Protoparvovirus Cell Entry

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
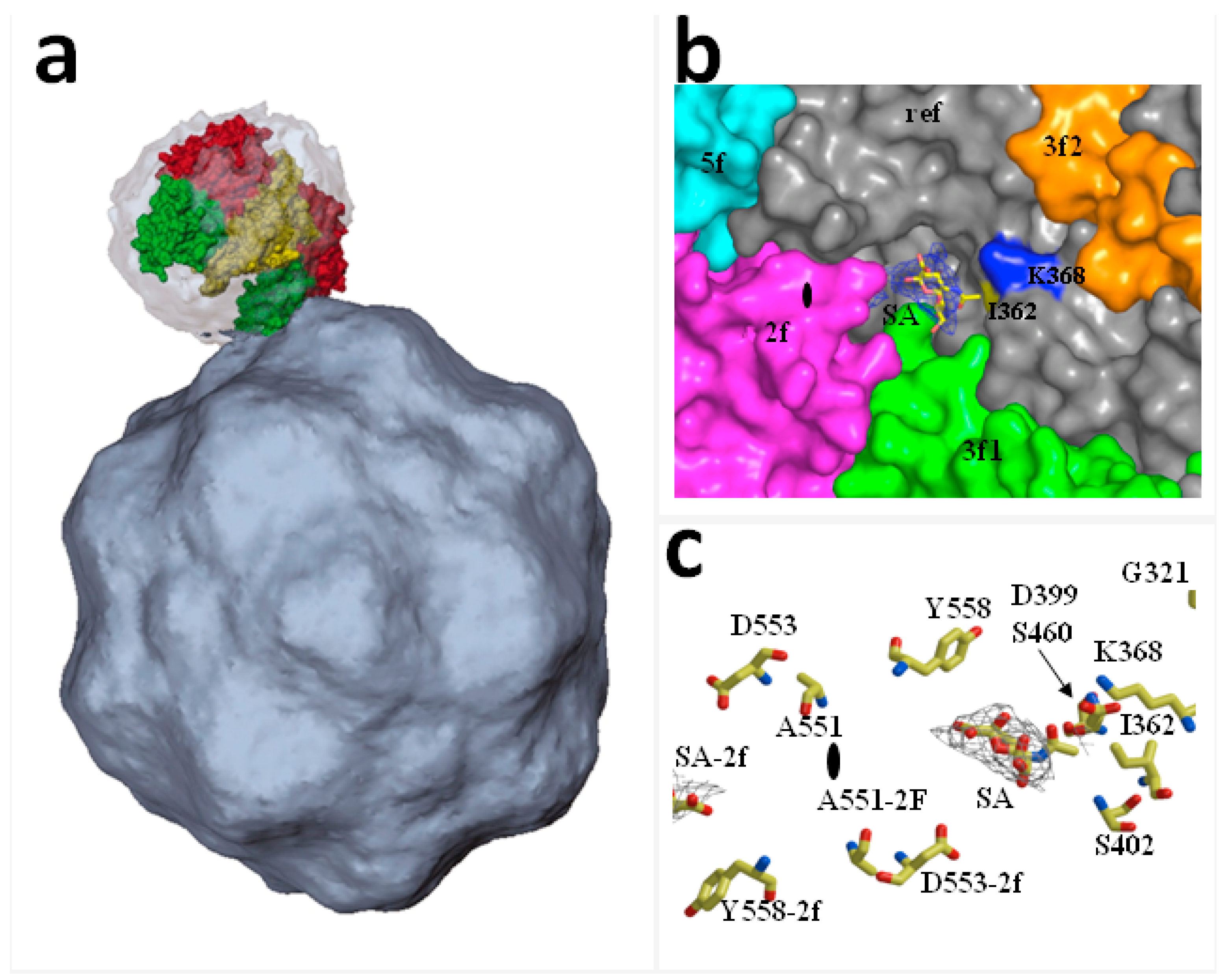
2. Functional Receptors for Protoparvovirus Entry
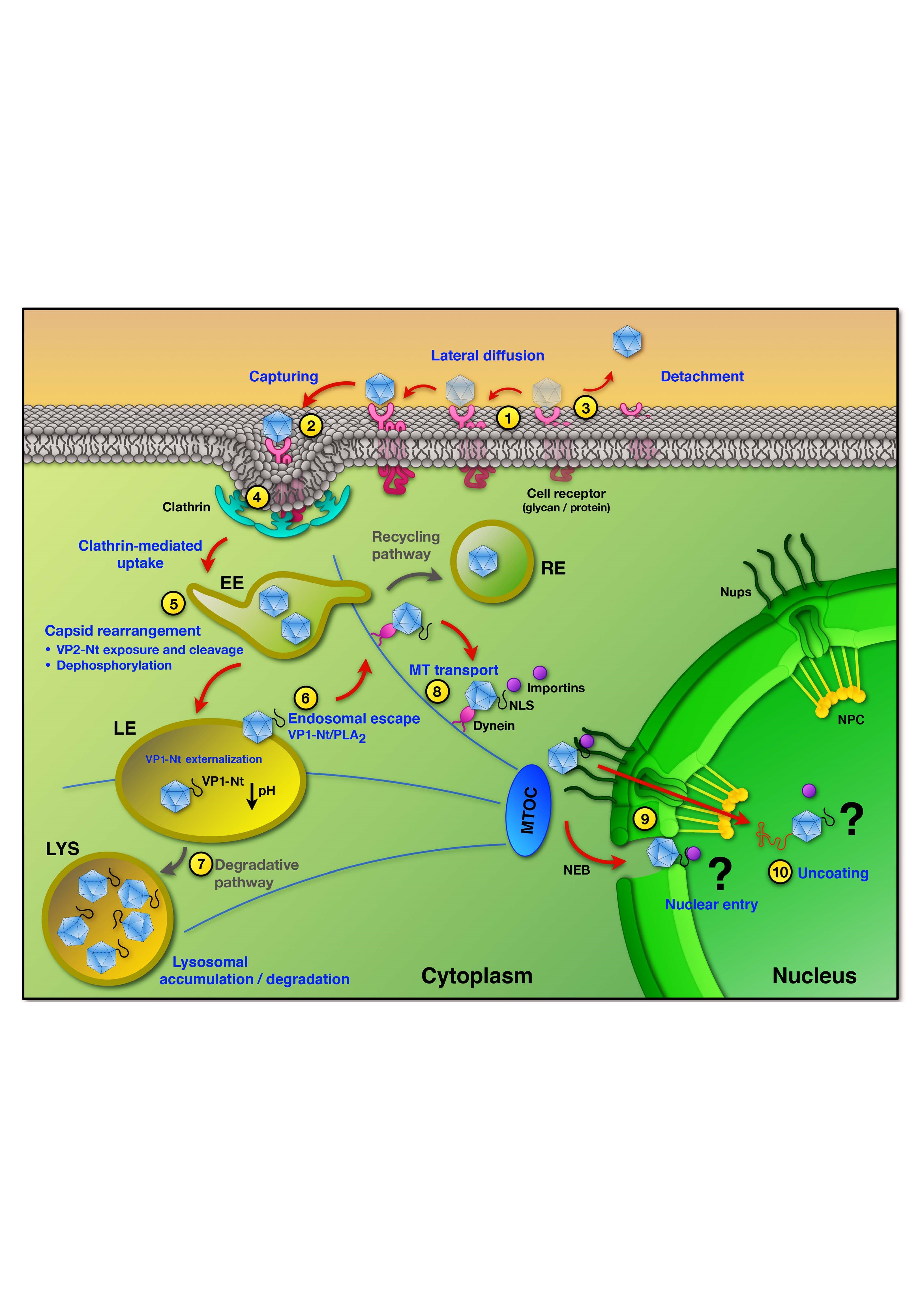
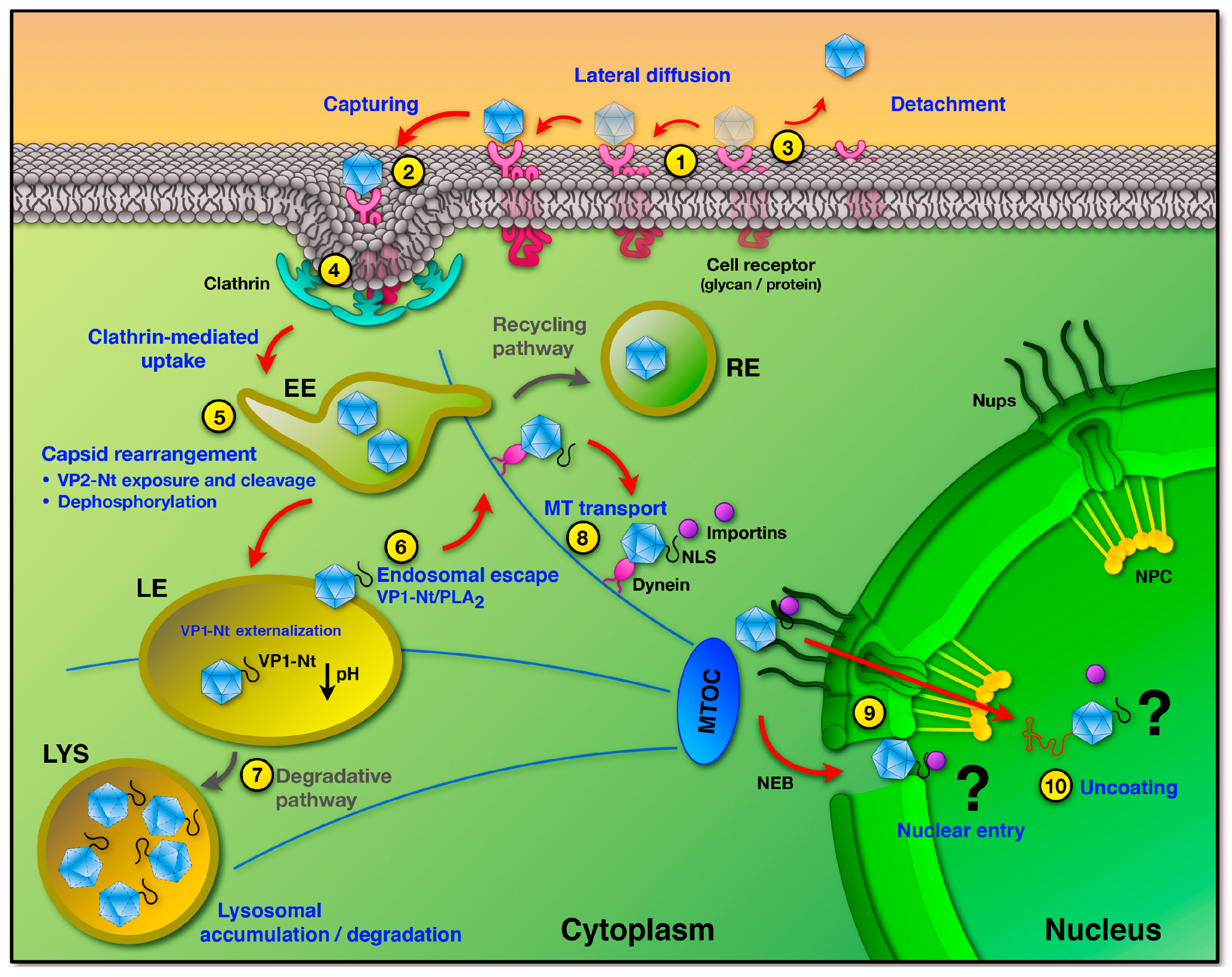
3. Uptake into Host Cells
4. Endosomal Trafficking and Capsid Rearrangements
5. Endosomal Escape
6. Trafficking and Interactions in the Cytosol
6.1. Interactions with the Cell Cytoskeleton and Motor Proteins
6.2. Interactions with the Ubiquitin-Proteasome Machinery
6.3. Other Interactions in the Cytoplasm
7. Nuclear Entry and Uncoating
8. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Kailasan, S.; Agbandje-McKenna, M.; Parrish, C.R. Parvovirus family conundrum: What makes a killer? Annu. Rev. Virol. 2015, 2, 425–450. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. The autonomously replicating parvoviruses of vertebrates. Adv. Virus Res. 1987, 33, 91–174. [Google Scholar] [CrossRef] [PubMed]
- Tijssen, P.; Agbandje-McKenna, M.; Almendral, J.M.; Bergoin, M.; Flegel, T.W.; Hedman, K.; Kleinschmidt, J.; Li, Y.; Pintel, D.J.; Tattersall, P. Parvoviridae. In Virus Taxonomy; King, A., Lefkowitz, E., Adams, M.J., Carstens, E.B., Eds.; Elsevier: London, UK, 2012; pp. 405–425. [Google Scholar] [CrossRef]
- Tsao, J.; Chapman, M.S.; Agbandje, M.; Keller, W.; Smith, K.; Wu, H.; Luo, M.; Smith, T.J.; Rossmann, M.G.; Compans, R.W. The three-dimensional structure of canine parvovirus and its functional implications. Science 1991, 251, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Agbandje-McKenna, M.; Llamas-Saiz, A.L.; Wang, F.; Tattersall, P.; Rossmann, M.G. Functional implications of the structure of the murine parvovirus, minute virus of mice. Structure 1998, 6, 1369–1381. [Google Scholar] [CrossRef]
- Kontou, M.; Govindasamy, L.; Nam, H.-J.; Bryant, N.; Llamas-Saiz, A.L.; Foces-Foces, C.; Hernando, E.; Rubio, M.-P.; McKenna, R.; Almendral, J.M.; et al. Structural determinants of tissue tropism and in vivo pathogenicity for the parvovirus minute virus of mice. J. Virol. 2005, 79, 10931–10943. [Google Scholar] [CrossRef] [PubMed]
- Zádori, Z.; Szelei, J.; Lacoste, M.C.; Li, Y.; Gariépy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Lombardo, E.; Ramírez, J.C.; Garcia, J.; Almendral, J.M. Complementary roles of multiple nuclear targeting signals in the capsid proteins of the parvovirus minute virus of mice during assembly and onset of infection. J. Virol. 2002, 76, 7049–7059. [Google Scholar] [CrossRef] [PubMed]
- Vihinen-Ranta, M.; Wang, D.; Weichert, W.S.; Parrish, C.R. The VP1 N-terminal sequence of canine parvovirus affects nuclear transport of capsids and efficient cell infection. J. Virol. 2002, 76, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Farr, G.A.; Zhang, L.-G.; Tattersall, P. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc. Natl. Acad. Sci. USA 2005, 102, 17148–17153. [Google Scholar] [CrossRef] [PubMed]
- Maroto, B.; Valle, N.; Saffrich, R.; Almendral, J.M. Nuclear export of the nonenveloped parvovirus virion is directed by an unordered protein signal exposed on the capsid surface. J. Virol. 2004, 78, 10685–10694. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D. The parvovirus life cycle: An introduction to molecular interactions important for infection. In Parvoviruses; Kerr, J.R., Cotmore, S.F., Bloom, M.E., Linden, R.M., Parrish, C.R., Eds.; Hodder Arnold: London, UK, 2006; pp. 143–156. [Google Scholar] [CrossRef]
- Bashir, T.; Horlein, R.; Rommelaere, J.; Willwand, K. Cyclin A activates the DNA polymerase delta—Dependent elongation machinery in vitro: A parvovirus DNA replication model. Proc. Natl. Acad. Sci. USA 2000, 97, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ranedo, J.; Hernando, E.; Riolobos, L.; Domínguez, C.; Kann, M.; Almendral, J.M. The mammalian cell cycle regulates parvovirus nuclear capsid assembly. PLoS Pathog. 2015, 11, e1004920. [Google Scholar] [CrossRef] [PubMed]
- Riolobos, L.; Valle, N.; Hernando, E.; Maroto, B.; Kann, M.; Almendral, J.M. Viral oncolysis that targets Raf-1 signaling control of nuclear transport. J. Virol. 2010, 84, 2090–2099. [Google Scholar] [CrossRef] [PubMed]
- Nüesch, J.P.F.; Lacroix, J.; Marchini, A.; Rommelaere, J. Molecular pathways: Rodent parvoviruses—Mechanisms of oncolysis and prospects for clinical cancer treatment. Clin. Cancer Res. 2012, 18, 3516–3523. [Google Scholar] [CrossRef] [PubMed]
- Ventoso, I.; Berlanga, J.J.; Almendral, J.M. Translation control by protein kinase R restricts minute virus of mice infection: Role in parvovirus oncolysis. J. Virol. 2010, 84, 5043–5051. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Geletneky, K.; Nüesch, J.P.F.; Rommelaere, J. Tumor selectivity of oncolytic parvoviruses: From in vitro and animal models to cancer patients. Front. Bioeng. Biotechnol. 2015, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Vihinen-Ranta, M.; Suikkanen, S.; Parrish, C.R. Pathways of cell infection by parvoviruses and adeno-associated viruses. J. Virol. 2004, 78, 6709–6714. [Google Scholar] [CrossRef] [PubMed]
- Harbison, C.E.; Chiorini, J.A.; Parrish, C.R. The parvovirus capsid odyssey: From the cell surface to the nucleus. Trends Microbiol. 2008, 16, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 2007, 70, 183–232. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R. Structures and functions of parvovirus capsids and the process of cell infection. Curr. Top. Microbiol. Immunol. 2010, 343, 149–176. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.L.; Murphy, W.J.; Wang, D.; O’Brien, S.J.; Parrish, C.R. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 2001, 75, 3896–3902. [Google Scholar] [CrossRef] [PubMed]
- Hueffer, K.; Parker, J.S.L.; Weichert, W.S.; Geisel, R.E.; Sgro, J.-Y.; Parrish, C.R. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 2003, 77, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Hueffer, K.; Palermo, L.M.; Parrish, C.R. Parvovirus infection of cells by using variants of the feline transferrin receptor altering clathrin-mediated endocytosis, membrane domain localization, and capsid-binding domains. J. Virol. 2004, 78, 5601–5611. [Google Scholar] [CrossRef] [PubMed]
- Palermo, L.M.; Hueffer, K.; Parrish, C.R. Residues in the apical domain of the feline and canine transferrin receptors control host-specific binding and cell infection of canine and feline parvoviruses. J. Virol. 2003, 77, 8915–8923. [Google Scholar] [CrossRef] [PubMed]
- Palermo, L.M.; Hafenstein, S.L.; Parrish, C.R. Purified feline and canine transferrin receptors reveal complex interactions with the capsids of canine and feline parvoviruses that correspond to their host ranges. J. Virol. 2006, 80, 8482–8492. [Google Scholar] [CrossRef] [PubMed]
- Kaelber, J.T.; Demogines, A.; Harbison, C.E.; Allison, A.B.; Goodman, L.B.; Ortega, A.N.; Sawyer, S.L.; Parrish, C.R. Evolutionary reconstructions of the transferrin receptor of Caniforms supports canine parvovirus being a re-emerged and not a novel pathogen in dogs. PLoS Pathog. 2012, 8, e1002666. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.B.; Kohler, D.J.; Ortega, A.; Hoover, E.A.; Grove, D.M.; Holmes, E.C.; Parrish, C.R. Host-specific parvovirus evolution in nature is recapitulated by in vitro adaptation to different carnivore species. PLoS Pathog. 2014, 10, e1004475. [Google Scholar] [CrossRef] [PubMed]
- Hafenstein, S.; Palermo, L.M.; Kostyuchenko, V.A.; Xiao, C.; Morais, M.C.; Nelson, C.D.S.; Bowman, V.D.; Battisti, A.J.; Chipman, P.R.; Parrish, C.R.; et al. Asymmetric binding of transferrin receptor to parvovirus capsids. Proc. Natl. Acad. Sci. USA 2007, 104, 6585–6589. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bueno, A.; Rubio, M.-P.; Bryant, N.; McKenna, R.; Agbandje-McKenna, M.; Almendral, J.M. Host-selected amino acid changes at the sialic acid binding pocket of the parvovirus capsid modulate cell binding affinity and determine virulence. J. Virol. 2006, 80, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, S.; Bergström, T. Glycoconjugate glycans as viral receptors. Ann. Med. 2005, 37, 154–172. [Google Scholar] [CrossRef] [PubMed]
- Barbis, D.P.; Chang, S.F.; Parrish, C.R. Mutations adjacent to the dimple of the canine parvovirus capsid structure affect sialic acid binding. Virology 1992, 191, 301–308. [Google Scholar] [CrossRef]
- Johnson, F.B.; Thacker, T.C. Binding of bovine parvovirus to erythrocyte membrane sialylglycoproteins. J. Gen. Virol. 1998, 79, 2163–2169. [Google Scholar] [CrossRef]
- Johnson, F.B.; Fenn, L.B.; Owens, T.J.; Faucheux, L.J.; Blackburn, S.D. Attachment of bovine parvovirus to sialic acids on bovine cell membranes. J. Gen. Virol. 2004, 85, 2199–2207. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Cline, S.E.; Hemming, J.P.; Johnson, F.B. Attachment of bovine parvovirus to O-linked α 2,3 neuraminic acid on glycophorin A. Arch. Virol. 2005, 150, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Linser, P.; Bruning, H.; Armentrout, R.W. Specific binding sites for a parvovirus, minute virus of mice, on cultured mouse cells. J. Virol. 1977, 24, 211–221. [Google Scholar] [PubMed]
- Spalholz, B.A.; Tattersall, P. Interaction of minute virus of mice with differentiated cells: strain-dependent target cell specificity is mediated by intracellular factors. J. Virol. 1983, 46, 937–943. [Google Scholar] [PubMed]
- Rubio, M.-P.; López-Bueno, A.; Almendral, J.M. Virulent variants emerging in mice infected with the apathogenic prototype strain of the parvovirus minute virus of mice exhibit a capsid with low avidity for a primary receptor. J. Virol. 2005, 79, 11280–11290. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.; Fernandes, S.; Tijssen, P. Multiple pathways involved in porcine parvovirus cellular entry and trafficking toward the nucleus. J. Virol. 2010, 84, 7782–7792. [Google Scholar] [CrossRef] [PubMed]
- Allaume, X.; El-Andaloussi, N.; Leuchs, B.; Bonifati, S.; Kulkarni, A.; Marttila, T.; Kaufmann, J.K.; Nettelbeck, D.M.; Kleinschmidt, J.; Rommelaere, J.; et al. Retargeting of rat parvovirus H-1PV to cancer cells through genetic engineering of the viral capsid. J. Virol. 2012, 86, 3452–3465. [Google Scholar] [CrossRef] [PubMed]
- Segovia, J.C.; Gallego, J.M.; Bueren, J.A.; Almendral, J.M. Severe leukopenia and dysregulated erythropoiesis in SCID mice persistently infected with the parvovirus minute virus of mice. J. Virol. 1999, 73, 1774–1784. [Google Scholar] [PubMed]
- Nam, H.-J.; Gurda-Whitaker, B.; Gan, W.Y.; Ilaria, S.; McKenna, R.; Mehta, P.; Alvarez, R.A.; Agbandje-McKenna, M. Identification of the sialic acid structures recognized by minute virus of mice and the role of binding affinity in virulence adaptation. J. Biol. Chem. 2006, 281, 25670–25677. [Google Scholar] [CrossRef] [PubMed]
- Antonietti, J.P.; Sahli, R.; Beard, P.; Hirt, B. Characterization of the cell type-specific determinant in the genome of minute virus of mice. J. Virol. 1988, 62, 552–557. [Google Scholar] [PubMed]
- Ball-Goodrich, L.J.; Tattersall, P. Two amino acid substitutions within the capsid are coordinately required for acquisition of fibrotropism by the lymphotropic strain of minute virus of mice. J. Virol. 1992, 66, 3415–3423. [Google Scholar] [PubMed]
- Gardiner, E.M.; Tattersall, P. Mapping of the fibrotropic and lymphotropic host range determinants of the parvovirus minute virus of mice. J. Virol. 1988, 62, 2605–2613. [Google Scholar] [PubMed]
- Tattersall, P.; Bratton, J. Reciprocal productive and restrictive virus-cell interactions of immunosuppressive and prototype strains of minute virus of mice. J. Virol. 1983, 46, 944–955. [Google Scholar] [PubMed]
- Halder, S.; Cotmore, S.; Heimburg-Molinaro, J.; Smith, D.F.; Cummings, R.D.; Chen, X.; Trollope, A.J.; North, S.J.; Haslam, S.M.; Dell, A.; et al. Profiling of glycan receptors for minute virus of mice in permissive cell lines towards understanding the mechanism of cell recognition. PLoS ONE 2014, 9, e86909. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bueno, A.; Segovia, J.C.; Bueren, J.A.; O’Sullivan, M.G.; Wang, F.; Tattersall, P.; Almendral, J.M. Evolution to pathogenicity of the parvovirus minute virus of mice in immunodeficient mice involves genetic heterogeneity at the capsid domain that determines tropism. J. Virol. 2008, 82, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Dudleenamjil, E.; Lin, C.-Y.; Dredge, D.; Murray, B.K.; Robison, R.A.; Johnson, F.B. Bovine parvovirus uses clathrin-mediated endocytosis for cell entry. J. Gen. Virol. 2010, 91, 3032–3041. [Google Scholar] [CrossRef] [PubMed]
- Seksek, O.; Biwersi, J.; Verkman, A.S. Translational diffusion of macromolecule-sized solutes in cytoplasm and nucleus. J. Cell Biol. 1997, 138, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Pillay, C.S.; Elliott, E.; Dennison, C. Endolysosomal proteolysis and its regulation. Biochem. J. 2002, 363, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Parrish, C.R. Cellular uptake and infection by canine parvovirus involves rapid dynamin-regulated clathrin-mediated endocytosis, followed by slower intracellular trafficking. J. Virol. 2000, 74, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Vendeville, A.; Ravallec, M.; Jousset, F.-X.; Devise, M.; Mutuel, D.; López-Ferber, M.; Fournier, P.; Dupressoir, T.; Ogliastro, M. Densovirus infectious pathway requires clathrin-mediated endocytosis followed by trafficking to the nucleus. J. Virol. 2009, 83, 4678–4689. [Google Scholar] [CrossRef] [PubMed]
- Quattrocchi, S.; Ruprecht, N.; Bönsch, C.; Bieli, S.; Zürcher, C.; Boller, K.; Kempf, C.; Ros, C. Characterization of the early steps of human parvovirus B19 infection. J. Virol. 2012, 86, 9274–9284. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Garcin, P.O.; Panté, N. The minute virus of mice exploits different endocytic pathways for cellular uptake. Virology 2015, 482, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.; Puthenveedu, M.A. Clathrin-mediated endocytosis. In Vesicle Trafficking in Cancer; Yarden, Y., Tarcic, G., Eds.; Springer: New York, NY, USA, 2013; pp. 1–31. [Google Scholar] [CrossRef]
- Boulant, S.; Stanifer, M.; Lozach, P.-Y. Dynamics of virus-receptor interactions in virus binding, signaling, and endocytosis. Viruses 2015, 7, 2794–2815. [Google Scholar] [CrossRef] [PubMed]
- Cureton, D.K.; Harbison, C.E.; Cocucci, E.; Parrish, C.R.; Kirchhausen, T. Limited transferrin receptor clustering allows rapid diffusion of canine parvovirus into clathrin endocytic structures. J. Virol. 2012, 86, 5330–5340. [Google Scholar] [CrossRef] [PubMed]
- Garcin, P.O.; Panté, N. Cell migration is another player of the minute virus of mice infection. Virology 2014, 468–470, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Garcin, P.O.; Nabi, I.R.; Panté, N. Galectin-3 plays a role in minute virus of mice infection. Virology 2015, 481, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Mani, B.; Baltzer, C.; Valle, N.; Almendral, J.M.; Kempf, C.; Ros, C. Low pH-dependent endosomal processing of the incoming parvovirus minute virus of mice virion leads to externalization of the VP1 N-terminal sequence (N-VP1), N-VP2 cleavage, and uncoating of the full-length genome. J. Virol. 2006, 80, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Ros, C.; Burckhardt, C.J.; Kempf, C. Cytoplasmic trafficking of minute virus of mice: low-pH requirement, routing to late endosomes, and proteasome interaction. J. Virol. 2002, 76, 12634–12645. [Google Scholar] [CrossRef] [PubMed]
- Vihinen-Ranta, M.; Kalela, A.; Mäkinen, P.; Kakkola, L.; Marjomäki, V.; Vuento, M. Intracellular route of canine parvovirus entry. J. Virol. 1998, 72, 802–806. [Google Scholar] [PubMed]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Hensens, O.D.; Monaghan, R.L.; Huang, L.; Albers-Schonberg, G. Structure of the sodium and potassium ion activated adenosine triphosphatase inhibitor L-681,110. J. Am. Chem. Soc. 1983, 105, 3672–3679. [Google Scholar] [CrossRef]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Turner, H. Infectious entry pathway for canine parvovirus. Virology 1992, 186, 368–376. [Google Scholar] [CrossRef]
- Tullis, G.E.; Burger, L.R.; Pintel, D.J. The minor capsid protein VP1 of the autonomous parvovirus minute virus of mice is dispensable for encapsidation of progeny single-stranded DNA but is required for infectivity. J. Virol. 1993, 67, 131–141. [Google Scholar] [PubMed]
- Weichert, W.S.; Parker, J.S.L.; Wahid, A.T.M.; Chang, S.-F.; Meier, E.; Parrish, C.R. Assaying for structural variation in the parvovirus capsid and its role in infection. Virology 1998, 250, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; D’Abramo, A.M.; Ticknor, C.M.; Tattersall, P. Controlled conformational transitions in the MVM virion expose the VP1 N-Terminus and viral genome without particle disassembly. Virology 1999, 254, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Hafenstein, S.; Tattersall, P. Depletion of virion-associated divalent cations induces parvovirus minute virus of mice to eject its genome in a 3′-to-5′ direction from an otherwise intact viral particle. J. Virol. 2010, 84, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Hernando, E.; Llamas-Saiz, A.L.; Foces-Foces, C.; McKenna, R.; Portman, I.; Agbandje-McKenna, M.; Almendral, J.M. Biochemical and physical characterization of parvovirus minute virus of mice virus-like particles. Virology 2000, 267, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Suikkanen, S.; Aaltonen, T.; Nevalainen, M.; Välilehto, O.; Lindholm, L.; Vuento, M.; Vihinen-Ranta, M. Exploitation of microtubule cytoskeleton and dynein during parvoviral traffic toward the nucleus. J. Virol. 2003, 77, 10270–10279. [Google Scholar] [CrossRef] [PubMed]
- Farr, G.A.; Cotmore, S.F.; Tattersall, P. VP2 cleavage and the leucine ring at the base of the fivefold cylinder control pH-dependent externalization of both the VP1 N terminus and the genome of minute virus of mice. J. Virol. 2006, 80, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.; Carreira, A.; Riolobos, L.; Almendral, J.M.; Mateu, M.G. Role of interfacial amino acid residues in assembly, stability, and conformation of a spherical virus capsid. Proc. Natl. Acad. Sci. USA 2004, 101, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, M.; Pérez, R.; Rodríguez-Huete, A.; Grueso, E.; Almendral, J.M.M.; Mateu, M.G. A slender tract of glycine residues is required for translocation of the VP2 protein N-terminal domain through the parvovirus MVM capsid channel to initiate infection. Biochem. J. 2013, 455, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Farr, G.A.; Tattersall, P. A conserved leucine that constricts the pore through the capsid fivefold cylinder plays a central role in parvoviral infection. Virology 2004, 323, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Organtini, L.J.; Grossman, A.; Domeier, P.P.; Cifuente, J.O.; Makhov, A.M.; Conway, J.F.; D’Abramo, A.; Cotmore, S.F.; Tattersall, P.; et al. Cryo-EM maps reveal five-fold channel structures and their modification by gatekeeper mutations in the parvovirus minute virus of mice (MVM) capsid. Virology 2017, 510, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Tattersall, P.; Shatkin, A.J.; Ward, D.C. Sequence homology between the structural polypeptides of minute virus of mice. J. Mol. Biol. 1977, 111, 375–394. [Google Scholar] [CrossRef]
- Tullis, G.E.; Burger, L.R.; Pintel, D.J. The trypsin-sensitive RVER domain in the capsid proteins of minute virus of mice is required for efficient cell binding and viral infection but not for proteolytic processing in vivo. Virology 1992, 191, 846–857. [Google Scholar] [CrossRef]
- Paradiso, P.R.; Williams, K.R.; Costantino, R.L. Mapping of the amino terminus of the H-1 parvovirus major capsid protein. J. Virol. 1984, 52, 77–81. [Google Scholar] [PubMed]
- Paradiso, P.R. Infectious process of the parvovirus H-1: Correlation of protein content, particle density, and viral infectivity. J. Virol. 1981, 39, 800–807. [Google Scholar] [PubMed]
- Vincent, R. Racaniello Fields Virology. In Picornaviridae: The Viruses and Their Replication; Knipe, D., Howley, P., Eds.; Lippincot Williams & Sons: New York, NY, USA, 2001; pp. 685–722. [Google Scholar]
- Tattersall, P.; Cawte, P.J.; Shatkin, A.J.; Ward, D.C. Three structural polypeptides coded for by minite virus of mice, a parvovirus. J. Virol. 1976, 20, 273–289. [Google Scholar] [PubMed]
- Sánchez-Martínez, C.; Grueso, E.; Carroll, M.; Rommelaere, J.; Almendral, J.M. Essential role of the unordered VP2 n-terminal domain of the parvovirus MVM capsid in nuclear assembly and endosomal enlargement of the virion fivefold channel for cell entry. Virology 2012, 432, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B. Penetration of nonenveloped viruses into the cytoplasm. Annu. Rev. Cell Dev. Biol. 2007, 23, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Odegard, A.L.; Kwan, M.H.; Walukiewicz, H.E.; Banerjee, M.; Schneemann, A.; Johnson, J.E. Low endocytic pH and capsid protein autocleavage are critical components of Flock House virus cell entry. J. Virol. 2009, 83, 8628–8637. [Google Scholar] [CrossRef] [PubMed]
- Chandran, K.; Farsetta, D.L.; Nibert, M.L. Strategy for nonenveloped virus entry: A hydrophobic conformer of the reovirus membrane penetration protein micro 1 mediates membrane disruption. J. Virol. 2002, 76, 9920–9933. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.; López-Bueno, A.; Mateu, M.G.; Chipman, P.R.; Nelson, C.D.S.; Parrish, C.R.; Almendral, J.M.; Rossmann, M.G. Minute virus of mice, a parvovirus, in complex with the Fab fragment of a neutralizing monoclonal antibody. J. Virol. 2007, 81, 9851–9858. [Google Scholar] [CrossRef] [PubMed]
- Suikkanen, S.; Antila, M.; Jaatinen, A.; Vihinen-Ranta, M.; Vuento, M. Release of canine parvovirus from endocytic vesicles. Virology 2003, 316, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Canaan, S.; Zádori, Z.; Ghomashchi, F.; Bollinger, J.; Sadilek, M.; Moreau, M.E.; Tijssen, P.; Gelb, M.H. Interfacial enzymology of parvovirus phospholipases A2. J. Biol. Chem. 2004, 279, 14502–14508. [Google Scholar] [CrossRef] [PubMed]
- Lyi, S.M.; Tan, M.J.A.; Parrish, C.R. Parvovirus particles and movement in the cellular cytoplasm and effects of the cytoskeleton. Virology 2014, 456–457, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Luby-Phelps, K.; Taylor, D.L.; Lanni, F. Probing the structure of cytoplasm. J. Cell Biol. 1986, 102, 2015–2022. [Google Scholar] [CrossRef] [PubMed]
- Luby-Phelps, K. Cytoarchitecture and physical properties of cytoplasm: Volume, viscosity, diffusion, intracellular surface area. Int. Rev. Cytol. 1999, 192, 189–221. [Google Scholar] [CrossRef]
- Lagache, T.; Dauty, E.; Holcman, D. Physical principles and models describing intracellular virus particle dynamics. Curr. Opin. Microbiol. 2009, 12, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.L.; Kraikivski, P.; Slepchenko, B.M. Diffusion in cytoplasm: Effects of excluded volume due to internal membranes and cytoskeletal structures. Biophys. J. 2009, 97, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, D. Particle-tracking microrheology of living cells: Principles and applications. Annu. Rev. Biophys. 2009, 38, 301–326. [Google Scholar] [CrossRef] [PubMed]
- Vihinen-Ranta, M.; Yuan, W.; Parrish, C.R. Cytoplasmic trafficking of the canine parvovirus capsid and its role in infection and nuclear transport. J. Virol. 2000, 74, 4853–4859. [Google Scholar] [CrossRef] [PubMed]
- Suikkanen, S.; Sääjärvi, K.; Hirsimäki, J.; Välilehto, O.; Reunanen, H.; Vihinen-Ranta, M.; Vuento, M. Role of recycling endosomes and lysosomes in dynein-dependent entry of canine parvovirus. J. Virol. 2002, 76, 4401–4411. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Bidirectional transport along microtubules. Curr. Biol. 2004, 14, R525–R537. [Google Scholar] [CrossRef] [PubMed]
- Sripada, S.; Dayaraj, C. Viral interactions with intermediate filaments: Paths less explored. Cell Health Cytoskelet. 2010, 2, 1–7. [Google Scholar] [CrossRef]
- Fay, N.; Panté, N. The intermediate filament network protein, vimentin, is required for parvoviral infection. Virology 2013, 444, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Nüesch, J.P.F.F.; Lachmann, S.; Rommelaere, J. Selective alterations of the host cell architecture upon infection with parvovirus minute virus of mice. Virology 2005, 331, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Paine, S.; Sheppard, P.W.; Mayer, R.J.; Roelofs, J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010, 20, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Luo, H. Interplay between the virus and the ubiquitin–proteasome system: Molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 2016, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ros, C.; Kempf, C. The ubiquitin-proteasome machinery is essential for nuclear translocation of incoming minute virus of mice. Virology 2004, 324, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Nykky, J.; Vuento, M.; Gilbert, L. Role of mitochondria in parvovirus pathology. PLoS ONE 2014, 9, e86124. [Google Scholar] [CrossRef] [PubMed]
- Riolobos, L.; Reguera, J.; Mateu, M.G.; Almendral, J.M. Nuclear transport of trimeric assembly intermediates exerts a morphogenetic control on the icosahedral parvovirus capsid. J. Mol. Biol. 2006, 357, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Timney, B.L.; Raveh, B.; Mironska, R.; Trivedi, J.M.; Kim, S.J.; Russel, D.; Wente, S.R.; Sali, A.; Rout, M.P. Simple rules for passive diffusion through the nuclear pore complex. J. Cell Biol. 2016, 215, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef] [PubMed]
- Panté, N.; Kann, M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol. Biol. Cell 2002, 13, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Fay, N.; Panté, N. Nuclear entry of DNA viruses. Front. Microbiol. 2015, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Vihinen-Ranta, M.; Kakkola, L.; Kalela, A.; Vilja, P.; Vuento, M. Characterization of a nuclear localization signal of canine parvovirus capsid proteins. Eur. J. Biochem. 1997, 250, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.; Bouchard-Lévesque, V.; Fernandes, S.; Tijssen, P. Classic nuclear localization signals and a novel nuclear localization motif are required for nuclear transport of porcine parvovirus capsid proteins. J. Virol. 2014, 88, 11748–11759. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; Mills, R.E.; Lange, C.J.; Stewart, M.; Devine, S.E.; Corbett, A.H. Classical nuclear localization signals: Definition, function, and interaction with importin α. J. Biol. Chem. 2007, 282, 5101–5105. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, E.; Ramírez, J.C.; Agbandje-McKenna, M.; Almendral, J.M. A β-stranded motif drives capsid protein oligomers of the parvovirus minute virus of mice into the nucleus for viral assembly. J. Virol. 2000, 74, 3804–3814. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Behzad, A.R.; Carroll, J.B.; Panté, N.; Pante, N. Parvoviral nuclear import: Bypassing the host nuclear-transport machinery. J. Gen. Virol. 2006, 87, 3209–3213. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Panté, N. Pushing the envelope: Microinjection of Minute virus of mice into Xenopus oocytes causes damage to the nuclear envelope. J. Gen. Virol. 2005, 86, 3243–3252. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Marr, A.K.; Garcin, P.; Pante, N.; Panté, N. Nuclear Envelope Disruption Involving Host Caspases Plays a Role in the Parvovirus Replication Cycle. J. Virol. 2011, 85, 4863–4874. [Google Scholar] [CrossRef] [PubMed]
- Porwal, M.; Cohen, S.; Snoussi, K.; Popa-Wagner, R.; Anderson, F.; Dugot-Senant, N.; Wodrich, H.; Dinsart, C.; Kleinschmidt, J.A.; Panté, N.; et al. Parvoviruses cause nuclear envelope breakdown by activating key enzymes of mitosis. PLoS Pathog. 2013, 9, e1003671. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Mutations at the base of the icosahedral five-fold cylinders of minute virus of mice induce 3′-to-5′ genome uncoating and critically impair entry functions. J. Virol. 2012, 86, 69–80. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ros, C.; Bayat, N.; Wolfisberg, R.; Almendral, J.M. Protoparvovirus Cell Entry. Viruses 2017, 9, 313. https://doi.org/10.3390/v9110313
Ros C, Bayat N, Wolfisberg R, Almendral JM. Protoparvovirus Cell Entry. Viruses. 2017; 9(11):313. https://doi.org/10.3390/v9110313
Chicago/Turabian StyleRos, Carlos, Nooshin Bayat, Raphael Wolfisberg, and José M. Almendral. 2017. "Protoparvovirus Cell Entry" Viruses 9, no. 11: 313. https://doi.org/10.3390/v9110313
APA StyleRos, C., Bayat, N., Wolfisberg, R., & Almendral, J. M. (2017). Protoparvovirus Cell Entry. Viruses, 9(11), 313. https://doi.org/10.3390/v9110313