K.; Cripe, T.; Hingorani, P.; Ahern, C.H.; Weigel, B.J.; Zwiebel, J.; et al. A phase I trial and viral clearance study of reovirus (Reolysin) in children with relapsed or refractory extra-cranial solid tumors: A Children’s Oncology Group Phase I Consortium report. Pediatr. Blood Cancer 2015, 62, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Toolan, H.W.; Rhode, S.L., III; Gierthy, J.F. Inhibition of 7,12-dimethylbenz(a)anthracene-induced tumors in Syrian hamsters by prior infection with H-1 parvovirus. Cancer Res. 1982, 42, 2552–2555. [Google Scholar] [PubMed]
- Toolan, H.W.; Saunders, E.L.; Southam, C.M.; Moore, A.E.; Levin, A.G. H-1 virus viremia in the human. Proc. Soc. Exp. Biol. Med. 1965, 119, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Geletneky, K.; Nüesch, J.P.; Rommelaere, J. Tumor selectivity of oncolytic parvoviruses: From in vitro and animal models to cancer patients. Front. Bioeng. Biotechnol. 2015, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Nuesch, J.P.F.; Lacroix, J.; Marchini, A.; Rommelaere, J. Molecular pathways: Rodent parvoviruses—Mechanisms of oncolysis and prospects for clinical cancer treatment. Clin. Cancer Res. 2012, 18, 3516–3523. [Google Scholar] [CrossRef] [PubMed]
- El-Andaloussi, N.; Endele, M.; Leuchs, B.; Bonifati, S.; Kleinschmidt, J.; Rommelaere, J.; Marchini, A. Novel adenovirus-based helper system to support production of recombinant parvovirus. Cancer Gene Ther. 2011, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Weiss, N.; Stroh-Dege, A.; Rommelaere, J.; Dinsart, C.; Salome, N. An in-frame deletion in the NS protein-coding sequence of parvovirus H-1PV efficiently stimulates export and infectivity of progeny virions. J. Virol. 2012, 86, 7554–7564. [Google Scholar] [CrossRef] [PubMed]
- De Veas, R.G.; Schweigerer, L.; Medina, M.A. Modulation of the proteolytic balance plasminogen activator/plasminogen activator inhibitor by enhanced N-myc oncogene expression or application of genistein. Eur. J. Cancer 1998, 34, 1736–1740. [Google Scholar] [CrossRef]
- Castro, F.; Dirks, W.G.; Fahnrich, S.; Hotz-Wagenblatt, A.; Pawlita, M.; Schmitt, M. High-throughput SNP-based authentication of human cell lines. Int. J. Cancer 2013, 132, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Pawlita, M. High-throughput detection and multiplex identification of cell contaminations. Nucleic Acids Res. 2009, 37, e119. [Google Scholar] [CrossRef] [PubMed]
- Wrzesinski, C.; Tesfay, L.; Salome, N.; Jauniaux, J.C.; Rommelaere, J.; Cornelis, J.; Dinsart, C. Chimeric and pseudotyped parvoviruses minimize the contamination of recombinant stocks with replication-competent viruses and identify a dna sequence that restricts parvovirus h-1 in mouse cells. J. Virol. 2003, 77, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Leuchs, B.; Roscher, M.; Muller, M.; Kurschner, K.; Rommelaere, J. Standardized large-scale H-1PV production process with efficient quality and quantity monitoring. J. Virol. Methods 2016, 229, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Kestler, J.; Neeb, B.; Struyf, S.; van Damme, J.; Cotmore, S.F.; D’Abramo, A.; Tattersall, P.; Rommelaere, J.; Dinsart, C.; Cornelis, J.J. Cis requirements for the efficient production of recombinant DNA vectors based on autonomous parvoviruses. Hum. Gene Ther. 1999, 10, 1619–1632. [Google Scholar] [CrossRef] [PubMed]
- Rayet, B.; Lopez-Guerrero, J.A.; Rommelaere, J.; Dinsart, C. Induction of programmed cell death by parvovirus H-1 in U937 cells: Connection with the tumor necrosis factor alpha signalling pathway. J. Virol. 1998, 72, 8893–8903. [Google Scholar] [PubMed]
- Herrero y Calle, M.; Cornelis, J.J.; Herold-Mende, C.; Rommelaere, J.; Schlehofer, J.R.; Geletneky, K. Parvovirus H-1 infection of human glioma cells leads to complete viral replication and efficient cell killing. Int. J. Cancer 2004, 109, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, J.; Leuchs, B.; Li, J.; Hristov, G.; Deubzer, H.E.; Kulozik, A.E.; Rommelaere, J.; Schlehofer, J.R.; Witt, O. Parvovirus H1 selectively induces cytotoxic effects on human neuroblastoma cells. Int. J. Cancer 2010, 127, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Hristov, G.; Krämer, M.; Li, J.; El-Andaloussi, N.; Mora, R.; Daeffler, L.; Zentgraf, H.; Rommelaere, J.; Marchini, A. Through its nonstructural protein NS1, parvovirus H-1 induces apoptosis via accumulation of reactive oxygen species. J. Virol. 2010, 84, 5909–5922. [Google Scholar] [CrossRef] [PubMed]
- Toolan, H.W. Lack of oncogenic effect of the H-viruses for hamsters. Nature 1967, 214, 1036. [Google Scholar] [CrossRef] [PubMed]
- Faisst, S.; Faisst, S.R.; Dupressoir, T.; Plaza, S.; Pujol, A.; Jauniaux, J.C.; Rhode, S.L.; Rommelaere, J. Isolation of a fully infectious variant of parvovirus H-1 supplanting the standard strain in human cells. J. Virol. 1995, 69, 4538–4543. [Google Scholar] [PubMed]
- Olijslagers, S.; Dege, A.Y.; Dinsart, C.; Voorhoeve, M.; Rommelaere, J.; Noteborn, M.H.; Cornelis, J.J. Potentiation of a recombinant oncolytic parvovirus by expression of Apoptin. Cancer Gene Ther. 2001, 8, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, J.; Schlund, F.; Leuchs, B.; Adolph, K.; Sturm, D.; Bender, S.; Hielscher, T.; Pfister, S.M.; Witt, O.; Rommelaere, J.; et al. Oncolytic effects of parvovirus H-1 in medulloblastoma are associated with repression of master regulators of early neurogenesis. Int. J. Cancer 2014, 134, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, H.; Stroh-Dege, A.Y.; Weiss, N.; Condurat, L.; Geiss, C.; Pilet, J.; Cornet-Bartolomé, C.; Rommelaere, J.; Salomé, N.; Dinsart, C. Mutations in the NS protein-coding sequence of protoparvovirus H-1PV enhance the fitness of the virus and show key benefits regarding the transduction efficiency of H-1PV-based vectors. Viruses 2017. submitted. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Culture | Del H-1PV | DM H-1PV | wt H-1PV |
|---|---|---|---|
| CAL-72 | 10 | 1 | 0.5 |
| H-OS | 5 | 5 | 1 |
| MG-63 | n. r. | 25 | 10 |
| SaOS-2 | >10 | 10 | 5 |
| U2-OS | n. r. | 5 | 5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geiss, C.; Kis, Z.; Leuchs, B.; Frank-Stöhr, M.; Schlehofer, J.R.; Rommelaere, J.; Dinsart, C.; Lacroix, J. Preclinical Testing of an Oncolytic Parvovirus: Standard Protoparvovirus H-1PV Efficiently Induces Osteosarcoma Cell Lysis In Vitro. Viruses 2017, 9, 301. https://doi.org/10.3390/v9100301
Geiss C, Kis Z, Leuchs B, Frank-Stöhr M, Schlehofer JR, Rommelaere J, Dinsart C, Lacroix J. Preclinical Testing of an Oncolytic Parvovirus: Standard Protoparvovirus H-1PV Efficiently Induces Osteosarcoma Cell Lysis In Vitro. Viruses. 2017; 9(10):301. https://doi.org/10.3390/v9100301
Chicago/Turabian StyleGeiss, Carsten, Zoltán Kis, Barbara Leuchs, Monika Frank-Stöhr, Jörg R. Schlehofer, Jean Rommelaere, Christiane Dinsart, and Jeannine Lacroix. 2017. "Preclinical Testing of an Oncolytic Parvovirus: Standard Protoparvovirus H-1PV Efficiently Induces Osteosarcoma Cell Lysis In Vitro" Viruses 9, no. 10: 301. https://doi.org/10.3390/v9100301
APA StyleGeiss, C., Kis, Z., Leuchs, B., Frank-Stöhr, M., Schlehofer, J. R., Rommelaere, J., Dinsart, C., & Lacroix, J. (2017). Preclinical Testing of an Oncolytic Parvovirus: Standard Protoparvovirus H-1PV Efficiently Induces Osteosarcoma Cell Lysis In Vitro. Viruses, 9(10), 301. https://doi.org/10.3390/v9100301