Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection



{kind=link}
{kind=link}
{kind=link}
Abstract
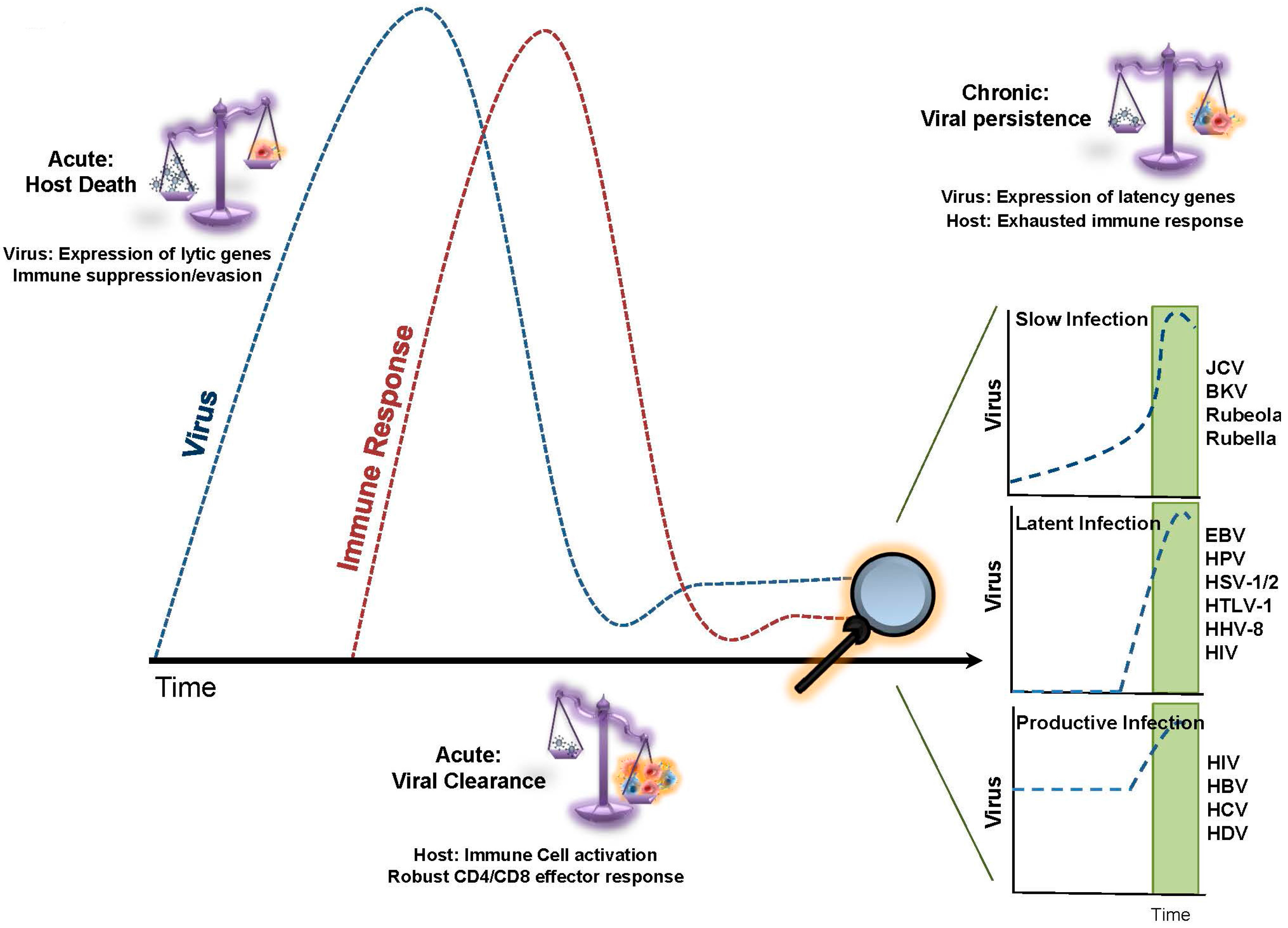
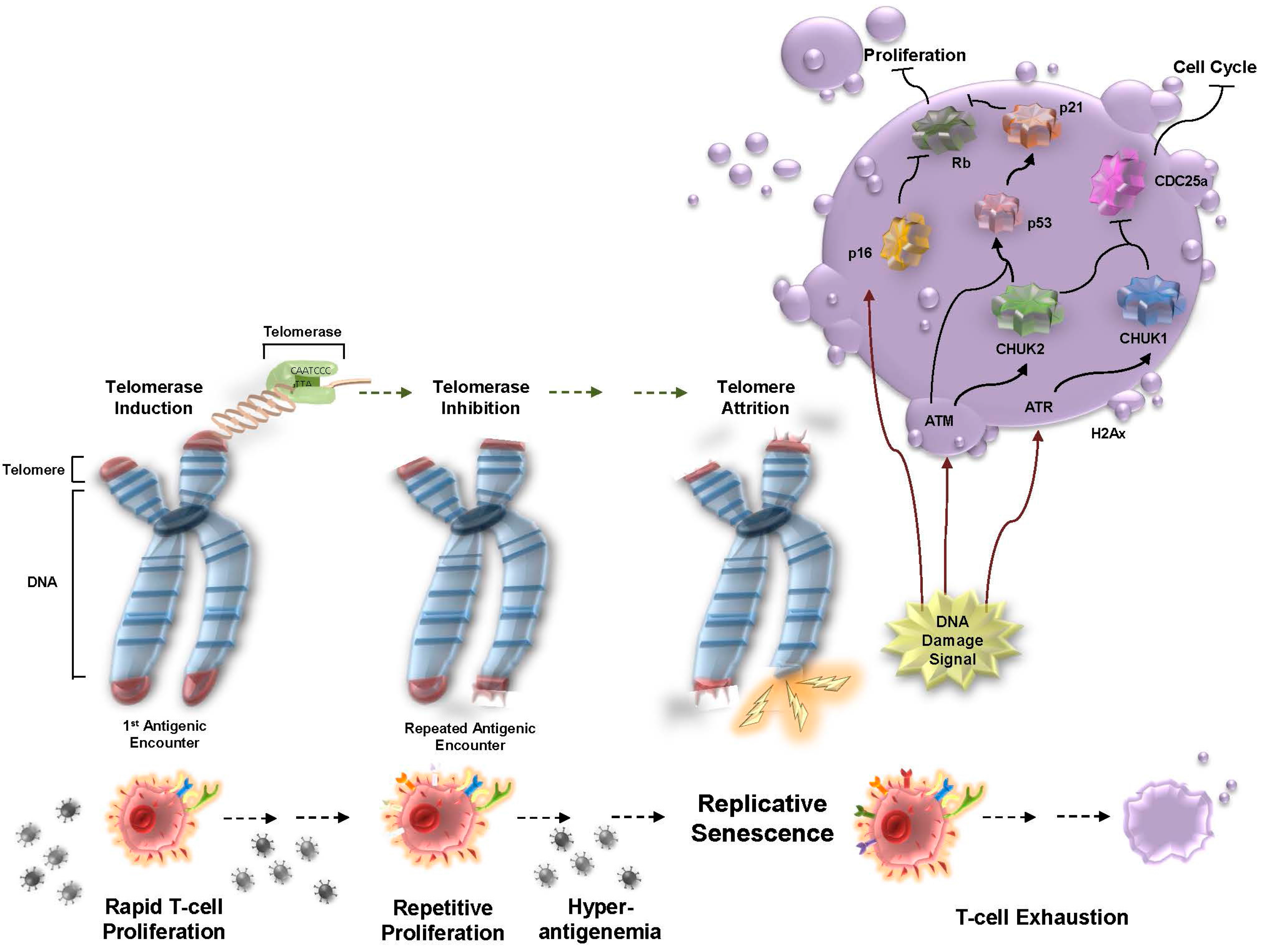
1. Replicative Senescence in Chronic Viral Infection
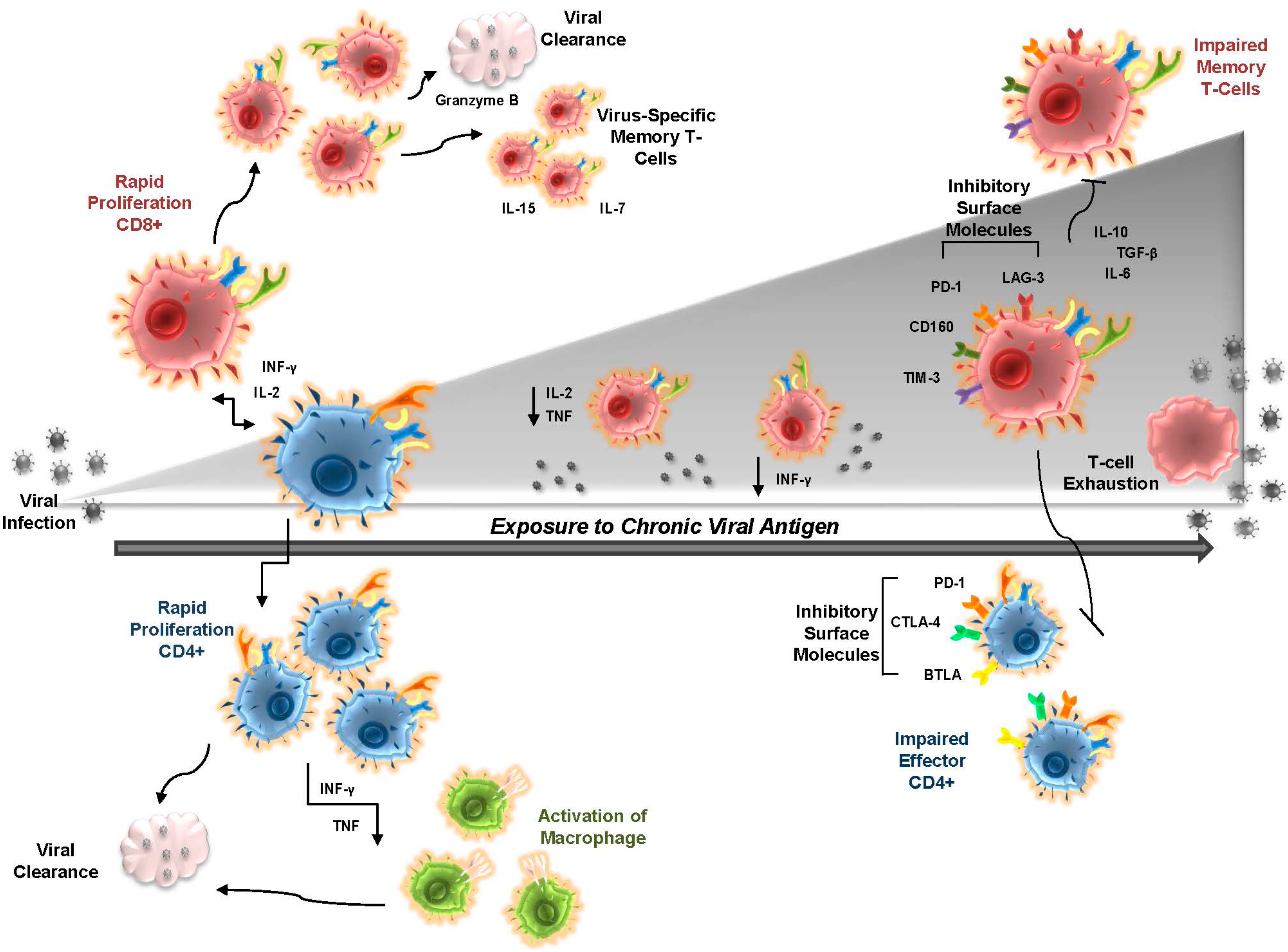
2. T-cell Exhaustion in Chronic Viral Infection
3. Telomere Dynamics and T-cell Exhaustion during Chronic/Productive Viral Infections
4. Telomere Dynamics and T-cell Exhaustion during Chronic/Latent Viral Infections
5. Concluding Remarks
Acknowledgments
Author contributions
Conflicts of Interest
References
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Albrecht, T.; Porter, D.D. Persistent Viral Infections. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Kahan, S.M.; Wherry, E.J.; Zajac, A.J. T cell exhaustion during persistent viral infections. Virology 2015, 479–480, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naive to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.T.; Ernst, B.; Kieper, W.C.; LeRoy, E.; Sprent, J.; Surh, C.D. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J. Exp. Med. 2002, 195, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Blackburn, S.D.; Blattman, J.N.; Wherry, E.J. Viral antigen and extensive division maintain virus-specific CD8 T cells during chronic infection. J. Exp. Med. 2007, 204, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Rufer, N.; Dragowska, W.; Thornbury, G.; Roosnek, E.; Lansdorp, P.M. Telomere length dynamics in human lymphocyte subpopulations measured by flow cytometry. Nat. Biotechnol. 1998, 16, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Soares, M.V.; Zilch, C.F.; Akbar, A.N.; Beverley, P.C. Virus-induced CD8+ T cell clonal expansion is associated with telomerase up-regulation and telomere length preservation: A mechanism for rescue from replicative senescence. J. Immunol. 1999, 162, 4521–4526. [Google Scholar] [PubMed]
- Hathcock, K.S.; Kaech, S.M.; Ahmed, R.; Hodes, R.J. Induction of telomerase activity and maintenance of telomere length in virus-specific effector and memory CD8+ T cells. J. Immunol. 2003, 170, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, F.J.; Soares, M.V.; Annels, N.; Hislop, A.; Ivory, K.; Lowdell, M.; Salmon, M.; Rickinson, A.; Akbar, A.N. The flow cytometric analysis of telomere length in antigen-specific CD8+ T cells during acute Epstein-Barr virus infection. Blood 2001, 97, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, G.; Pehrson, P.O.; Larsson, S.; Ahrlund-Richter, L.; Britton, S. Telomere loss in peripheral blood mononuclear cells may be moderately accelerated during highly active antiretroviral therapy (HAART). J. Acquir. Immune Defic. Syndr. 1999, 22, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, L.; Ricoul, M.; Pottier, G.; Murnane, J.P. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol. Cancer Res. 2005, 3, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Hemann, M.T.; Strong, M.A.; Hao, L.Y.; Greider, C.W. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001, 107, 67–77. [Google Scholar] [CrossRef]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef]
- Weng, N.P.; Levine, B.L.; June, C.H.; Hodes, R.J. Human naive and memory T lymphocytes differ in telomeric length and replicative potential. Proc. Natl. Acad. Sci. USA 1995, 92, 11091–11094. [Google Scholar] [CrossRef] [PubMed]
- Barsov, E.V. Telomerase and primary T cells: Biology and immortalization for adoptive immunotherapy. Immunotherapy 2011, 3, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Weng, N.P. Telomere and adaptive immunity. Mech. Ageing Dev. 2008, 129, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, H.F.; Effros, R.B. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin. Immunol. 2002, 105, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Moro-Garcia, M.A.; Alonso-Arias, R.; Lopez-Larrea, C. Molecular mechanisms involved in the aging of the T-cell immune response. Curr. Genom. 2012, 13, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D.; Blaser, C.; Brawand, P.; Raulet, D.H.; Hanke, T.; Pircher, H. Viral infections induce abundant numbers of senescent CD8 T cells. J. Immunol. 2001, 167, 4838–4843. [Google Scholar] [CrossRef] [PubMed]
- Effros, R.B. Replicative senescence of CD8 T cells: Potential effects on cancer immune surveillance and immunotherapy. Cancer Immunol. Immunother. 2004, 53, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Hooijberg, E.; Ruizendaal, J.J.; Snijders, P.J.; Kueter, E.W.; Walboomers, J.M.; Spits, H. Immortalization of human CD8+ T cell clones by ectopic expression of telomerase reverse transcriptase. J. Immunol. 2000, 165, 4239–4245. [Google Scholar] [CrossRef] [PubMed]
- Bandres, E.; Merino, J.; Vazquez, B.; Inoges, S.; Moreno, C.; Subira, M.L.; Sanchez-Ibarrola, A. The increase of IFN-γ production through aging correlates with the expanded CD8(+high)CD28(−)CD57(+) subpopulation. Clin. Immunol. 2000, 96, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Weekes, M.P.; Wills, M.R.; Mynard, K.; Hicks, R.; Sissons, J.G.; Carmichael, A.J. Large clonal expansions of human virus-specific memory cytotoxic T lymphocytes within the CD57+ CD28− CD8+ T-cell population. Immunology 1999, 98, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28− and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Verges, S.; Milush, J.M.; Pandey, S.; York, V.A.; Arakawa-Hoyt, J.; Pircher, H.; Norris, P.J.; Nixon, D.F.; Lanier, L.L. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood 2010, 116, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, F.J.; Franzese, O.; Finney, H.M.; Fletcher, J.M.; Belaramani, L.L.; Salmon, M.; Dokal, I.; Webster, D.; Lawson, A.D.; Akbar, A.N. The loss of telomerase activity in highly differentiated CD8+CD28−CD27− T cells is associated with decreased Akt (Ser473) phosphorylation. J. Immunol. 2007, 178, 7710–7719. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Franzese, O.; Macaulay, R.; Libri, V.; Azevedo, R.I.; Kiani-Alikhan, S.; Plunkett, F.J.; Masters, J.E.; Jackson, S.; Griffiths, S.J.; et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood 2009, 113, 6619–6628. [Google Scholar] [CrossRef] [PubMed]
- Larbi, A.; Fulop, T. From “truly naive” to “exhausted senescent” T cells: When markers predict functionality. Cytom. A 2014, 85, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Dagarag, M.; Evazyan, T.; Rao, N.; Effros, R.B. Genetic manipulation of telomerase in HIV-specific CD8+ T cells: Enhanced antiviral functions accompany the increased proliferative potential and telomere length stabilization. J. Immunol. 2004, 173, 6303–6311. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, G.M.; Wills, M.R.; Appay, V.; O’Callaghan, C.; Murphy, M.; Smith, N.; Sissons, P.; Rowland-Jones, S.; Bell, J.I.; Moss, P.A. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J. Virol. 2000, 74, 8140–8150. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Dunbar, P.R.; Callan, M.; Klenerman, P.; Gillespie, G.M.; Papagno, L.; Ogg, G.S.; King, A.; Lechner, F.; Spina, C.A.; et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 2002, 8, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Akbar, A.N. KLRG1—More than a marker for T cell senescence. Age 2009, 31, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Murnane, J.P. Telomeres, chromosome instability and cancer. Nucleic Acids Res. 2006, 34, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Plentz, R.R.; Schlegelberger, B.; Flemming, P.; Gebel, M.; Kreipe, H.; Manns, M.P.; Rudolph, K.L.; Wilkens, L. Telomere shortening correlates with increasing aneuploidy of chromosome 8 in human hepatocellular carcinoma. Hepatology 2005, 42, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Beverley, P.C.; Salmon, M. Will telomere erosion lead to a loss of T-cell memory? Nat. Rev. Immunol. 2004, 4, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Perillo, N.L.; Naeim, F.; Walford, R.L.; Effros, R.B. The in vitro senescence of human T lymphocytes: Failure to divide is not associated with a loss of cytolytic activity or memory T cell phenotype. Mech. Ageing Dev. 1993, 67, 173–185. [Google Scholar] [CrossRef]
- Spaulding, C.; Guo, W.; Effros, R.B. Resistance to apoptosis in human CD8+ T cells that reach replicative senescence after multiple rounds of antigen-specific proliferation. Exp. Gerontol. 1999, 34, 633–644. [Google Scholar] [CrossRef]
- Vallejo, A.N.; Schirmer, M.; Weyand, C.M.; Goronzy, J.J. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J. Immunol. 2000, 165, 6301–6307. [Google Scholar] [CrossRef] [PubMed]
- Strauss, G.; Knape, I.; Melzner, I.; Debatin, K.M. Constitutive caspase activation and impaired death-inducing signaling complex formation in CD95-resistant, long-term activated, antigen-specific T cells. J. Immunol. 2003, 171, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ramachandran, S.; Mann, M.; Popkin, D.L. Role of lymphocytic choriomeningitis virus (LCMV) in understanding viral immunology: Past, present and future. Viruses 2012, 4, 2650–2669. [Google Scholar] [CrossRef] [PubMed]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Shankar, P.; Xu, Z.; Harnisch, B.; Chen, G.; Lange, C.; Lee, S.J.; Valdez, H.; Lederman, M.M.; Lieberman, J. Most antiviral CD8 T cells during chronic viral infection do not express high levels of perforin and are not directly cytotoxic. Blood 2003, 101, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Suresh, M.; Glass, A.; Galvan, M.; Chow, K.; Whitmire, J.K.; Walsh, C.M.; Clark, W.R.; Ahmed, R. A role for perforin in downregulating T-cell responses during chronic viral infection. J. Virol. 1999, 73, 2527–2536. [Google Scholar] [PubMed]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhong, S.; Ma, Z.; Kong, H.; Medvec, A.; Ahmed, R.; Freeman, G.J.; Krogsgaard, M.; Riley, J.L. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl. Acad. Sci. USA 2013, 110, E2480–E2489. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.J.; Chatzidakis, I.; Tsoukatou, D.; Boumpas, D.T.; Garinis, G.A.; Mamalaki, C. Programmed death-1 shapes memory phenotype CD8 T cell subsets in a cell-intrinsic manner. J. Immunol. 2013, 190, 6104–6114. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.; Angelosanto, J.M.; Kao, C.; Doering, T.A.; Odorizzi, P.M.; Barnett, B.E.; Wherry, E.J. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 2014, 40, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Park, J.S.; Jeong, Y.H.; Son, J.; Ban, Y.H.; Lee, B.H.; Chen, L.; Chang, J.; Chung, D.H.; Choi, I.; et al. PD-1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD-L1 expressed on CD8+ T cells. J. Immunol. 2015, 194, 5801–5811. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: From pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. 2017, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Mehraj, V.; Kaufmann, D.E.; Li, T.; Routy, J.P. Elevation and persistence of CD8 T-cells in HIV infection: The Achilles heel in the ART era. J. Int. AIDS Soc. 2016, 19, 20697. [Google Scholar] [CrossRef] [PubMed]
- El-Far, M.; Halwani, R.; Said, E.; Trautmann, L.; Doroudchi, M.; Janbazian, L.; Fonseca, S.; van Grevenynghe, J.; Yassine-Diab, B.; Sekaly, R.P.; et al. T-cell exhaustion in HIV infection. Curr. HIV/AIDS Rep. 2008, 5, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, A.; Qiu, C.; Wang, W.; Yang, Y.; Qiu, C.; Liu, A.; Zhu, L.; Yuan, S.; Hu, H.; et al. The upregulation of LAG-3 on T cells defines a subpopulation with functional exhaustion and correlates with disease progression in HIV-infected subjects. J. Immunol. 2015, 194, 3873–3882. [Google Scholar] [CrossRef] [PubMed]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Petrovas, C.; Casazza, J.P.; Brenchley, J.M.; Price, D.A.; Gostick, E.; Adams, W.C.; Precopio, M.L.; Schacker, T.; Roederer, M.; Douek, D.C.; et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006, 203, 2281–2292. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Badley, A.D. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis. 2010, 1, e99. [Google Scholar] [CrossRef] [PubMed]
- Holm, G.H.; Zhang, C.; Gorry, P.R.; Peden, K.; Schols, D.; de Clercq, E.; Gabuzda, D. Apoptosis of bystander T cells induced by human immunodeficiency virus type 1 with increased envelope/receptor affinity and coreceptor binding site exposure. J. Virol. 2004, 78, 4541–4551. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Mudd, J.C.; Lederman, M.M. CD8 T cell persistence in treated HIV infection. Curr. Opin. HIV AIDS 2014, 9, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Konstantinou, D.; Deutsch, M. The spectrum of HBV/HCV coinfection: Epidemiology, clinical characteristics, viralinteractions and management. Ann. Gastroenterol. 2015, 28, 221–228. [Google Scholar] [PubMed]
- Monteiro, J.; Batliwalla, F.; Ostrer, H.; Gregersen, P.K. Shortened telomeres in clonally expanded CD28-CD8+ T cells imply a replicative history that is distinct from their CD28+CD8+ counterparts. J. Immunol. 1996, 156, 3587–3590. [Google Scholar] [PubMed]
- Gianesin, K.; Noguera-Julian, A.; Zanchetta, M.; Del Bianco, P.; Petrara, M.R.; Freguja, R.; Rampon, O.; Fortuny, C.; Camos, M.; Mozzo, E.; et al. Premature aging and immune senescence in HIV-infected children. AIDS 2016, 30, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.D.; Weng, N.; Levine, B.L.; June, C.H.; Lane, H.C.; Hodes, R.J. Telomere length, telomerase activity, and replicative potential in HIV infection: Analysis of CD4+ and CD8+ T cells from HIV-discordant monozygotic twins. J. Exp. Med. 1997, 185, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Lichterfeld, M.; Mou, D.; Cung, T.D.; Williams, K.L.; Waring, M.T.; Huang, J.; Pereyra, F.; Trocha, A.; Freeman, G.J.; Rosenberg, E.S.; et al. Telomerase activity of HIV-1-specific CD8+ T cells: Constitutive up-regulation in controllers and selective increase by blockade of PD ligand 1 in progressors. Blood 2008, 112, 3679–3687. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.H.; Hsu, J.L.; Su, I.J.; Huang, W. Genomic instability caused by hepatitis B virus: Into the hepatoma inferno. Front. Biosci. (Landmark Ed.) 2011, 16, 2586–2597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, J.; Ming, W.; Lee, Y.P.; Santella, R.M. Telomere length in hepatocellular carcinoma and paired adjacent non-tumor tissues by quantitative PCR. Cancer Investig. 2007, 25, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Horikawa, I.; Nishimoto, A.; Ohmura, H.; Ito, H.; Hirohashi, S.; Shay, J.W.; Oshimura, M. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet. Cytogenet. 1997, 93, 56–62. [Google Scholar] [CrossRef]
- Barnes, E.; Ward, S.M.; Kasprowicz, V.O.; Dusheiko, G.; Klenerman, P.; Lucas, M. Ultra-sensitive class I tetramer analysis reveals previously undetectable populations of antiviral CD8+ T cells. Eur. J. Immunol. 2004, 34, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Satra, M.; Dalekos, G.N.; Kollia, P.; Vamvakopoulos, N.; Tsezou, A. Telomerase reverse transcriptase mRNA expression in peripheral lymphocytes of patients with chronic HBV and HCV infections. J. Viral Hepat. 2005, 12, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Yu, D.; Li, B.; Luo, O.Y.; Xu, T.; Cao, Y.; Ding, Y. Paired assessment of liver telomere lengths in hepatocellular cancer is a reliable predictor of disease persistence. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Gelson, W.T.; Das, A.; Fletcher, J.M.; Davies, S.E.; Curran, M.D.; Vowler, S.L.; Maini, M.K.; Akbar, A.N.; Alexander, G.J. CD4+ T-lymphocyte telomere length is related to fibrosis stage, clinical outcome and treatment response in chronic hepatitis C virus infection. J. Hepatol. 2010, 53, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wan, S.; Hann, H.W.; Myers, R.E.; Hann, R.S.; Au, J.; Chen, B.; Xing, J.; Yang, H. Relative telomere length: A novel non-invasive biomarker for the risk of non-cirrhotic hepatocellular carcinoma in patients with chronic hepatitis B infection. Eur. J. Cancer 2012, 48, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Grady, B.P.; Nanlohy, N.M.; van Baarle, D. HCV monoinfection and HIV/HCV coinfection enhance T-cell immune senescence in injecting drug users early during infection. Immun. Ageing 2016, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Barili, V.; Montanini, B.; Acerbi, G.; Ferracin, M.; Guerrieri, F.; Salerno, D.; Boni, C.; Massari, M.; Cavallo, M.C.; et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017, 23, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Ngom, P.T.; Solon, J.; Moore, S.E.; Morgan, G.; Prentice, A.M.; Aspinall, R. Thymic function and T cell parameters in a natural human experimental model of seasonal infectious diseases and nutritional burden. J. Biomed. Sci. 2011, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Shankar, A.; Shah, M.; Rushbrook, S.; Gelson, W.; Davies, S.; Akbar, A.; Alexander, G.J. gamma-H2AX+CD8+ T lymphocytes cannot respond to IFN-α, IL-2 or IL-6 in chronic hepatitis C virus infection. J. Hepatol. 2013, 58, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Vukmanovic-Stejic, M.; Dunne, P.J.; Birch, K.E.; Cook, J.E.; Jackson, S.E.; Salmon, M.; Rustin, M.H.; Akbar, A.N. Cytomegalovirus-specific CD4+ T cells in healthy carriers are continuously driven to replicative exhaustion. J. Immunol. 2005, 175, 8218–8225. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.R.; Vukmanovic-Stejic, M.; Fletcher, J.M.; Soares, M.V.; Cook, J.E.; Orteu, C.H.; Jackson, S.E.; Birch, K.E.; Foster, G.R.; Salmon, M.; et al. Telomere erosion in memory T cells induced by telomerase inhibition at the site of antigenic challenge in vivo. J. Exp. Med. 2004, 199, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Boettler, T.; Panther, E.; Bengsch, B.; Nazarova, N.; Spangenberg, H.C.; Blum, H.E.; Thimme, R. Expression of the interleukin-7 receptor alpha chain (CD127) on virus-specific CD8+ T cells identifies functionally and phenotypically defined memory T cells during acute resolving hepatitis B virus infection. J. Virol. 2006, 80, 3532–3540. [Google Scholar] [CrossRef] [PubMed]
- Mojumdar, K.; Vajpayee, M.; Chauhan, N.K.; Singh, A.; Singh, R.; Kurapati, S. Loss of CD127 & increased immunosenescence of T cell subsets in HIV infected individuals. Indian J. Med. Res. 2011, 134, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef] [PubMed]
- Bucks, C.M.; Norton, J.A.; Boesteanu, A.C.; Mueller, Y.M.; Katsikis, P.D. Chronic antigen stimulation alone is sufficient to drive CD8+ T cell exhaustion. J. Immunol. 2009, 182, 6697–6708. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Nicot, C. Regulation of telomerase and telomeres: Human tumor viruses take control. J. Natl. Cancer Inst. 2008, 100, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cosyns, M.; Levin, M.J.; Hayward, A.R. Cytokine production in varicella zoster virus-stimulated limiting dilution lymphocyte cultures. Clin. Exp. Immunol. 1994, 98, 128–133. [Google Scholar] [CrossRef] [PubMed]
- James, S.F.; Traina-Dorge, V.; Deharo, E.; Wellish, M.; Palmer, B.E.; Gilden, D.; Mahalingam, R. T cells increase before zoster and PD-1 expression increases at the time of zoster in immunosuppressed nonhuman primates latently infected with simian varicella virus. J. Neurovirol. 2014, 20, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Hamrah, P.; Gate, D.; Mott, K.R.; Mantopoulos, D.; Zheng, L.; Town, T.; Jones, C.; von Andrian, U.H.; Freeman, G.J.; et al. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus 1. J. Virol. 2011, 85, 4184–4197. [Google Scholar] [CrossRef] [PubMed]
- O’Bryan, J.M.; Woda, M.; Co, M.; Mathew, A.; Rothman, A.L. Telomere length dynamics in human memory T cells specific for viruses causing acute or latent infections. Immun. Ageing 2013, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, D.U.; Proietti, F.A.; Ribas, J.G.; Araujo, M.G.; Pinheiro, S.R.; Guedes, A.C.; Carneiro-Proietti, A.B. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin. Microbiol. Rev. 2010, 23, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Goon, P.K.; Biancardi, A.; Fast, N.; Igakura, T.; Hanon, E.; Mosley, A.J.; Asquith, B.; Gould, K.G.; Marshall, S.; Taylor, G.P.; et al. Human T cell lymphotropic virus (HTLV) type-1-specific CD8+ T cells: Frequency and immunodominance hierarchy. J. Infect. Dis. 2004, 189, 2294–2298. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Yamano, Y.; Brennan, M.B.; Mora, C.A.; Jacobson, S. Increased HTLV-I proviral load and preferential expansion of HTLV-I Tax-specific CD8+ T cells in cerebrospinal fluid from patients with HAM/TSP. Ann. Neurol. 2001, 50, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Yoshimitsu, M.; Fujiwara, H.; Masamoto, I.; Horai, S.; White, Y.; Akimoto, M.; Suzuki, S.; Matsushita, K.; Uozumi, K.; et al. PD-1/PD-L1 expression in human T-cell leukemia virus type 1 carriers and adult T-cell leukemia/lymphoma patients. Leukemia 2009, 23, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Arima, N.; Toji, S.; Masamoto, I.; Akimoto, M.; Hamada, H.; Che, X.F.; Fujiwara, H.; Matsushita, K.; Tokunaga, M.; et al. Reduced frequency, diversity, and function of human T cell leukemia virus type 1-specific CD8+ T cell in adult T cell leukemia patients. J. Immunol. 2006, 177, 5718–5726. [Google Scholar] [CrossRef] [PubMed]
- Takamori, A.; Hasegawa, A.; Utsunomiya, A.; Maeda, Y.; Yamano, Y.; Masuda, M.; Shimizu, Y.; Tamai, Y.; Sasada, A.; Zeng, N.; et al. Functional impairment of Tax-specific but not cytomegalovirus-specific CD8+ T lymphocytes in a minor population of asymptomatic human T-cell leukemia virus type 1-carriers. Retrovirology 2011, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Sabouri, A.H.; Usuku, K.; Hayashi, D.; Izumo, S.; Ohara, Y.; Osame, M.; Saito, M. Impaired function of human T-lymphotropic virus type 1 (HTLV-1)-specific CD8+ T cells in HTLV-1-associated neurologic disease. Blood 2008, 112, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Abdelbary, N.H.; Abdullah, H.M.; Matsuzaki, T.; Hayashi, D.; Tanaka, Y.; Takashima, H.; Izumo, S.; Kubota, R. Reduced Tim-3 expression on human T-lymphotropic virus type I (HTLV-I) Tax-specific cytotoxic T lymphocytes in HTLV-I infection. J. Infect. Dis. 2011, 203, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Ezinne, C.C.; Yoshimitsu, M.; White, Y.; Arima, N. HTLV-1 specific CD8+ T cell function augmented by blockade of 2B4/CD48 interaction in HTLV-1 infection. PLoS ONE 2014, 9, e87631. [Google Scholar] [CrossRef] [PubMed]
- Yasuma, K.; Yasunaga, J.; Takemoto, K.; Sugata, K.; Mitobe, Y.; Takenouchi, N.; Nakagawa, M.; Suzuki, Y.; Matsuoka, M. HTLV-1 bZIP Factor Impairs Anti-viral Immunity by Inducing Co-inhibitory Molecule, T Cell Immunoglobulin and ITIM Domain (TIGIT). PLoS Pathog. 2016, 12, e1005372. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Leal, F.E.; Hasenkrug, A.M.; Jha, A.R.; Carvalho, K.I.; Eccles-James, I.G.; Bruno, F.R.; Vieira, R.G.; York, V.A.; Chew, G.M.; et al. HTLV-1 tax specific CD8+ T cells express low levels of Tim-3 in HTLV-1 infection: Implications for progression to neurological complications. PLoS Negl. Trop. Dis. 2011, 5, e1030. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Yoshimitsu, M.; Akimoto, M.; White, Y.; Matsushita, K.; Soeda, S.; Shimeno, H.; Kubota, R.; Izumo, S.; Arima, N. Programmed death-1 (PD-1)/PD-1 ligand pathway-mediated immune responses against human T-lymphotropic virus type 1 (HTLV-1) in HTLV-1-associated myelopathy/tropical spastic paraparesis and carriers with autoimmune disorders. Hum. Immunol. 2011, 72, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Enose-Akahata, Y.; Matsuura, E.; Oh, U.; Jacobson, S. High expression of CD244 and SAP regulated CD8 T cell responses of patients with HTLV-I associated neurologic disease. PLoS Pathog. 2009, 5, e1000682. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.B.; Porto, A.F.; Muniz, A.L.; de Jesus, A.R.; Magalhaes, E.; Melo, A.; Dutra, W.O.; Gollob, K.J.; Carvalho, E.M. Exacerbated inflammatory cellular immune response characteristics of HAM/TSP is observed in a large proportion of HTLV-I asymptomatic carriers. BMC Infect. Dis. 2004, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Nauroth, J.M.; Graber, J.; Yao, K.; Jacobson, S.; Calabresi, P.A. Memory lineage relationships in HTLV-1-specific CD8+ cytotoxic T cells. J. Neuroimmunol. 2006, 176, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Datta, A.; Brown, M.; Pouliquen, J.F.; Couppie, P.; Kazanji, M.; Nicot, C. Increased expression of telomere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia. Int. J. Cancer 2006, 119, 2090–2097. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Nicot, C. Telomere attrition induces a DNA double-strand break damage signal that reactivates p53 transcription in HTLV-I leukemic cells. Oncogene 2008, 27, 1135–1141. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 Blockade Can Restore Functions of T-Cells in Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma In Vitro. PLoS ONE 2015, 10, e0136476. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.; Sauce, D.; Deback, C.; Arnaud, L.; Mathian, A.; Miyara, M.; Boutolleau, D.; Parizot, C.; Dorgham, K.; Papagno, L.; et al. Exhausted cytotoxic control of Epstein-Barr virus in human lupus. PLoS Pathog. 2011, 7, e1002328. [Google Scholar] [CrossRef] [PubMed]
- Loebel, M.; Strohschein, K.; Giannini, C.; Koelsch, U.; Bauer, S.; Doebis, C.; Thomas, S.; Unterwalder, N.; von Baehr, V.; Reinke, P.; et al. Deficient EBV-specific B- and T-cell response in patients with chronic fatigue syndrome. PLoS ONE 2014, 9, e85387. [Google Scholar] [CrossRef] [PubMed]
- Pender, M.P.; Csurhes, P.A.; Burrows, J.M.; Burrows, S.R. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin. Transl. Immunol. 2017, 6, e126. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Annels, N.E.; Callan, M.F.; Tan, L.; Rickinson, A.B. Changing patterns of dominant TCR usage with maturation of an EBV-specific cytotoxic T cell response. J. Immunol. 2000, 165, 4831–4841. [Google Scholar] [CrossRef] [PubMed]
- Van Baarle, D.; Hovenkamp, E.; Callan, M.F.; Wolthers, K.C.; Kostense, S.; Tan, L.C.; Niesters, H.G.; Osterhaus, A.D.; McMichael, A.J.; van Oers, M.H.; et al. Dysfunctional Epstein-Barr virus (EBV)-specific CD8+ T lymphocytes and increased EBV load in HIV-1 infected individuals progressing to AIDS-related non-Hodgkin lymphoma. Blood 2001, 98, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Van Baarle, D.; Nanlohy, N.M.; Otto, S.; Plunkett, F.J.; Fletcher, J.M.; Akbar, A.N. Progressive telomere shortening of Epstein-Barr virus-specific memory T cells during HIV infection: Contributor to exhaustion? J. Infect. Dis. 2008, 198, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, F.J.; Franzese, O.; Belaramani, L.L.; Fletcher, J.M.; Gilmour, K.C.; Sharifi, R.; Khan, N.; Hislop, A.D.; Cara, A.; Salmon, M.; et al. The impact of telomere erosion on memory CD8+ T cells in patients with X-linked lymphoproliferative syndrome. Mech. Ageing Dev. 2005, 126, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Guihot, A.; Dupin, N.; Marcelin, A.G.; Gorin, I.; Bedin, A.S.; Bossi, P.; Galicier, L.; Oksenhendler, E.; Autran, B.; Carcelain, G. Low T cell responses to human herpesvirus 8 in patients with AIDS-related and classic Kaposi sarcoma. J. Infect. Dis. 2006, 194, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vely, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbe, C.; et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [PubMed]
- Unemori, P.; Leslie, K.S.; Hunt, P.W.; Sinclair, E.; Epling, L.; Mitsuyasu, R.; Effros, R.B.; Dock, J.; Dollard, S.G.; Deeks, S.G.; et al. Immunosenescence is associated with presence of Kaposi’s sarcoma in antiretroviral treated HIV infection. AIDS 2013, 27, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Ahmed, R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2009, 106, 8623–8628. [Google Scholar] [CrossRef] [PubMed]
- Cid-Arregui, A.; Juarez, V.; zur Hausen, H. A synthetic E7 gene of human papillomavirus type 16 that yields enhanced expression of the protein in mammalian cells and is useful for DNA immunization studies. J. Virol. 2003, 77, 4928–4937. [Google Scholar] [CrossRef] [PubMed]
- Youde, S.J.; Dunbar, P.R.; Evans, E.M.; Fiander, A.N.; Borysiewicz, L.K.; Cerundolo, V.; Man, S. Use of fluorogenic histocompatibility leukocyte antigen-A*0201/HPV 16 E7 peptide complexes to isolate rare human cytotoxic T-lymphocyte-recognizing endogenous human papillomavirus antigens. Cancer Res. 2000, 60, 365–371. [Google Scholar] [PubMed]
- Nilges, K.; Hohn, H.; Pilch, H.; Neukirch, C.; Freitag, K.; Talbot, P.J.; Maeurer, M.J. Human papillomavirus type 16 E7 peptide-directed CD8+ T cells from patients with cervical cancer are cross-reactive with the coronavirus NS2 protein. J. Virol. 2003, 77, 5464–5474. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Stanley, M.A. Analysis of HLA-DR expression on keratinocytes in cervical neoplasia. Int. J. Cancer 1994, 56, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, F.; Terme, M.; Nizard, M.; Badoual, C.; Bureau, M.F.; Freyburger, L.; Clement, O.; Marcheteau, E.; Gey, A.; Fraisse, G.; et al. Mucosal imprinting of vaccine-induced CD8(+) T cells is crucial to inhibit the growth of mucosal tumors. Sci. Transl. Med. 2013, 5, 172ra120. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Song, Y.; Lu, Y.L.; Sun, J.Z.; Wang, H.W. Increased expression of programmed death (PD)-1 and its ligand PD-L1 correlates with impaired cell-mediated immunity in high-risk human papillomavirus-related cervical intraepithelial neoplasia. Immunology 2013, 139, 513–522. [Google Scholar] [CrossRef] [PubMed]
- James, E.A.; DeVoti, J.A.; Rosenthal, D.W.; Hatam, L.J.; Steinberg, B.M.; Abramson, A.L.; Kwok, W.W.; Bonagura, V.R. Papillomavirus-specific CD4+ T cells exhibit reduced STAT-5 signaling and altered cytokine profiles in patients with recurrent respiratory papillomatosis. J. Immunol. 2011, 186, 6633–6640. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.E.; Latchman, Y.E.; Balint, J.P.; Lee, J.H.; Gabitzsch, E.S.; Jones, F.R. An HPV-E6/E7 immunotherapy plus PD-1 checkpoint inhibition results in tumor regression and reduction in PD-L1 expression. Cancer Gene Ther. 2015, 22, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Badoual, C.; Hans, S.; Merillon, N.; van Ryswick, C.; Ravel, P.; Benhamouda, N.; Levionnois, E.; Nizard, M.; Si-Mohamed, A.; Besnier, N.; et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013, 73, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.T.; Kyo, S.; Laimins, L.A. Telomerase activation by human papillomavirus type 16 E6 protein: Induction of human telomerase reverse transcriptase expression through Myc and GC-rich Sp1 binding sites. J. Virol. 2001, 75, 5559–5566. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Roberts, J.; Dakic, A.; Zhang, Y.; Schlegel, R. HPV E7 contributes to the telomerase activity of immortalized and tumorigenic cells and augments E6-induced hTERT promoter function. Virology 2008, 375, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Plug-DeMaggio, A.W.; Sundsvold, T.; Wurscher, M.A.; Koop, J.I.; Klingelhutz, A.J.; McDougall, J.K. Telomere erosion and chromosomal instability in cells expressing the HPV oncogene 16E6. Oncogene 2004, 23, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Wang, J.; Zheng, B.; Fang, X.; Angstrom, T.; Liu, C.; Li, X.; Erlandsson, F.; Bjorkholm, M.; Nordenskjord, M.; et al. Telomere attrition predominantly occurs in precursor lesions during in vivo carcinogenic process of the uterine cervix. Oncogene 2004, 23, 7441–7447. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sturgis, E.M.; Dahlstrom, K.R.; Wen, J.; Liu, H.; Wei, Q.; Li, G.; Liu, Z. Telomere length in peripheral blood lymphocytes contributes to the development of HPV-associated oropharyngeal carcinoma. Cancer Res. 2013, 73, 5996–6003. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Yang, J.; Liu, B.; Li, W.; Hu, G.; Qiu, H.; Huang, L.; Xiong, H.; Yuan, X. Combined effects of leukocyte telomere length, p53 polymorphism and human papillomavirus infection on esophageal squamous cell carcinoma in a Han Chinese population. Cancer Epidemiol. 2014, 38, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Amos, C.I.; Zhu, Y.; Zhao, H.; Grossman, B.H.; Shay, J.W.; Luo, S.; Hong, W.K.; Spitz, M.R. Telomere dysfunction: A potential cancer predisposition factor. J. Natl. Cancer Inst. 2003, 95, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Charmoy, M.; Chennupati, V.; Pousse, L.; Ferreira, D.P.; Calderon-Copete, S.; Danilo, M.; Alfei, F.; Hofmann, M.; Wieland, D.; et al. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 2016, 45, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Cecchinato, V.; Tryniszewska, E.; Ma, Z.M.; Vaccari, M.; Boasso, A.; Tsai, W.P.; Petrovas, C.; Fuchs, D.; Heraud, J.M.; Venzon, D.; et al. Immune activation driven by CTLA-4 blockade augments viral replication at mucosal sites in simian immunodeficiency virus infection. J. Immunol. 2008, 180, 5439–5447. [Google Scholar] [CrossRef] [PubMed]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009, 458, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, H.T.; Tsai, H.F.; Liao, H.J.; Lin, Y.J.; Chen, L.; Chen, P.J.; Hsu, P.N. PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PLoS ONE 2012, 7, e39179. [Google Scholar] [CrossRef] [PubMed]
- Ivashko, I.N.; Kolesar, J.M. Pembrolizumab and nivolumab: PD-1 inhibitors for advanced melanoma. Am. J. Health Syst. Pharm. 2016, 73, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.K.; Salem, M.E.; Nimeiri, H.; Iqbal, S.; Singh, P.; Ciombor, K.; Polite, B.; Deming, D.; Chan, E.; Wade, J.L.; et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 446–453. [Google Scholar] [CrossRef]
- Ma, S.D.; Xu, X.; Jones, R.; Delecluse, H.J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 blockade inhibits Epstein-Barr virus-induced lymphoma growth in a cord blood humanized-mouse model. PLoS Pathog. 2016, 12, e1005642. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Carrette, F.; Barraza, M.L.; Otero, D.C.; Magana, J.; Bosenberg, M.W.; Swain, S.L.; Bradley, L.M. PSGL-1 is an immune checkpoint regulator that promotes T cell exhaustion. Immunity 2016, 44, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T.; Dumitriu, B. Telomere dynamics in mice and humans. Semin. Hematol. 2013, 50, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M. The discovery of HTLV-1, the first pathogenic human retrovirus. Proc. Natl. Acad. Sci. USA 2015, 112, 15525–15529. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellon, M.; Nicot, C. Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses 2017, 9, 289. https://doi.org/10.3390/v9100289
Bellon M, Nicot C. Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses. 2017; 9(10):289. https://doi.org/10.3390/v9100289
Chicago/Turabian StyleBellon, Marcia, and Christophe Nicot. 2017. "Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection" Viruses 9, no. 10: 289. https://doi.org/10.3390/v9100289
APA StyleBellon, M., & Nicot, C. (2017). Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses, 9(10), 289. https://doi.org/10.3390/v9100289